

Abstract
Labeling proteins with long-lifetime emitting lanthanide (III) chelate reporters enables sensitive, time-resolved luminescence bioaffinity assays. Heterodimers of trimethoprim (TMP) covalently linked to various cs124-sensitized, polyaminocarboxylate chelates stably retain lanthanide ions and exhibit quantum yields of europium emission up to 20% in water. A time-resolved, luminescence resonance energy transfer (LRET) assay showed that TMP-polyaminocarboxylates bind to Escherichia coli dihydrofolate reductase (eDHFR) fusion proteins with nanomolar affinity in purified solutions and in bacterial lysates. The ability to selectively impart terbium or europium luminescence to fusion proteins in complex physiological mixtures bypasses the need for specific antibodies and simplifies sample preparation.
INTRODUCTION
Ligand-sensitized complexes of lanthanide cations have unique photophysical properties, including large Stokes shifts (>150 nm), multiple, narrow emission bands (<10 nm at half-maximum) and long emission lifetimes (μs-ms). These properties offer distinct advantages for luminescence-based biological assays and microscopy, most notably high signal-to-background ratios that are obtained by temporally discriminating lanthanide luminescence from short-lifetime (<10 ns) scattering and autofluorescence background.(1–2) Direct conjugation of terbium or europium complexes to oligonucleotides, antibodies and proteins has enabled the development of sensitive luminescence resonance energy transfer (LRET1, also known as time-resolved fluorescence resonance energy transfer, or TR-FRET) assays of biomolecular interactions in purified biochemical preparations, cellular extracts, and on cell surfaces.(3–5) Recently, we showed that interactions between a terbium complex-labeled protein and a green fluorescent protein (GFP) fusion could be microscopically imaged in living cells by detecting long-lived, terbium-to-GFP LRET in time-resolved mode.(6)
Because lanthanide absorption transitions are parity-forbidden, direct excitation is inefficient, and Tb3+ or Eu3+ complexes typically incorporate the metal ion into an organic chelating ligand that contains a sensitizing chromophore. When excited with ultraviolet (UV) light in the absorption band, the chromophore undergoes intersystem crossing to its triplet excited state followed by intramolecular energy transfer to the emissive level of the chelated metal.(1) In general, lanthanide complexes used in time-resolved screening or cellular imaging applications must meet several requirements: (i) the molecule must be kinetically stable; (ii) the molecule must have a high extinction coefficient and quantum yield of emission (i.e., good brightness); (iii) the excitation wavelength should be as long as possible; (iv) the molecule must be resistant to photobleaching; and (v) bioconjugates of the molecule must be easy to prepare. Kinetic stability is particular important for cellular imaging or screening applications in cell lysates where endogenous chelators could scavenge metal from the complex.(2) For microscopy, near-UV (>340 nm) absorption is preferred because typical optics won’t transmit below this limit.(7) While hundreds of luminescent lanthanide chelates have been prepared over the past decades, relatively few meet most or all of the aforementioned criteria.(1–2)
Polyaminocarboxylates, diethylene triamine pentaacetic acid (DTPA) and and triethylene tetraamine hexaacetic acid (TTHA), have proven to be robust scaffolds for developing bright and photostable luminescent chelates of Tb3+ and Eu3+. Selvin and coworkers have developed various DTPA and TTHA chelates that utilize 7-aminoquinolinone (carbostyril 124, or cs124) as the sensitizing chromophore.(5, 8–13) Conjugable analogs include an amide-linked, amine-reactive or thiol-reactive moiety at one end of the polyaminocarboxylate chain, opposite the sensitizer (Figure 1). The cs124-sensitized chelates have reasonable extinction coefficients (ε = ~10,500 M−1cm−1 at λmax = 341 nm) and overall quantum yields of emission up to 40% for Tb3+ and up to 28% for Eu3+.(12, 14) Variants that incorporate 7-amino-4-trifluoromethyl-2-(1H)-quinolinone (cs124CF3) were shown to be effective Eu3+ luminophores, being 2- to 4-fold brighter than their analogous cs124-sensitized DTPA and TTHA analogues with ~15 nm red-shifted excitation maxima.(15) However, cross-linkable DTPA-cs124 analogues are 8-dentate chelates and therefore suffer reduced brightness and higher kinetic instability because one or more water molecules are coordinated with the metal. This is not the case with TTHA-cs124 conjugates.(11) Mustaev and co-workers addressed this issue by developing DTPA-cs124 analogues that could be conjugated to biomolecules via modification of the chromophore moiety (Figure 1).(16) Chromophore-linked DTPA-cs124 conjugates were shown to be highly luminescent and exhibited greater resistance to EDTA challenge than Selvin-type DTPA-cs124 analogues. Analogous thiol-reactive, coumarin-sensitized, DTPA and TTHA chelates were reported by Heyduk and Heyduk, but these molecules only supported Eu3+ luminescence and were less bright than cs124-sensitized chelates.(17)
Figure 1.

Variants of carbostyril 124-polyaminocarboxylates and their modes of attachment to biomolecules.
In this article, we present results of our continued efforts to develop luminescent lanthanide protein labels for time-resolved LRET microscopy and screening applications. We previously showed that conjugates of the common antibiotic, trimethoprim (TMP) linked to Tb3+-complexed cs124-polyaminocarboxylates or a 2-hydroxyisophthalamide-based macrocycle (Lumi4®-Tb) bound to Escherichia coli dihydrofolate reductase (eDHFR) with high affinity, thereby offering an effective means of selectively imparting Tb3+ luminescence to fusion proteins.(18) Here, our original goal was to develop a TMP-linked, Eu3+ chelate that exhibited good brightness, high kinetic stability and appreciable absorption at 365 nm (for eventual microscopy applications). We prepared conjugates of TMP coupled to 3- and 4-acetic acid derivatives of cs124CF3 and cs124, respectively (scheme 1), and linked these moieties to DTPA and TTHA (scheme 2). We then assessed the photophysical properties of these chelates as well as our previously reported, Selvin-type, cs124-TTHA-TMP conjugate by measuring the relative quantum yields of Eu3+ and Tb3+ luminescence using DTPA-cs124 as a reference compound. We assessed comparative kinetic stability by measuring time-dependent decrease in lanthanide luminescence in the presence of a large excess of EDTA.
Scheme 1.

Synthesis of TMP conjugated to (A) cs124-CF3 (3) and (B) cs124 (5).
Scheme 2.

Synthesis of sensitized TMP-polyaminocarboxylate conjugates.
The previously developed cs124-TTHA-TMP (10) and the novel TTHA-cs124CF3-TMP (7) were the most efficient Eu3+ complexes with estimated overall quantum yields of ~20%, and they exhibited relatively high resistance to EDTA challenge. Affinity for eDHFR (KD = ~1.5 nM) was measured directly in purified biochemical preparations and in bacterial lysates by titrating a GFP-eDHFR fusion protein against a fixed concentration (20 nM) of selected, TMP-Tb3+ chelates and measuring the protein concentration-dependent increase in Tb3+-to-GFP LRET. The ability to selectively impart Tb3+ or Eu3+ luminescence to fusion proteins and to quantitatively measure binding events in cellular extracts simplifies LRET-based bioassays and eliminates the need for lanthanide chelate-conjugated antibodies. Moreover, the good brightness, kinetic stability and long-wavelength absorption of the Eu3+ complex of TTHA-cs124CF3-TMP (7) make it a potentially useful reporter label for live-cell microscopy.
EXPERIMENTAL PROCEDURES
Chemicals and instrumentation
The following chemicals were purchased from Sigma-Aldrich: TTHA, DTPA dianhydride, diaminobenzene, methyl bromoacetate, TMP, hydrobromic acid (HBr, 48%), 1, 8-Diazabicyclo[5.4.0]undec-7-ene (DBU), 2, 2′-(Ethylenedioxy)bis(ethylamine), diethyl-1, 3-acetonedicarboxylate, ethyl bromovalerate, di-tert-butyl dicarbonate, dichloromethane, trifluoroacetic acid (TFA), citric acid, O-Benzotriazole-N, N, N′, N′-tetramethyluronium hexafluorophosphate (HBTU), N-Hydroxybenzotriazole (HOBt), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), ethyl acetoacetate, ethyl 4, 4, 4-trifluoroacetoacetate, anhydrous dimethylformamide (DMF, in sure seal bottle), anhydrous dimethyl sulfoxide (DMSO, in sure seal bottle), EuCl3·6H2O, TbCl3·6H2O, ZnCl2, diisopropyl ethylamine (DIPEA), pyridine, acetic anhydride, triethylamine (TEA) and β-nicotinamide adenine dinucleotide phosphate (β-NADPH). Glacial acetic acid, NaOH, KOH, NaCl, NaHCO3, H2SO4, anhydrous Na2SO4, ethylenediaminetetracetic acid (EDTA) disodium salt dihydrate, acetonitrile, methanol, hexane, ethyl acetate, diethyl ether, BugBuster™, DNAse, Halt™ protease inhibitor cocktail, PMSF and imidazole were purchased from Thermo Fisher Scientific. Tris(hydroxymethyl) aminomethane hydrochloride (Tris·HCl) was purchased from Acros Organics. Distilled and deionized (18 MΩ cm−1) water was used throughout. All glassware was washed with a 10% nitric acid solution and thoroughly rinsed with deionized, distilled water. 1H NMR and 13C NMR spectra were recorded at the UIC Research Resources Center (RRC) in the deuterated solvents stated. 1H and 13C chemical shifts were referenced to internal solvent resonances and reported relative to SiMe4. Low-resolution and high-resolution electrospray (ESI) mass spectra were obtained at the UIC RRC. UV-Visible (UV-Vis) absorption spectra were recorded using a Cary 3000 spectrophotometer (Varian, Inc.). Fluorescence emission spectra were recorded using a Fluoromax 3 fluorimeter (Horiba–Jobin Yvon, Inc.). Time-resolved luminescence intensity was measured using a 96-well plate reader (Perkin Elmer, Victor 3V) with 340 nm excitation (60 nm bandpass) and indicated emission filters (10 nm bandpass).
Purification
Reverse-phase high-performance liquid chromatography (HPLC) was performed using a Beckman System Gold instrument equipped with an analytical scale pump (model 128), a UV-Vis detector (model 168) and a C18 analytical column (GraceVydac, cat. no. 218TP54, 5 μm, 4.6 mm i.d. × 250 mm). A 20 min linear gradient, from 0 to 30%, 3–33% or 5–25% solvent B (solvent A = 0.1M triethylammonium acetate (pH 6.5), solvent B = CH3CN) was used (Table 1). An initial hold of 3 min was applied for all runs.
Table 1.
HPLC purification solvents, retention times and detected m/z ratios for chelates prepared in this study.
| compound | linear gradient | retention time (min) | ESI MS [M+H]+ |
|---|---|---|---|
| cs124CF3-TMP (3) | 3–33% CH3CN/TEAA | 22 | 775 |
| cs124-TMP (5) | 5–25% CH3CN/TEAA | 18 | 707 |
| DTPA-cs124CF3-TMP (6) | 0–30% CH3CN/TEAA | 22 | 1151 |
| TTHA-cs124CF3-TMP (7) | 0–30% CH3CN/TEAA | 20 | 1252 |
| DTPA-cs124-TMP (8) | 0–30% CH3CN/TEAA | 18 | 1083 |
| TTHA-cs124-TMP (9) | 0–30% CH3CN/TEAA | 17 | 1184 |
| cs124-TTHA-TMP (10) | 0–30% CH3CN/TEAA | 18 | 1140 |
Synthesis
Compound 1 (Scheme 1)
Ethyl-4, 4, 4-trifluoroacetoacetate (2.2 mL, 15 mmol) and KOH (0.86 g, 15 mmol) were mixed in 7 mL of DMF and stirred at 40 °C for 20 min. To this mixture, 1.5 mL of methylbromoacetate was added and the solution was stirred overnight at room temperature under nitrogen. 300 mg (5.2 mmol) of KOH was then added, and stirred at 60 °C for 1 h. This mixture was diluted by the addition of 20 mL of water and was then extracted with chloroform. The organic layer was collected, dried over anhydrous sodium sulfate, and then evaporated in vacuo, first at 30 °C, and then at 75 °C for 45 min. The residue obtained was dissolved in 3.5 mL of DMSO, and then 0.76 g (7 mmol) of diaminobenzene was added and the solution stirred at 110 °C for 6 h. This solution was cooled overnight and diluted with 30 mL of 0.05M aqueous NaOH and extracted with ether (2 × 50 mL). The aqueous phase obtained after ether extraction containing product 1, was acidified by the addition of citric acid to pH 3–3.5; the precipitate was collected by centrifugation, washed with water, dried. Recrystallization from ethyl acetate resulted in the isolation of pure compound 1. Yield 3%. ESIMS (m/z) 287 [M+H]+, 309 [M+Na]+, 285 [M−H]−. 1H NMR (500 MHz, DMF-d7, ppm): δ = 7.65 (d, 1H), 6.87 (d, 2H), 6.36 (s, br, 2H), 4.10 (s, 2H). 13C NMR (125 MHz, DMF-d7, ppm): δ = 171.99, 161.81, 152.07, 141.07, 134.82, 126.16, 124.85, 122.00, 112.05, 105.40, 97.18, 31.99.
Compound 2 was prepared as previously reported.(18)
Compound 3 (Scheme 1)
To a stirred solution of compound 1 (11.44 mg, 0.04 mmol) in CH3CN (7 mL) was added HBTU (18.2 mg, 0.05 mmol), HOBt (6.5 mg, 0.05 mmol) and DIPEA (0.02 mL, 0.12 mmol) and the mixture was stirred for 30 min at room temperature under nitrogen. Then, compound 2 (20 mg, 0.04 mmol) in CH3CN (3 mL) was added to the reaction mixture and stirred for 3 h. After evaporation of CH3CN, 15 mL water was added and the mixture was extracted with ethyl acetate (4 × 50 mL). The organic extracts were washed with saturated NaHCO3 solution, dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude reaction mixture was purified on reverse-phase HPLC to obtain the pure compound 3. ESIMS (m/z) 775 [M+H]+, 797 [M+Na]+, 773 [M−H]−. HRMS: Pred. m/z for C36H45N8F3O8 775.3385 meas. m/z 775.3382 [M+H]+.
Compound 4 (Scheme 1)
A suspension of 1.36 g (10 mmol) of ZnCl2 in 5 mL of DMSO was supplemented with 1.08 g (10 mmol) of diaminobenzene and 2.02 g (10 mmol) of diethyl-1, 3-acetonedicarboxylate. The reaction mixture was stirred at 100 °C for 24 h. This mixture was diluted with 8 mL of ethanol, poured into 150 mL of ice-cold 0.1M citric acid, and left for 3–5 h at 4 °C. The residue was filtered and successively washed with water (4 × 15 mL), and with hot CH3CN (2 × 10 mL), and then dried in vacuo. Yield 53%. From this 200 mg was taken and dissolved in 3 mL dioxane. To this 4 mL of aqueous 2M NaOH was added and stirred at 50 °C for 5 h, and then 15 mL water was added and extracted with ether (2 × 40 mL). The product (4) was precipitated from the aqueous phase by the addition of 10% H2SO4 to pH 2.5, collected by centrifugation, washed few times with water and dried in vacuo. Yield 37%. ESIMS (m/z) 219 [M+H]+. 1H NMR (500 MHz, DMSO-d6, ppm): δ = 11.24 (s, 1H), 7.26 (d, 1H), 6.41 (d, 1H), 6.35 (s, 1H), 5.98 (s, 1H), 5.77 (s, br, 2H), 3.65 (s, 2H). 13C NMR (125 MHz, DMSO-d6, ppm): δ = 171.97, 162.65, 151.27, 145.40, 141.39, 125.95, 116.60, 111.07, 110.33, 97.42, 38.28.
Compound 5 (Scheme 1)
To a stirred solution of compound 4 (19 mg, 0.09 mmol) in DMF (1 mL) was added PyBop (45.24 mg, 0.09 mmol), and TEA (0.08 mL, 0.58 mmol) and allowed to stir for 30 min at room temperature under nitrogen. Then, compound 2 (15 mg, 0.03 mmol) in DMF (0.5 mL) was added to the reaction mixture and stirred for 12 h. The reaction mixture was purified on reverse-phase HPLC to obtain the pure compound 5. ESIMS (m/z) 707 [M+H]+, 705 [M−H]−. HRMS: Pred. m/z for C35H46N8O8 354.1792 meas. m/z 354.1797 [M+2H]2+.
General procedure for the synthesis of compounds 6, 7, 8 and 9 (Scheme 2)
5 equiv. of DTPA dianhydride/TTHA dianhydride was dissolved in DMF and 20 equiv. of TEA under nitrogen. 1 equiv. of compound 3/compound 5 in DMF was added and stirred at room temperature for 8–10 h. A few drops of water were added to quench the reaction. The products were purified by reverse-phase HPLC.
Compound 10 (Scheme 2)
2 equiv. of TTHA dianhydride was dissolved in DMSO and 20 equiv. of TEA under nitrogen. 0.7 equiv. of cs-124 was added and stirred at room temperature for 10 min. 1 equiv. of compound 2 in DMSO was added and stirred for 3–8 h. A few drops of water were added to quench the reaction. The product was purified by reverse-phase HPLC.
Addition of metals
Compound concentration was obtained using measured absorptions and reported extinction coefficients for the fluorophores (cs124, ε = 10,500 M−1cm−1 at λ = 341 nm; cs124CF3, ε = 21,000 M−1cm−1 at λ = 341 nm).(12, 15) EuCl3·6H2O or TbCl3·6H2O was added to the chelate in a 1:1.5 molar ratio at typically >5 μM concentration in TBS buffer (50 mM Tris·HCl, 150 mM NaCl, pH 7.6) and allowed to stand for 30 min at room temperature before use.
Quantum yield measurements
The luminescence quantum yield of lanthanide chelates in TBS (pH = 7.6) was determined by standard methods using DTPA-cs124 as a reference from the equation:
| (1) |
where, ΦS and ΦR equal the quantum yields of the sample and reference, respectively. GradS and GradR equal the slopes of plots of integrated luminescence intensity vs. absorption (0.02 < Abs < 0.1) for either the sample or reference, and ηS or ηR equal the refractive indices of the respective sample or reference solutions.(19) The excitation wavelength was 330 nm in all cases. The results were reported as relative to cs124 (Table 2), so ΦR was taken to equal 1. The quantum yield of Tb3+ emission for DTPA-cs124 was measured using quinine sulfate as a reference (Φ = 0.54 in 0.1M H2SO4). Each sample was measured at least twice, and results did not vary by more than ±15%.
Table 2.
Relative quantum yields of emission for prepared chelates of Tb3+ and Eu3+.
| lanthanide | compound | quantum yield |
|---|---|---|
| Tb3+ | DTPA-cs-124a | 1.00 |
| DTPA-cs-124-TMP (8) | 0.23 | |
| TTHA-cs-124-TMP (9) | 0.20 | |
| cs-124-TTHA-TMP (10) | 0.90 | |
| Eu3+ | DTPA-cs-124a | 1.00 |
| DTPA-cs-124-CF3-TMP (6) | 0.56 | |
| TTHA-cs-124-CF3-TMP (7) | 2.02 | |
| DTPA-cs-124-TMP (8) | 0.77 | |
| TTHA-cs-124-TMP (9) | 1.56 | |
| cs-124-TTHA-TMP (10) | 1.98 |
reference compound
EDTA challenge assay
In order to measure the stability of the TMP-lanthanide complex conjugates, each metallated compound was diluted to 50 nM in TBS, pH = 7.4 in a 1.5 mL centrifuge tube. EDTA was added from a 0.02 M stock solution (H2O/NaOH, pH = 8.0) to a final concentration of 10 mM, and the samples were vortexed for 1 min. The samples were then aliquotted (100 μL) into 96-well plates, and the Tb3+ or Eu3+ emission was measured over time using a time-resolved luminescence plate reader (time delay = 100 μs; measurement window = 1400 μs; λ ex = 340/60 nm; Tb3+, λ em = 545/10 nm; Eu3+, λ em = 615/10 nm). Lanthanide luminescence was normalized to the value measured at the first time point (10 min.) and plotted vs. time using Kaleidagraph (Synergy Software).
Protein expression and purification
GFP-eDHFR was purified from the E. coli strain BL21 DE3 (pLysS) transformed with pRSETb-EGFP-eDHFR.(18) A 5mL luria broth (LB) overnight culture was used to inoculate 500 mL of LB (Ampicillin, 100 mg/mL; chloramphenicol, 34 mg/mL). The 500 mL culture was grown at 37 °C, shaking at 200 rpm, to an OD600 of ~0.6, at which time expression of the protein was induced by the addition of IPTG to a final concentration of 1 mM. After growth for another 4 h, the cells were harvested by centrifugation. The pellet was lysed in 30 mL of lysis buffer (1X BugBuster™ Protein Extraction Reagent (Novagen), 10μg/ml DNAse, 1mM PMSF, 100mM HEPES, 10mM imidazole, pH 7.5). Samples were placed on an orbital shaker for 30 min., then centrifuged (15000 rpm, 15 min, 4 °C) and applied to a column containing 2.0 mL of HisLink™ Protein Purification Resin (Promega). Following purification, the protein was concentrated (to ~10 μM), dialyzed with phosphate buffer (10mM K2HPO4, KH2PO4, pH 7.4), and stored at −80 °C.
Cell lysate preparation
Cell lysates containing GFP-eDHFR were obtained from 5.0 mL LB cultures of E. coli strain BL21 DE3 (pLysS) transformed with pRSETb-EGFP-eDHFR and treated to induce protein expression as described above. After growth and expression, the cells were harvested by centrifugation. The cell pellets were resuspended in 10mM phosphate buffer (pH 7.4, with Halt™ Protease and Phosphatase Inhibitor, Thermo Scientific, Inc.) to a final concentration of 0.1mg/μL. The cells were then lysed by sonicating for 1 min (2, 15 s on/off cycles) and re-centrifuged (15000 rpm, 15 min, 4 °C). The GFP-eDHFR concentration in the cell supernatant was determined by measuring the absorption of GFP at 484 nm (ε484=56,000 M−1cm−1).
Binding affinity assay
The binding affinity of compounds 9 and 10 complexed with Tb3+ for eDHFR was determined by measuring Tb3+-to-GFP LRET in time-resolved mode. TMP conjugates (20nM), were titrated in 96-well plates with either bacterial cell lysates containing GFP-eDHFR or with purified protein at concentrations ranging from ~0.5 nM to 1000 nM in assay buffer (50 mM K2HPO4, KH2PO4, 18 mM β-mercaptoethanol, 20 μM β-NADPH, pH 7.2). Titrations were done in triplicate for each sample. Time-resolved luminescence (time delay = 100 μs; measurement window = 1400 μs, λ ex = 340/60 nm) was measured at λ em = 520/10 nm (LRET-sensitized GFP emission signal) and at λ em = 615/10 nm (Tb3+ donor signal). For each well, the percent change in the donor-normalized LRET signal was calculated from the following equation:
| (2) |
where, (520/615)S represents the ratios of the emission signals at the indicated wavelengths for the sample well and (520/615)−ctrl. represents the average emission ratio for three wells containing TMP-conjugate (20 nM) and no GFP-eDHFR. Using Kaleidagraph (Synergy Software), ΔLRETN% was plotted against protein concentration and dissociation constants were obtained by fitting the data to the following equation:
| (3) |
where Lmin is the ΔLRETN% of the TMP-Tb3+ complex with no receptor, Lmax is the maximum ΔLRETN% signal observed at saturating receptor concentration, [L]T is the total amount of lanthanide complex used, and [P] T is the total amount of protein used.
RESULTS AND DISCUSSION
Syntheses
The compounds cs124,(20) DTPA-cs124,(12) TTHA dianhydride(21) and an amine-derivatized TMP (2)(18) were prepared according to previously reported procedures. Compounds 1 and 4 were obtained by condensing diaminobenzene with ethyl-4, 4, 4-trifluoroacetoacetate and diethyl-1, 3-acetonedicarboxylate, respectively.(16) Compound 3 and 5 were prepared by peptide coupling between compounds 1 or 3 and 2 (Scheme 1). DTPA-based chelates (6, 8) were prepared by reacting an excess of commercially available DTPA dianhydride with 3 or 5 in DMF at room temperature. TTHA dianhydride was similarly reacted with 3 or 5 to form compounds 7 and 9. Compound 10 was synthesized in a three-component reaction by treating TTHA dianhydride (2 equivalents) with 0.7 equivalents of cs-124, followed by the addition of 1 equivalent of compound 2 in DMSO (Scheme 2). All of the polyaminocarboxylate compounds were purified by reverse-phase HPLC and identified by mass spectrometry (Table 1), UV-Vis absorption spectroscopy, and lanthanide luminescence properties.
Absorption spectra
The absorption spectra of representative TMP conjugates of cs124- and cs124CF3-polyaminocarboxylates complexed with Eu3+ are shown in Figure 2 and compared to DTPA-cs124. The compounds were assumed to retain the molar absorptivities of the sensitizing chromophores (10,500 M−1cm−1 @ 341 nm for cs124, 21,000 M−1cm−1 @ 341 nm for cs124CF3).(12, 15) This assumption was supported by the observation that ~1.3 equivalents of Tb3+ or Eu3+ was sufficient to saturate lanthanide luminescence emission of all the chelates. The absorption spectrum of cs124-TTHA-TMP (10) is nearly identical to that of DTPA-cs124, but with a slight red shift of 2 nm. Consistent with previous studies by Mustaev and coworkers, we observed a red shift of 6-nm for the chromophore-linked, cs124 and cs124CF3 analogs.(16) Thus, TTHA-cs124CF3-TMP (7) has a long-wavelength absorption maximum at 348 nm with a shoulder at 366 nm and DTPA-cs124-TMP (8) has a long-wavelength maximum at 348 nm. The analogous compounds DTPA-cs124CF3-TMP (6) and TTHA-cs124-TMP (9) exhibited identically shaped spectra to compounds 7 and 8, respectively (not shown). The two-fold greater absorptivity and longer-wavelength maxima of compounds 6 and 7 allow for efficient excitation with high-power UV LED sources that emit at 365 nm, an important aspect for microscopy.(7)
Figure 2.

UV absorption spectra of representative trimethoprim-polyaminocarboxylate Eu3+ complexes and the reference compound (DTPA-cs124) described in this study.
Emission spectra
All compounds displayed the narrow emission spectra typical of luminescent lanthanide chelates (Figure 3). The emission spectra and relative peak heights of Tb3+ emission with all compounds were essentially the same, matching the spectrum shown for 10 (Figure 3D). However, the relative intensities of the different Eu3+ emission lines are dependent on the chelate (DTPA or TTHA) but are independent of the sensitizer (cs124 or cs124CF3).(12, 15) The Eu3+ complexes of the TTHA conjugates, 7 (Figure 3B), 9 (not shown, identical to 7) and 10 (Figure 3C) have a greater fraction of their luminescence in the sharply spiked peak at 613 nm, whereas the DTPA compounds all have relatively higher emission intensity in the peaks centered around 580 nm and 700 nm, as seen in the emission spectrum for 6 (Figure 3A). The Eu3+ emission pattern of 10 varies slightly from that of 7 or 9, reflecting a different coordination environment resulting from amide bonds at both ends of TTHA. For spectroscopic or microscopic detection of Eu3+ luminescence through a narrow-pass emission filter at 613 nm, TTHA chelates are more detectable, as a lesser proportion of the emission intensity is excluded.
Figure 3.

Normalized emission spectra (λex = 330 nm) of Eu3+ complexes of (A) DTPA-cs124CF3− TMP (6); (B) TTHA-cs124CF3− TMP (7); (C) cs124-TTHA-TMP (10); and (D) Tb3+ complex of 10.
Relative quantum yields of emission
We measured overall luminescent quantum yields for both Tb3+ and Eu3+ complexes of our heterodimeric TMP-linked chelates using appropriately metallated DTPA-cs124 as a reference standard (Table 2). We chose DTPA-cs124 as the reference because its quantum yields for Tb3+ and Eu3+ luminescence were reported previously (0.32 and 0.10, respectively),(14) and it offered a similar spectral profile and intensity for comparison. We also measured the overall quantum yield of DTPA-cs124(Tb3+) using quinine sulfate as a reference and obtained a similar result (0.29) to the reported literature value (0.32).
The quantum yield data revealed some unexpected trends based on prior literature and the molecular architecture of the chelates. For Tb3+ luminescence, both chromophore-linked TMP conjugates, 8 and 9, exhibited quantum yields of only ~20% relative to DTPA-cs124, whereas a similarly structured DTPA-cs124-isothiocyante was reported by Mustaev and co-workers to be ~80% as bright as DTPA-cs124.(16) Similarly, for Eu3+ luminescence, 8 and 6 had relative quantum yields of 0.77 and 0.56, respectively; substantially lower than the 1.4-fold greater Eu3+ luminescence reported for DTPA-cs124-isothiocyanate relative to that of DTPA-cs124.(16) While the previous study did not directly measure quantum yields, the identical absorptivities observed for chromophore-linked vs. conventional cs124 analogs allow for direct comparison. While the aforementioned compounds showed low quantum yields relative to DTPA-cs124 (and thus lower brightness), the Eu3+ complexes of 7 and 10 emitted with twice the efficiency of DTPA-cs124 (Table 2). Thus, assuming a ~10% overall Eu3+ quantum yield for DTPA-cs124,(14) we estimate a ~20% Eu3+ quantum yield for compounds 7 and 10. The higher quantum yield, greater absorptivity and longer wavelength absorption make compound 7 a potentially useful label for cell-based microscopy.
Relative stability of the lanthanide chelates
Besides photophysical characteristics, stable retention of the metal ion is an important property for studies in living cells or cell extracts where endogenous chelators could scavenge metal from the lanthanide probes. To compare relative stability, we measured Tb3+ and Eu3+ emission intensity over a period of 3 h in the presence of EDTA for the metal complexes of the TMP-polyaminocarboxylate conjugates and of DTPA-cs124 (Figure 4). In the presence of 10 mM of EDTA, the Eu3+ emission intensity for nearly all compounds rapidly decreases and stabilizes after ~75 min suggesting equilibrium was achieved (compound 9 did not appear to equilibrate within 3 h). The Eu3+ complex of 10 was the most stable, retaining ~55% of its initial intensity after 3 h, while DTPA-cs124(Eu3+) was the least stable (~9% initial intensity). A similar pattern was seen for Tb3+ complexes, with TTHA analogues exhibiting greater stability than DTPA chelates, although neither of the TTHA compounds achieved equilibrium with EDTA within the 3 h observation period.
Figure 4.

Relative emission intensity over time in the presence of EDTA for trimethoprim-polyaminocarboxylate conjugates and DTPA-cs124. (A) Eu3+ complexes. (B) Tb3+ complexes.
Protein binding affinity measurements
For both in vitro and cell-based applications, TMP conjugates must necessarily bind with high affinity to eDHFR fusion proteins. To determine the dissociation constants for binding of select TMP-polyaminocarboxylate Tb3+ complex conjugates to eDHFR, we performed a time-resolved LRET assay that detected binding to a GFP-eDHFR fusion protein as Tb3+-sensitized, GFP emission. The assay was performed in two formats: 1) purified GFP-eDHFR was titrated against a fixed concentration (20 nM) of either 9 or 10 in assay buffer; and 2) a lysate of E. coli expressing GFP-eDHFR was diluted directly into assay buffer without purification and titrated against 20 nM of each compound. Using a time-resolved luminescence plate reader, we measured sensitized, long-lifetime (>100 μs) GFP emission at 520 nm (the LRET signal) and the Tb3+ donor emission signal (615 nm). The percent change in the 520 nm/615 nm emission ratio was plotted vs. GFP-eDHFR concentration, revealing characteristic binding isotherms (Figure 5). Nonlinear, least-squares fits of the data showed that the dissociation constants for binding to GFP-eDHFR equaled ~1.5 nM for both compounds under each assay condition. The ability to quantitatively measure binding events in a complex mixture, coupled with the large dynamic range of the LRET signal (~5,000 % for compound 10 in lysate/buffer) suggests that TMP-lanthanide complex conjugates could be used for high throughput screening or quantification of biomolecular interactions, especially in cases where highly pure biochemical preparations cannot be obtained.
Figure 5.
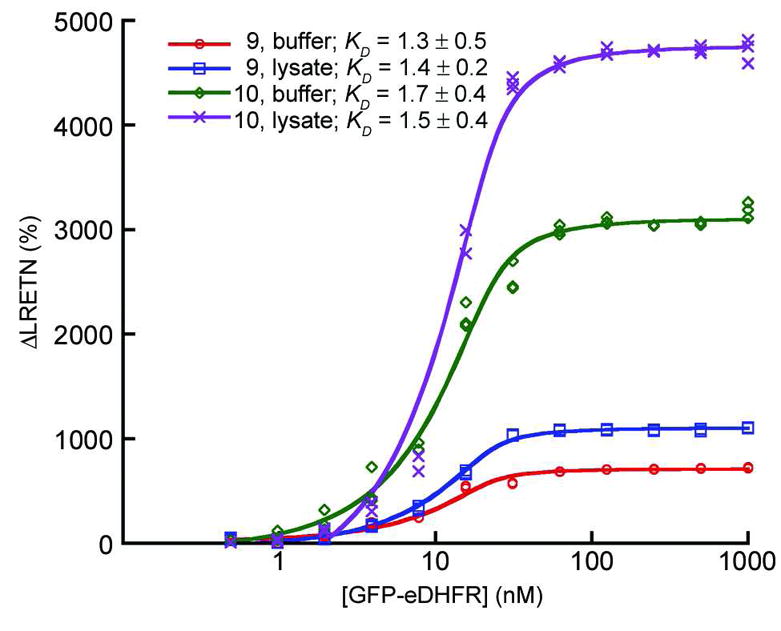
Intramolecular, luminescence resonance energy transfer (LRET) between eDHFR-bound Tb3+ complexes of TTHA-cs124-TMP (9) or cs124-TTHA-TMP (10) and GFP. Increasing concentrations of either purified eDHFR-GFP or a bacterial lysate containing the protein were titrated against a constant concentration (20 nM) of each compound. Sensitized GFP emission (520 nm) and Tb3+ emission (615 nm) was detected after a time delay of 100 μs, upon pulsed excitation with near-UV light (~340 nm). The y-axis represents the percent change in the 520/615 emission ratio. Lines represent nonlinear least squares fit to the data.
Conclusions
Currently, homogeneous, LRET-based assays of protein-protein or protein-DNA/RNA interactions require direct conjugation of lanthanide reporters to purified proteins or to specific antibodies. However, antibodies are not always available for a particular target, and many proteins cannot be purified. In such cases, and especially for specific protein labeling in living cells, the high-affinity interaction between TMP and eDHFR provides an effective means for imparting lanthanide luminescence to recombinant fusion proteins. Of the TMP-polyaminocarboxylate conjugates described here, the Tb3+ and Eu3+ complexes of cs124-TTHA-TMP (10) and the Eu3+ complex of TTHA-cs124CF3-TMP (7) have the requisite brightness and kinetic stability to be effective reporters for time-resolved bioassays. Compound 7 is particularly promising as an Eu3+ reporter for cell-based microscopy because of its relatively long-wavelength absorption and high molar extinction coefficient. Paired with a suitable Tb3+ complex, 7 (Eu3+) could be used for multiplexed lanthanide luminescence imaging.
Acknowledgments
This study was supported by the National Institutes of Health (National Institute of General Medical Sciences Grant R01GM081030).
Footnotes
Abbreviations: diethylenetriaminepentaacetic acid (DTPA); Escherichia coli dihydrofolate reductase (eDHFR); green fluorescent protein (GFP); luminescence resonance energy transfer (LRET); trimethoprim (TMP); triethylenetetraaminehexaacetic acid (TTHA).
References
- 1.Bunzli JC. Lanthanide luminescence for biomedical analyses and imaging. Chem Rev. 2010;110:2729–55. doi: 10.1021/cr900362e. [DOI] [PubMed] [Google Scholar]
- 2.Hovinen J, Guy PM. Bioconjugation with stable luminescent lanthanide(III) chelates comprising pyridine subunits. Bioconjug Chem. 2009;20:404–21. doi: 10.1021/bc800370s. [DOI] [PubMed] [Google Scholar]
- 3.Leblanc V, Delaunay V, Claude Lelong J, Gas F, Mathis G, Grassi J, May E. Homogeneous time-resolved fluorescence assay for identifying p53 interactions with its protein partners, directly in a cellular extract. Anal Biochem. 2002;308:247–54. doi: 10.1016/s0003-2697(02)00229-4. [DOI] [PubMed] [Google Scholar]
- 4.Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, Bazin H, Tinel N, Durroux T, Prezeau L, Trinquet E, Pin JP. Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods. 2008;5:561–7. doi: 10.1038/nmeth.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selvin PR, Hearst JE. Luminescence energy transfer using a terbium chelate: improvements on fluorescence energy transfer. Proc Natl Acad Sci U S A. 1994;91:10024–8. doi: 10.1073/pnas.91.21.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajapakse HE, Gahlaut N, Mohandessi S, Yu D, Turner JR, Miller LW. Time-resolved luminescence resonance energy transfer imaging of protein-protein interactions in living cells. Proc Natl Acad Sci U S A. 2010;107:13582–7. doi: 10.1073/pnas.1002025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gahlaut N, Miller LW. Time-resolved microscopy for imaging lanthanide luminescence in living cells. Cytometry A. 2010;77:1113–25. doi: 10.1002/cyto.a.20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Selvin PR. Thiol-reactive luminescent chelates of terbium and europium. Bioconjug Chem. 1999;10:311–5. doi: 10.1021/bc980113w. [DOI] [PubMed] [Google Scholar]
- 9.Ge P, Selvin PR. Thiol-reactive luminescent lanthanide chelates: part 2. Bioconjug Chem. 2003;14:870–6. doi: 10.1021/bc034029e. [DOI] [PubMed] [Google Scholar]
- 10.Ge P, Selvin PR. Carbostyril derivatives as antenna molecules for luminescent lanthanide chelates. Bioconjug Chem. 2004;15:1088–94. doi: 10.1021/bc049915j. [DOI] [PubMed] [Google Scholar]
- 11.Ge P, Selvin PR. New 9- or 10-dentate luminescent lanthanide chelates. Bioconjug Chem. 2008;19:1105–11. doi: 10.1021/bc800030z. [DOI] [PubMed] [Google Scholar]
- 12.Li M, Selvin PR. Luminescent Polyaminocarboxylate Chelates of Terbium and Europium: The Effect of Chelate Structure. J Am Chem Soc. 1995;117:8132–8138. [Google Scholar]
- 13.Li M, Selvin PR. Amine-reactive forms of a luminescent diethylenetriaminepentaacetic acid chelate of terbium and europium: Attachment to DNA and energy transfer measurements. Bioconjugate Chemistry. 1997;8:127–132. doi: 10.1021/bc960085m. [DOI] [PubMed] [Google Scholar]
- 14.Xiao M, Selvin PR. Quantum yields of luminescent lanthanide chelates and farred dyes measured by resonance energy transfer. J Am Chem Soc. 2001;123:7067–73. doi: 10.1021/ja0031669. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Selvin PR. Synthesis of 7-amino-4-trifluoromethyl-2-(1H)-quinolinone and its use as an antenna molecule for luminescent europium polyaminocarboxylates chelates. Journal of Photochemistry and Photobiology A: Chemistry. 2000;135:27–32. [Google Scholar]
- 16.Krasnoperov LN, Marras SA, Kozlov M, Wirpsza L, Mustaev A. Luminescent probes for ultrasensitive detection of nucleic acids. Bioconjug Chem. 2010;21:319–27. doi: 10.1021/bc900403n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heyduk E, Heyduk T. Thiol-reactive, luminescent Europium chelates: luminescence probes for resonance energy transfer distance measurements in biomolecules. Anal Biochem. 1997;248:216–27. doi: 10.1006/abio.1997.2148. [DOI] [PubMed] [Google Scholar]
- 18.Rajapakse HE, Reddy DR, Mohandessi S, Butlin NG, Miller LW. Luminescent terbium protein labels for time-resolved microscopy and screening. Angew Chem Int Ed Engl. 2009;48:4990–2. doi: 10.1002/anie.200900858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demas JN, Crosby GA. The measurement of photoluminescence quantum yields. A review. J Phys Chem. 1971;75:991–1024. [Google Scholar]
- 20.Barraja P, Diana P, Montalbano A, Dattolo G, Cirrincione G, Viola G, Vedaldi D, Dall’Acqua F. Pyrrolo[2, 3-h]quinolinones: a new ring system with potent photoantiproliferative activity. Bioorg Med Chem. 2006;14:8712–28. doi: 10.1016/j.bmc.2006.07.061. [DOI] [PubMed] [Google Scholar]
- 21.Zitha-Bovens E, Muller RN, Laurent S, Elst LV, Geraldes CFGC, Bekkum Hv, Peters JA. Structure and Dynamics of Lanthanide Complexes of Triethylenetetramine-N, N, N′, N″, N‴, N‴-hexaacetic Acid (H6ttha) and of Diamides H4ttha(NHR) Derived from H6ttha as Studied by NMR, NMRD, and EPR. Helv Chim Acta. 2005:88. [Google Scholar]