

Abstract
The PDL1: PD1 costimulatory pathway plays an important role in the inhibition of alloimmune responses as well as in the induction and maintenance of peripheral tolerance. It has recently been demonstrated that PDL1 can also bind B7.1 to inhibit T cell responses in vitro. Using the bm12 into B6 heart transplant model, we investigated the functional significance of this interaction in alloimmune responses in vivo. PD1 blockade unlike PDL1 blockade failed to accelerate bm12 allograft rejection suggesting a role for an additional binding partner for PDL1 other than PD1 in transplant rejection. PDL1 blockade was able to accelerate allograft rejection in B7.2-deficient recipients but not B7.1-deficient recipients, indicating that PDL1 interaction with B7.1 was important in inhibiting rejection. Administration of the novel 2H11 anti-PDL1 mAb, which only blocks PDL1: B7.1 interaction, aggravated chronic injury of bm12 allografts in B6 recipients. Aggravated chronic injury was associated with an increased frequency of alloreactive IFN-γ-, IL-4-, and IL-6-producing splenocytes and a decreased percentage of regulatory T cells in the recipients. Using an in vitro cell culture assay, blockade of the interaction of PDL1 on dendritic cells with B7.1 on T cells increased IFN-γ production from alloreactive CD4+ T cells, whereas blockade of dendritic cell B7.1 interaction with T cell PDL1 did not. These data indicate that PDL1 interaction with B7.1 plays an important role in the inhibition of alloimmune responses in vivo and suggests a dominant direction for PDL1 and B7.1 interaction.
Keywords: Costimulation, Transplantation, MHC, Knockout mice
INTRODUCTION
The programmed death (PD) 1: PDL (PD-ligand) 1/PDL2 pathway is an important negative costimulatory pathway that plays a key role in the inhibition of alloimmune responses and the induction and maintenance of peripheral tolerance (1-4). In vitro studies have shown that engagement of PD1 by its ligands inhibits proliferation and cytokine production by antigen-specific CD4+ and CD8+ T cells (5). In transplant models, administration of blocking anti-PDL1 mAb enhances alloantigen-driven T-cell expansion in vivo, promotes Th1 differentiation and accelerates graft rejection (6). Furthermore, administration of a PDL1-Ig fusion protein that triggers PD1 negative signaling prevents allograft rejection and facilitates tolerance induction when combined with anti-CD154 or suboptimal doses of rapamycin (7). These studies demonstrate the negative regulatory function of the PD1 pathway in organ transplantation. A number of lines of evidence have suggested a second receptor for PDL1 aside from PD1. Recently, B7.1 has been identified as a second binding partner for PDL1 (8). The affinity of B7.1 for PD-L1 is intermediate between the affinities of B7.1 for CD28 and CTLA-4, but three fold less than the affinity of PDL1 for PD1. The PDL1: B7.1 interaction was found to induce an inhibitory signal into T cells in vitro (8). This signal could occur either by ligation of PDL1 on CD4+ T cells with B7.1, or ligation of B7.1 on CD4+ T cells with PDL1 (9). These results demonstrate a specific and significant bidirectional interaction between B7.1 and PDL1 that inhibits T cell responses.
In light of the identification of the PDL1: B7.1 interaction, a reassessment of the roles of B7.1 and PDL1 in regulating alloimmune responses is necessary. In the present study, an anti-PDL1 antibody (10F.2H11) that solely blocks the PDL1: B7.1 interaction is used to investigate the function of this pathway in a murine cardiac transplant model. Furthermore, the mechanism of PDL1: B7.1 mediated regulation of alloimmune responses is explored.
MATERIALS AND METHODS
Mice
C57BL/6 (H2b, B6), B6.C-H2bm12/KhEgJ (bm12), B6.129S4-Cd80tm1Shr/J (B7.1KO), and B6.129S4-Cd86tm1Shr/J (B7.2KO) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). PD1−/− (10), PDL1−/− (11) mice on the B6 background, and PDL1-/- on the BALB/c background (12) were maintained as a breeding colony in our animal facility. All mice were 8–12 weeks of age and housed in accordance with institutional and National Institutes of Health guidelines.
Heterotopic heart transplantation
Vascularized heart grafts were placed in an intra-abdominal location using microsurgical techniques as described by Corry et al. (13). Graft function was assessed by palpation of the heartbeat. Rejection was determined by complete cessation of palpable heartbeat and was confirmed by direct visualization after laparotomy. Graft survival is shown as the median survival time (MST) in days.
Antibodies and in vivo treatment protocol
The anti-mouse PDL1 mAb (MIH6) was generated by M. Azuma, which blocks both PDL1: PD1 and PDL1: B7.1 interactions (14). The anti-mouse PD1 mAb (J43) was generated by H. Yagita. We have previously demonstrated the blocking properties of the mAbs against PD-L1 and PD1 (15, 16). Both mAbs were manufactured and purified from the original hybridomas by a commercial source, Bio-X-Cell Culture (West Lebanon, NH). The anti-PDL1 mAb 10F.2H11 (2H11) solely blocks PDL1: B7.1 interaction (9). PDL1 and PD1 mAbs were given i.p. according to the following protocol: 0.5 mg of mAb on the day of transplantation and 0.25 mg on days 2, 4, 6, and 8 after transplantation.
ELISPOT
Splenocytes harvested at 10 -14 days after transplantation from WT B6, PD1KO, B7.1KO and B7.2KO recipients of bm12 heart allografts were restimulated with irradiated donor-type splenocytes. The ELISPOT assay (R&D Systems, Minneapolis, MN) was adapted to measure the frequency of alloreactive T cells producing interferon (IFN)-γ and interleukin (IL)-4 as described previously(17). The frequencies of cytokine-secreting alloreactive cells were expressed as the number of cytokine-producing cells per 0.5 ×106 responder cells.
Flow Cytometry
Splenocytes from WT B6, PD1KO, B7.1KO and B7.2KO recipients of bm 12 hearts at 10 days after transplantation were stained with fluorochrome-labeled monoclonal antibodies (mAbs) against CD4, CD8, CD62 ligand (CD62L), CD44, CD25, and FoxP3 (BD Biosciences, San Jose, CA). Intracellular FoxP3 staining was performed using the Cytofix/Cytoperm intracellular staining kit. Flow cytometry was performed with a FACSCalibur system (BD Biosciences) and analyzed using FlowJo software, assessing regulatory T cells (CD4+CD25+FoxP3+) as well as CD4+ and CD8+ effector memory (CD44high CD62Llow phenotype) and central memory (CD44high CD62Lhigh phenotype) cells.
In vitro Culture Assay
B6 wild-type skin was transplanted into Balb/c WT and PDL1KO recipients in order to sensitize T cells to alloantigen. Fourteen days after transplantation, spleens and lymph nodes from recipients were harvested and CD4+ T cells were isolated by magnetic activated sorting by a CD4+ T cell Isolation Kit (Miltenyi Biotec). Sensitized CD4+ T cells (0.1 × 106) were then cultured with irradiated CD11c+ cells from Flt-3-ligand injected B6 WT or PDL1KO, which were purified by MACS- (130-052-001; Miltenyi Biotec). The purity of CD4+ and CD11c+ cells was estimated to be greater than 95% by FACS. 2H11 or IgG control was added to wells at the optimized concentration of 40 μg/ml. Cell-free supernatants of individual wells were removed after 48 h of incubation and analyzed by a multiplexed cytokine bead–based immunoassay using a preconfigured 21-plex mouse cytokine detection kit (Millipore, Billerica, MA) as described above.
Morphology
Cardiac graft samples from transplanted mice were harvested at 14 and 56 days after transplantation, then fixed in 10% formalin, embedded in paraffin, coronally sectioned, and stained with H&E for evaluation of the degree of rejection according to International Society of Heart and Lung Transplantation (ISHLT) guidelines and elastin to evaluate vasculopathy according to the percentage of luminal occlusion by intimal thickening with a scoring system described previously (17) by light microscopy. An examiner blinded to the groups read all the samples. Digital photomicrographs were acquired using a Nikon TE300 microscope and SPOT software (Diagnostic Instruments, Sterling Heights MI). Images were adjusted for brightness, contrast and color balance only using Photoshop (Adobe Systems, San Jose, CA) software. Magnification 20x with bars of 200μm depicted on photos.
Immunohistochemistry was performed on 5-μm-thick, formalin-fixed, paraffin-embedded tissue sections with anti-CD3 (CMC363, Cell Marque, Rocklin, CA; 1:1500, EDTA) and anti-FoxP3 (no. 14-5773, eBioscience; 1:25, citrate) primary Abs. All stained slides were scanned using an Aperio ScanScope XT (Aperio Technologies, Vista, CA). Images were analyzed using ImageScope software (version 10.0.35.1800; Aperio Technologies) and a standard analysis algorithm (nuclear version 9.0; Aperio Technologies) with appropriate modifications for cell and nuclear sizes.
Statistical analysis
Graft survival was expressed graphically using the Kaplan-Meier method, and statistical differences in survival between the groups were assessed by the log-rank test. Student’s t-test was used for comparison of means. A p<0.05 was considered statistically significant.
RESULTS
PDL1 has an additional binding partner other than PD1 during alloimmune responses
We have previously demonstrated that blockade of PD1: PDL1 pathway by the PDL1 dual-blocker MIH6 mAb resulted in accelerated rejection in the bm12 into B6 cardiac transplant model (17). In this single MHC class II-mismatched transplant model, cardiac allografts are not acutely rejected but survive long term, and develop severe transplant arteriosclerosis within 8 weeks after transplantation (17, 18). To compare the effects of PD1 and PDL1 blockade, we use dual-blocker anti-PDL1 mAbs (MIH6) and anti-PD1 monoclonal antibodies following transplant of bm12 hearts into B6 recipients. PD1 blockade was not as effective at shortening graft survival as PDL1 blockade (MIH6) (Figure 1A). These results suggest that PDL1 may have an additional binding partner other than PD1. To test this hypothesis, we transplanted bm12 hearts into PD1-deficient B6 recipients and treated these recipients with the MIH6 anti-PDL1 mAb. Similar to anti-PD1 mAb treatment, PD1- deficient recipients did not accelerate bm12 graft rejection. Remarkably, PDL1 blockade accelerated allograft rejection in the PD1-deficient recipients (MST= 18.5 days, P=0.0183) similar to the rejection in wild-type recipients given anti-PDL1 (Figure 1A,1B). While neither PD1-deficient recipients nor wild-type recipients given anti-PD1 rejected the allograft within 8 weeks in the bm12 into B6 cardiac transplant model, administration of MIH6 anti-PDL1 antibody resulted in loss of graft loss by 40 days. These results are strongly suggestive of the existence of an alternative ligand for PDL1 other than PD1 that delivers a negative signal to T cells to down-regulate the alloimmune response.
Figure 1. Disparate allograft outcome following PD1 and PDL1 blockade in the single MHC class II-mismatched transplant model.
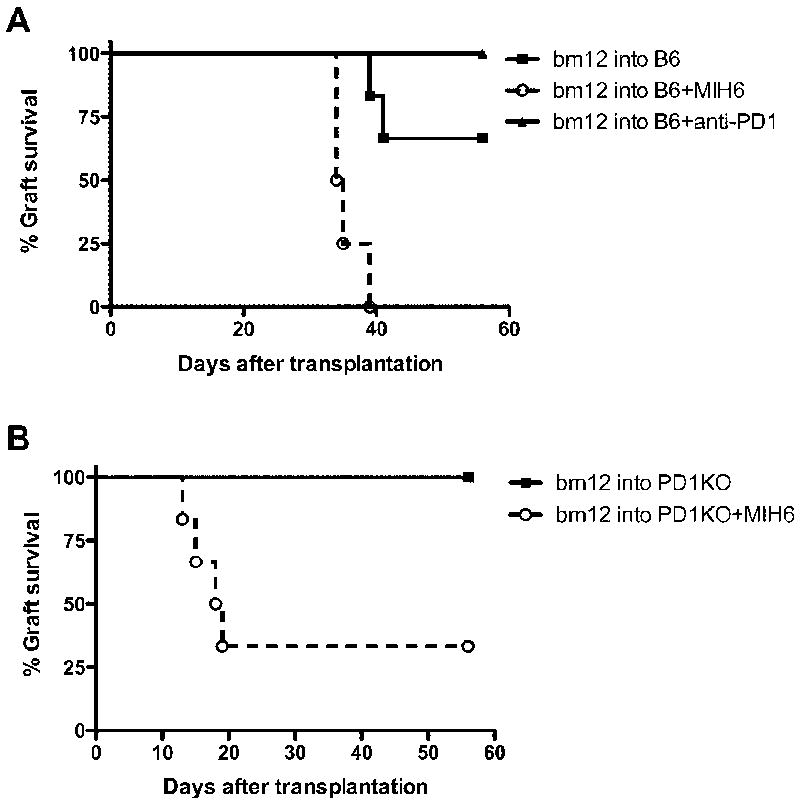
(A) bm12 cardiac grafts transplanted into B6 WT recipients had a median survival time of greater than 56 days. Anti-PD1 mAbs treatment had similar allograft survival times to untreated group; however, allograft rejection was remarkably accelerated when recipients were treated with dual-blockade MIH6 anti-PDL1 mAb. (B) Bm12 cardiac grafts transplanted into a PD1-deficient recipients had a median survival time of greater than 56 days. When PD1KO recipients were treated with anti-PDL1 mAb, (MIH6) allograft rejection was remarkably accelerated despite PD1 deficiency. N= 6 for each group.
B7.1 is an alternate ligand for PDL1 during alloimmune responses
To examine whether PDL1 binding to B7.1 in addition to PD1 can deliver an inhibitory costimulatory signal in vivo, we transplanted bm12 hearts into wild-type (WT), B7.1-, or B7.2-deficient B6 mice and treated these recipients with the dual-blocker anti-PDL1 mAb (MIH6) (Figure 2). Bm12 cardiac grafts transplanted into single B7.1KO (MST>56 days, n=6) or B7-2 KO (MST>56 days, n=6) recipients had comparable survival times to WT recipients (MST>56 days). In contrast, while PDL1 (MIH6) blockade accelerated allograft rejection in WT (MST=34.5 days, n=6, P=0.0032) and B7.2- deficient recipients (MST=10.5, n=6, P= 0.0007), it failed to do so in B7.1-deficient recipients. Grafts in MIH6-treated B7.1 recipients survived long-term in a similar fashion to those of untreated recipients (Figure 2). These data clearly indicate that PDL1 interaction with B7.1 functions to regulate alloimmune responses in vivo.
Figure 2. PDL1 blockade leads to accelerated cardiac allograft loss in B7.2-deficient but not in B7.1-deficient recipients.
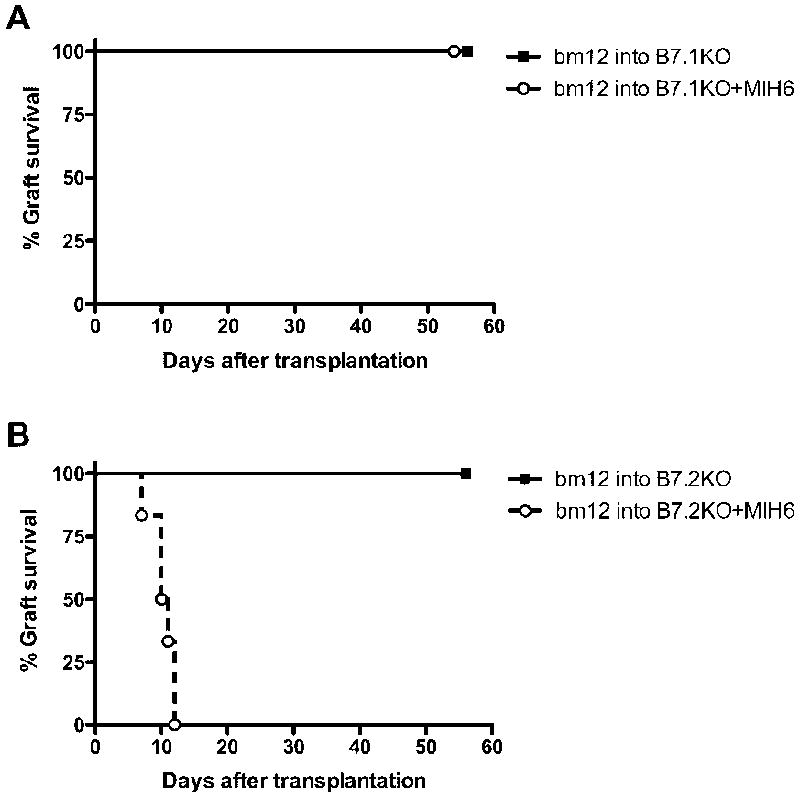
bm12 cardiac grafts transplanted into B6 B7.1- or B7.2-deficient recipients had a median survival time of greater than 56 days. When treated with MIH6 anti-PDL1 mAb, B7.2-deficient recipients rejected all allografts within 10 days (B), while allografts survived long-term in B7.1- deficient recipients (A). N= 6 for each group.
Blockade of PDL1: B7.1 interaction aggravates cardiac allograft vasculopathy and accelerates allograft rejection in PD1- or B7.2-deficient recipients
Molecular analysis of the PDL1 binding sites by chemical crosslinking revealed that the PDL1: B7.1 interface overlaps at least partially with the PDL1: PD1 interface (8). Consequently, blocking antibodies might interrupt PDL1: B7.1 as well as PDL1: PD1 interactions. The capacity of an array of anti-PDL1 mAbs to interfere with PDL1: B7.1 or PDL1: PD1 interactions has been tested by adhesion assays (8). 10F.2H11 anti-PDL1 mAb (2H11) was identified as the only mAb to solely block PDL1: B7.1 interaction (9). We took advantage of this novel 2H11 anti-PDL1 mAb to explore the unique role of PDL1: B7.1 interaction in regulating alloimmune responses.
We first transplanted bm12 hearts into WT B6 recipients and treated recipients with 2H11 mAb (Figure 3). Unlike the dual-blocker MIH6 anti-PDL1 mAb, all allografts treated with 2H11 survived long term (>56 days). However, the pathology of allografts treated with 2H11 mAb and harvested 56 days after transplantation showed significantly higher vasculopathy score (2.15 ± 0.30 Vs. 0.80± 0.18, P=0.0103) as compared to the untreated animals. The 2H11 treatment also intensified the allograft rejection score although this did not reach statistical significance (2.40 ± 0.40 vs. 1.33 ± 0.33, P=0.0686).
Figure 3. Blockade of PDL1: B7.1 pathway leads to enhanced cardiac allograft vasculopathy in the single MHC class II-mismatched transplant model.
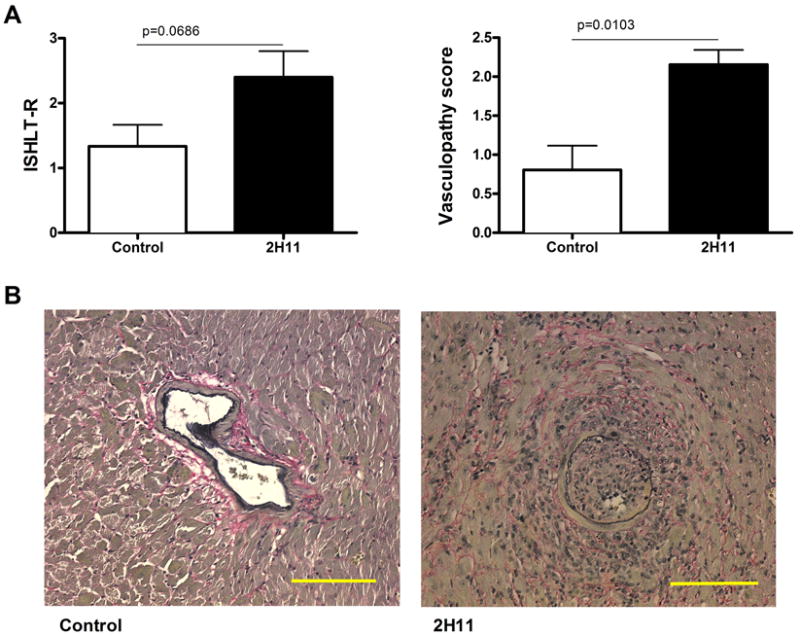
The hearts from bm12 mice were transplanted into B6 WT recipients and treated with 2H11 anti-PDL1 mAb, which exclusively blocks PDL1: B7.1 pathway. (A) 2H11 treatment significantly increased vasculopathy score by 56 days after transplantation as compared with no treatment. ISHLT grade was also increased, although differences did not reach statistical significance. Values shown represent mean ± SEM histological score obtained from 3 individual mice. (B) Representative photomicrographs of elastin staining show advanced vasculopathy in 2H11-treated allografts. Bars, 200 μm.
To ensure that the effect of 2H11 mAb treatment is due to blockade of PDL1: B7.1 interaction and not blockade of PDL1: PD1 pathway, we used PD1KO mice on B6 background as recipients of bm12 hearts. The administration of 2H11 anti-PDL1 mAb led to an accelerated bm12 allograft rejection in PD1KO recipients (MST=10 days), while controls survived long-term (>56 days) (Figure 4A). This data indicates that 2H11 functions by blocking the PDL1: B7.1 interaction.
Figure 4. Blockade of PDL1: B7.1 pathway accelerated cardiac allograft rejection in PD1 and B7.2 deficient recipients.
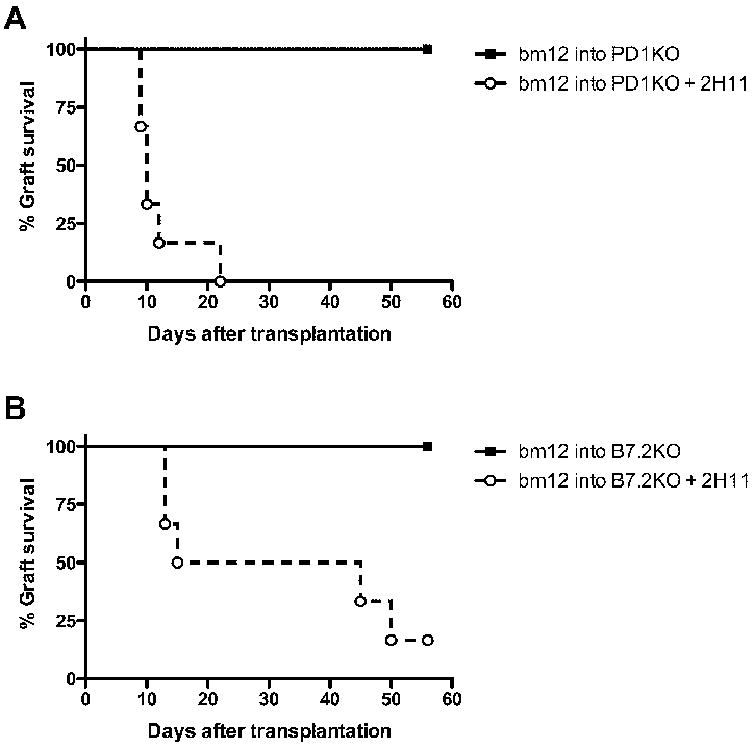
(A) bm12 hearts were transplanted into PD1KO recipients and the recipients were treated with 2H11 anti-PDL1 mAb. The survival time of cardiac allografts was significantly shortened as compared to controls. (B) bm12 hearts were transplanted into B7.2-deficient B6 recipients and the recipients treated with 2H11. Bm12 cardiac grafts transplanted into B6 B7.2-deficient recipients had a median survival time of greater than 56 days, while 2H11 precipitated allograft loss. N= 6 for each group.
To further confirm that 2H11 interferes with PDL1 binding to B7.1, we transplanted bm12 hearts into B7.1- and B7.2-deficient B6 recipients and treated with 2H11. While 2H11 accelerated allograft rejection in B7.2KO recipients (Figure 4B), it failed to do so in B7.1KO recipients, all of whose grafts survived long-term, similar to those in untreated WT recipients (data not shown). These data clearly show that 2H11 anti-PDL1 mAb can accelerate allograft rejection only when the PDL1 and B7.1 interaction is present. This confirms that PDL1 can interact with B7.1 to inhibit an aggressive alloimmune response in vivo.
Blockade of PDL1: B7.1 interaction enhances Th1 and Th2 alloimmune responses and alters regulatory T cell numbers
Analysis of the frequency of alloreactive cytokine-producing cells in B6 recipients of cardiac grafts from bm12 mice by ELISPOT at 10-14 days after transplantation showed that the frequency of alloreactive T cells producing IFN-γ in 2H11 mAb-treated recipients was increased by 2.7-fold over untreated recipients (Figure 5A). A similar increase was seen in IL-4 and IL-6-producing alloreactive T cells, with a 4.8-fold and 11.2-fold increase in 2H11-treated recipients, respectively, compared to control B6 WT recipients (Figure 5B, C).
Figure 5. PDL1: B7.1 blockade upregulates production of Th1, Th2 and proinflammatory cytokines in B6 recipients of bm12 hearts.
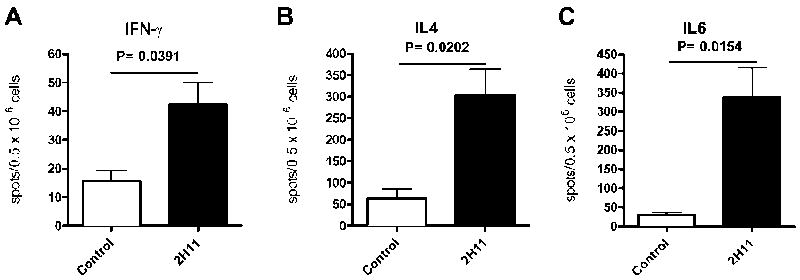
IFN-γ (A), IL-4 (B) and IL-6 (C) production by the splenocytes from B6 recipients of bm12 heart grafts assessed at 10 days post-Tx by ELISPOT. Data represent mean ± SEM of triplicate wells from three mice per group.
To investigate the effects of 2H11 on the regulatory / effector T cell balance, we compared the percentage of CD4+CD25+Foxp3+ regulatory T cells (Tregs), CD4+ and CD8+ effector/memory T cells in splenocytes from wild-type B6 recipients of bm12 cardiac grafts 10-14 days after transplantation in 2H11 mAb-treated or untreated recipients. The percentage of CD4+ and CD8 effector/memory phenotype (CD44highCD62Llow) was similar among the 2H11 mAb-treated or untreated recipients (Figure 6B, C). However, 2H11 mAb treatment did significantly decrease the percentage of CD4+CD25+Foxp3+ (forkhead box p3) Tregs (5.98±0.4252, n=5, P=0.0044) as compared to no treatment (9.81±0.88, n=5) as well as the absolute cell count of Tregs (3.66×106 ±0.49 vs 6.07×106 ±0.53, P=0.02970). These data suggest that the PDL1: B7.1 pathway may play a role in regulatory T cell homeostasis. However, the decrease in the percentage of CD4+CD25+Foxp3+ T cells in the spleen from 2H11 treated mice could be also due to a relocation of Tregs from spleens to the allografts. Therefore, we stained cardiac allografts for Foxp3 by immunohistochemistry to clarify this issue. The Foxp3 staining within cardiac allografts showed no significant difference between both groups (1.96 ±0.28 % vs. 2.39 ±0.28 %, P=0.3832), indicating that the reduction of Tregs in the spleen was unlikely to be related to relocation.
Figure 6. PDL1: B7.1 blockade decreases CD4+CD25+Foxp3+ regulatory T cells in spleens of B6 recipients of bm12 hearts without affecting effector T cells.
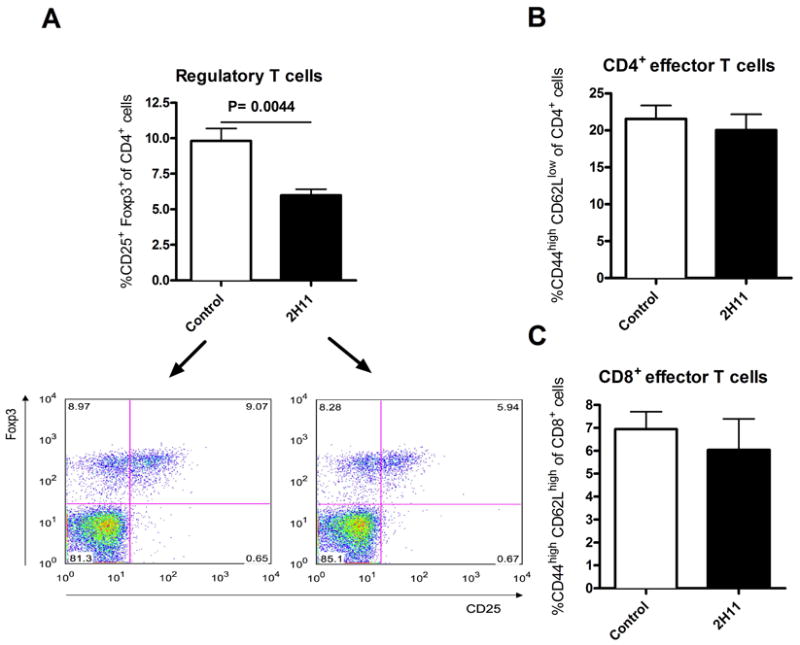
FACS analysis of splenocytes from B6 recipients of bm12 hearts with or without 2H11 treatment at 10 -14 days post-Tx showed that 2H11 treatment resulted in a significantly lower percentage of CD4+CD25+Foxp3+ regulatory T cells than controls (A). The percentage of CD4+ and CD8+ effector/memory phenotype (CD44highCD62Llow) was similar between the 2H11-treated and control recipients (B and C). Bar graphs show means ±SEM. All results are representative of at least three different sets of experiments.
The interaction between PDL1 on APCs and B7.1 on T cells plays a dominant role in bidirectional interactions between these two molecules during alloresponses
In order to dissect the function of the bidirectional interaction of PDL1 and B7.1, we used an in vitro culture assay in which a combination of APCs and T cells from WT and PDL1KO mice were used in the presence of either 2H11 or control IgG. B6 wild-type skin was transplanted into fully MHC mismatched Balb/c WT and PDL1KO recipients in order to sensitize T cells to alloantigen. Fourteen days after transplantation, spleens and lymph nodes from recipients were harvested and CD4+ T cells were isolated and cultured with irradiated CD11c+ cells from Flt-3L-injected B6 WT or PDL1KO mice. 2H11 was able to mildly increase IFN-γ production by CD4+ T cells when PDL1 was present on both APCs and T cells in vitro (Figure 7A), however no effect was seen when PDL1 was absent on both cells (Figure 7B). In addition, 2H11 did not enhance IFN-γ production when PDL1 was absent on CD11c+ cells alone (Figure 7D). Interestingly, 2H11 increased IFN-γ production when PDL1 was present on the APC side but absent in T cells (Figure 7C). These findings suggest a dominant direction of the PDL1: B7.1 interaction, whereas PDL1 on CD11c+ APCs transmits preferentially an inhibitory signal via B7.1 on T cells and this pathway is effectively blocked by 2H11 mAb.
Figure 7. The interaction of PDL1 on APCs with B7.1 on T cells plays a dominant role in bidirectional interactions between these two molecules.
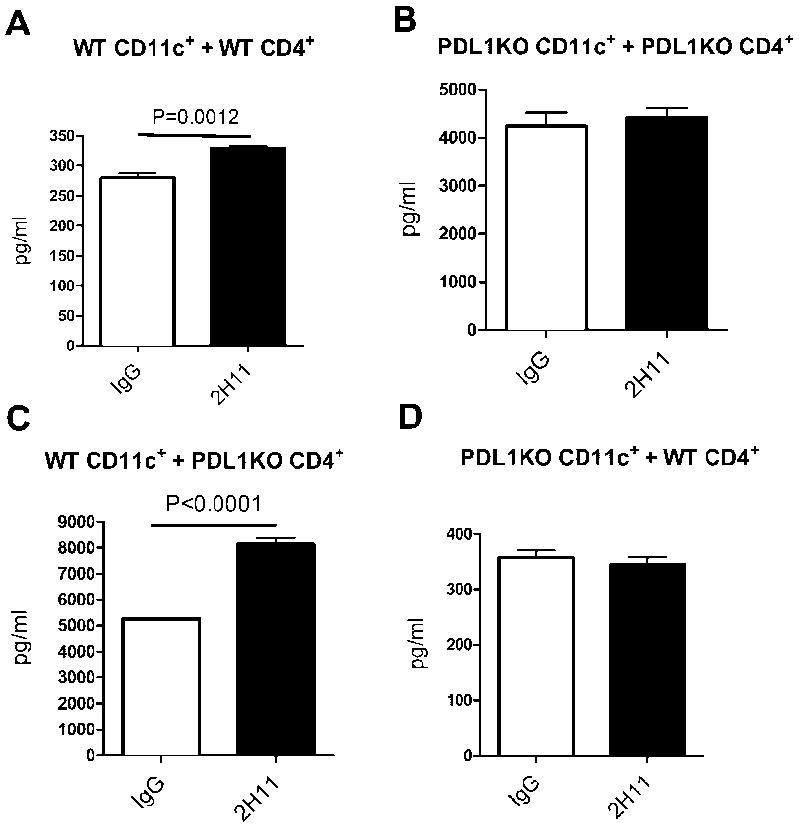
Fourteen days after B6 WT skin was transplanted into Balb/c WT or PDL1KO recipients, spleens and lymph nodes from recipients were harvested and CD4+ T cells were isolated. CD4+ T cells were then cultured with irradiated CD11c+ cells from Flt-3L-injected B6 WT or PDL1KO mice. Cell-free supernatants of individual wells were removed after 48 h of incubation with 2H11 or control IgG and analyzed by a Luminex for IFN-γ production. 2H11 mildly increased IFN-γ-production by CD4+ T cells when PDL1 was present on both APCs and T cells (A), however no effect was seen when PDL1 was absent on both cells (B). In addition, 2H11 did not enhance IFN-γ production when PDL1 was absent on CD11c+ cells alone (D). 2H11 increased IFN-γ production when PDL1 was present on the APC side but absent in T cells (C). Bar graphs show means ±SEM. All results are representative of at least three different sets of experiments with 3 mice.
DISCUSSION
PD1 has been defined as a negative regulator of immunity, limiting the activation of B and T cells and protecting against autoimmunity (19). The PD1 surface molecule is predominantly expressed on activated T and B cells, and is able to interact with at least two ligands: programmed death ligand-1 (PDL1) and another closely related molecule PDL2. Signaling through PD1 has been shown to have greater effect on cytokine production than on cellular proliferation, suppressing the production of IFN-γ, TNF-α and IL-2 via its effect on the PI3K signaling pathway (9). Therefore, further exploring this pathway might lead to potential promising targets for the induction of allograft tolerance. Indeed, our group has previously shown that PDL1 is required for peripheral tolerance and protection from chronic allograft rejection (20). Interestingly, when this pathway is blocked in vivo, several studies have found a greater effect of PDL1 blockade in precipitating rejection when compared to either PD1 or PDL2 blockade, and this has been initially attributed to differences in the expression of PDL1 and PDL2, in the half-life and/or affinities of the blocking antibodies (6, 20), In addition, the presence of a second stimulatory receptor for PDLs was also postulated (21-23).
We recently identified B7-1 as a second binding partner for PDL1, and showed that there are bidirectional interactions between B7-1 and PDL1 that can inhibit T cell responses in vitro (8). In this study we showed that PD1 blockade either by using monoclonal antibodies or genetically deficient mice was not as effective at shortening graft survival as PDL1 blockade (MIH6).in a single MHC class II mismatched cardiac transplant model. This finding suggests that PDL1 has an additional binding partner in addition to PD1 and that this interaction might be important for the fate of the alloimmune response. With the use of B7.1- or B7.2-deficient mice as recipients of bm12 hearts, we further showed that PDL1 blockade could only accelerates allograft rejection when B7.1 was present This confirmed the recent finding identifying B7.1 as an additional binding partner for PD1 (8).
Exploring the importance of the PDL1: B7.1 in transplantation in vivo raised numerous challenges, including the broad expression pattern of these receptors on T cells, APCs and nonhematopoietic cells as well as its potential bidirectional signaling characteristics. By using a dual blocker MIH6 anti-PDL1 mAb (Butte and Sharpe, unpublished data), we have previously shown that blocking PDL1: B7.1 and PDL1: PD1 significantly shortens allograft survival of bm12 hearts (17). Based on the special characteristics of a specific blocking mAbs 10F.2H11 that solely blocks PDL1: B7.1 interaction, we were able to further explore the function of this pathway in a cardiac transplant model. The 10F.2H11 mAb is a novel antibody that have been previously shown to block the interaction of 300.19 PD-L1 transfectants with B7-1Ig fusion protein but not PD-1Ig fusion protein in a cell adhesion assay (9). We found that blockade of PDL1: B7.1 by 2H11 mAbs remarkably aggregated allograft vasculopathy although there was no effect on the allograft survival in the bm12 into B6 cardiac transplant model. We also showed that this blockade significantly enhanced the alloimmune response by upregulating the frequency of IFN-γ-, I L-4- and IL-6-producing cells. These findings demonstrated that PDL1: B7.1 interaction was important of the inhibition of the alloimmune response in vivo. These data together with the results with PD1 blockade indicates that sole blockade of either PDL1: PD1 or PDL1: B7.1 interaction has a reduced effect on alloimmune responses as compared to the dual PDL1 blockade, suggesting some non-overlapping functions. However, to directly compare the contributions of the PDL1: PD1 and PDL1: B7.1 pathways requires the development of a specific anti-PDL1 mAb that exclusively blocks the PDL1: PD1 interaction. Moreover, both 2H11 (Figure 4A) and MIH6 anti-PDL1 (Figure 1B) Abs were able to accelerate allograft rejection in PD1-deficient recipients, indicating that blockade of PDL1: B7.1 interaction has a much more profound effect when PD1 is absent.
PD1 and PD-L1 are highly expressed on Treg cells, but the role of PD1 and PDL1 pathway in expansion or function of Tregs is not yet clear (24, 25). Recent studies have identified a key role for PD-L1 (26) and PD1 (27, 28) in promoting Treg cell development and function. In this study, we showed that blockade of PDL1: B7.1 might impair the generation of regulatory T cells in vivo, consequently enhances alloimmune immune responses and aggravates allograft vasculopathy. Our findings are consistent with recent publication by Yi et al. suggesting a similar importance of PDL1:B7.1 interaction in Treg generation (29). However, the mechanism by which PDL1: B7.1 controls Tregs needs further investigation.
Because both PDL1 and B7.1 are expressed on T cells, B cells, dendritic cells (DCs) and macrophages, there is the potential for bidirectional interactions between B7.1 and PDL1 on these cell types. Our findings in this study point to a potential role of PDL1 on CD11c+ APCs in transmitting an inhibitory signal via B7.1 on T cells, suggesting a potential dominant direction for PDL1 and B7.1 interaction. These data are consistent with a recent report from Park et al. showing that an inhibitory signal through B7.1, but not PDL1, on T cells is responsible in part for the tolerogenic effects of PDL1 and B7.1 interaction (30). However, due to the expression of PDL1 on a broad range of cells, our in-vitro experiments are limited by the use of selected cell combinations. For example, these studies are unable to address, the possible role of the interaction of PDL1 expressed on non-hematopoietic cells with B7.1 on T cells, which have recently been shown to be important in the alloimmune response in vivo (17). Moreover, additional effects of other hematopoietic cell interactions in vivo cannot be accounted for.
In summary, our present study demonstrates that PDL1 interaction with B7.1 inhibits alloimmune responses in vivo. Accumulating evidence shows that B7.1 is an additional binding partner for PDL1 and this interaction may also result in the suppression of an aggressive alloimmune response. These findings provide a rationale for a new therapeutic approach by targeting PDL1: B7.1 pathways to improve the long-term outcome of the allograft. Our results also have potentially important therapeutic implications about the use of Belatacept (LEA29Y). Belatacept is a second generation human CTLA4-Ig that selectively blocks B7 receptors on APCs and it was recently approved by the FDA for the prevention of acute rejection in kidney transplant patients. However, B7 blockade by Belatacept might adversely affect negative signaling by PDL1 and, therefore, impair long-term allograft survival. Further studies will be needed to elucidate the overall importance of this pathway in the maintenance of a tolerogenic state.
Acknowledgments
We thank Dr. Ibrahim Batal for technical help.
Abbreviations used in this paper
- Foxp3
forkhead box p3
- PD
programmed death
- PD-L
PD-ligand
- Treg
regulatory T cell
Footnotes
This study was supported by the following grants AST/Roche Basic Science Faculty Development Grant to JY; Research grant from the American Society of Transplantation to LVR; NIH AI-37691, AI-51559, PO1 AI-41521 to MHS and P01 AI56299 to AHS, MHS, BB and GJF; National Kidney Foundation Clinical Scientist Award to AC, and the American Society of Nephrology Student Scholar Grant to SC.
DISCLOSURES The authors have no financial conflicts of interest.
References
- 1.Dong VM, Womer KL, Sayegh MH. Transplantation tolerance: the concept and its applicability. Pediatr Transplant. 1999;3:181–192. doi: 10.1034/j.1399-3046.1999.00042.x. [DOI] [PubMed] [Google Scholar]
- 2.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 5.Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 6.Sandner SE, Clarkson MR, Salama AD, Sanchez-Fueyo A, Domenig C, Habicht A, Najafian N, Yagita H, Azuma M, Turka LA, Sayegh MH. Role of the programmed death-1 pathway in regulation of alloimmune responses in vivo. J Immunol. 2005;174:3408–3415. doi: 10.4049/jimmunol.174.6.3408. [DOI] [PubMed] [Google Scholar]
- 7.Gao W, Demirci G, Strom TB, Li XC. Stimulating PD-1-negative signals concurrent with blocking CD154 co-stimulation induces long-term islet allograft survival. Transplantation. 2003;76:994–999. doi: 10.1097/01.TP.0000085010.39567.FB. [DOI] [PubMed] [Google Scholar]
- 8.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keir ME, Freeman GJ, Sharpe AH. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J Immunol. 2007;179:5064–5070. doi: 10.4049/jimmunol.179.8.5064. [DOI] [PubMed] [Google Scholar]
- 11.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–5545. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 15.Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ, Auchincloss H, Jr, Sayegh MH. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–69. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsushima F, Iwai H, Otsuki N, Abe M, Hirose S, Yamazaki T, Akiba H, Yagita H, Takahashi Y, Omura K, Okumura K, Azuma M. Preferential contribution of B7-H1 to programmed death-1-mediated regulation of hapten-specific allergic inflammatory responses. Eur J Immunol. 2003;33:2773–2782. doi: 10.1002/eji.200324084. [DOI] [PubMed] [Google Scholar]
- 17.Yang J, Popoola J, Khandwala S, Vadivel N, Vanguri V, Yuan X, Dada S, Guleria I, Tian C, Ansari MJ, Shin T, Yagita H, Azuma M, Sayegh MH, Chandraker A. Critical role of donor tissue expression of programmed death ligand-1 in regulating cardiac allograft rejection and vasculopathy. Circulation. 2008;117:660–669. doi: 10.1161/CIRCULATIONAHA.107.741025. [DOI] [PubMed] [Google Scholar]
- 18.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M, Habicht A, Clarkson MR, Iacomini J, Glimcher LH, Sayegh MH, Ansari MJ. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205:3133–3144. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka K, Albin MJ, Yuan X, Yamaura K, Habicht A, Murayama T, Grimm M, Waaga AM, Ueno T, Padera RF, Yagita H, Azuma M, Shin T, Blazar BR, Rothstein DM, Sayegh MH, Najafian N. PDL1 is required for peripheral transplantation tolerance and protection from chronic allograft rejection. J Immunol. 2007;179:5204–5210. doi: 10.4049/jimmunol.179.8.5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong H, Chen X. Immunoregulatory role of B7-H1 in chronicity of inflammatory responses. Cell Mol Immunol. 2006;3:179–187. [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Gao JX, Wen J, Yin L, Li O, Zuo T, Gajewski TF, Fu YX, Zheng P, Liu Y. B7DC/PDL2 promotes tumor immunity by a PD-1-independent mechanism. J Exp Med. 2003;197:1721–1730. doi: 10.1084/jem.20022089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin T, Kennedy G, Gorski K, Tsuchiya H, Koseki H, Azuma M, Yagita H, Chen L, Powell J, Pardoll D, Housseau F. Cooperative B7-1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J Exp Med. 2003;198:31–38. doi: 10.1084/jem.20030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25+ regulatory cells from human peripheral blood express very high levels of CD25 ex vivo. Novartis Found Symp. 2003;252:67–88. doi: 10.1002/0470871628.ch6. discussion 88-91, 106-114. [DOI] [PubMed] [Google Scholar]
- 25.Krupnick AS, Gelman AE, Barchet W, Richardson S, Kreisel FH, Turka LA, Colonna M, Patterson GA, Kreisel D. Murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T cells. J Immunol. 2005;175:6265–6270. doi: 10.4049/jimmunol.175.10.6265. [DOI] [PubMed] [Google Scholar]
- 26.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J Neurosci Res. 2006;84:370–378. doi: 10.1002/jnr.20881. [DOI] [PubMed] [Google Scholar]
- 28.Wang C, Li Y, Proctor TM, Vandenbark AA, Offner H. Down-modulation of programmed death 1 alters regulatory T cells and promotes experimental autoimmune encephalomyelitis. J Neurosci Res. 2010;88:7–15. doi: 10.1002/jnr.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi T, Li X, Yao S, Wang L, Chen Y, Zhao D, Johnston HF, Young JS, Liu H, Todorov I, Forman SJ, Chen L, Zeng D. Host APCs augment in vivo expansion of donor natural regulatory T cells via B7H1/B7.1 in allogeneic recipients. J Immunol. 186:2739–2749. doi: 10.4049/jimmunol.1002939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, Yao S, Tsushima F, Narazaki H, Anand S, Liu Y, Strome SE, Chen L, Tamada K. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T cell tolerance. Blood. 2010;116:1292–1298. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]