

Abstract
Brain metastasis from breast cancer is an increasingly important clinical problem. Here we assessed the role of CD44hi/CD24lo cells and pathways that regulate them, in an experimental model of brain metastasis. Notch signaling (mediated by γ-secretase) has been shown to contribute to maintenance of the cancer stem cell (CSC) phenotype. Cells sorted for a reduced stem-like phenotype had a reduced ability to form brain metastases compared to unsorted or CD44hi/CD24lo cells (p<0.05; Kruskal Wallis). To assess the effect of γ-secretase inhibition, cells were cultured with DAPT and the CD44/CD24 phenotypes quantified. 231-BR cells with a CD44hi/CD24lo phenotype was reduced by ~15 % in cells treated with DAPT compared to DMSO-treated or untreated cells (p = 0.001, ANOVA). In vivo, mice treated with DAPT developed significantly fewer micro and macrometastases compared to vehicle treated or untreated mice (p = 0.011, Kruskal Wallis). Notch1 knockdown reduced the expression of CD44hi/CD24lo phenotype by ~20%. In vitro, Notch1 shRNA resulted in a reduction in cellular growth at 24 hour, 48 hour and 72 hour time points (p = 0.033, p = 0.002 and p = 0.009, ANOVA) and a ~60% reduction in Matrigel™ invasion was observed (p < 0.001, ANOVA). Cells transfected with shNotch1 formed significantly fewer macrometastases and micrometastases compared to scrambled shRNA or untransfected cells (p < 0.001; Kruskal Wallis). These data suggest that the CSC phenotype contributes to the development of brain metastases from breast cancer, and this may arise in part from increased Notch activity.
Keywords: breast cancer, Notch signaling, brain metastasis, cancer stem cell, gamma secretase inhibitor
INTRODUCTION
Brain metastasis from breast cancer is a growing problem, due in part to improved therapies for systemic metastatic disease and the inability of many conventional drugs to cross the blood brain barrier. Ten to sixteen percent of patients with metastatic breast cancer historically developed brain metastases, with a one year survival of 20% (1-2). Currently, one-third of metastatic breast cancer patients with either HER2 positive tumors or “triple negative” tumors (estrogen and progesterone receptor negative, HER2 normal) develop brain metastases (1, 3-6). In addition, much remains to be elucidated about the mechanisms involved in the formation of brain metastases from breast cancer, and the processes by which it is regulated. It is hoped that such information, combined with improved blood-brain barrier pharmacokinetics, will enable the identification and validation of novel therapeutics with increased potency in the brain (for review, (7)). Here we investigated the role of the cancer stem cell (CSC) phenotype and, in particular, the Notch pathway, in an experimental model of breast cancer metastasis to the brain.
Stem cells are defined as cells that have the ability to a) sustain themselves through self-renewal and b) generate mature cells through differentiation (8). Growing evidence suggests that CSCs have similar self-renewal capacity to that of normal stem cells, and that only a subset of tumor cells have the ability to grow and divide to repopulate a tumor. As such, the identification and study of these cells, in addition to the pathways that regulate their maintenance, may allow selective targeting of the core population of tumor promoting cells. In breast cancer, Al-Hajj et al. identified a population of CD44+CD24−/loLineage− cells in breast cancer patients (9). As few as 100 cells with this phenotype were able to initiate and form a tumor in vivo. In contrast, 20,000 cells with alternate phenotypes were unable to form tumors (9). In addition, measurement of aldehyde dehydrogenase (ALDH) activity has recently been utilized to identify normal stem cells and CSCs (10-11). ALDH1 levels have been found to correlate with high tumor grade, HER2 positivity, hormone receptor negativity and expression of the basal markers cytokeratin (CK) 5/6 and CK14 (12). In addition, expression of ALDH1 was shown to be an independent prognostic indicator of poor overall survival for breast cancer. Increasing evidence suggests that CSCs play an important role in mediating metastasis. Indeed ALDH-positive breast cancer cell lines demonstrated enhanced metastatic capabilities when injected into NOD/SCID mice (13).
Notch proteins are a family of four transmembrane, heterodimeric receptors (Notch1IC – Notch4IC), with five known ligands (Delta-like1, Delta-like3, Delta-like4, Jagged1 and Jagged2). In response to ligand binding, the intracellular domain of the receptor is proteolytically released in two stages, mediated by ADAM proteases (14-15) and a γ-secretase (presenilin; (16)). The cleaved intracellular domain translocates to the nucleus where it is involved in transcriptional activation of the CSL (CBF1-Suppressor of Hairless-Lag2) transcription factor family. Following Notch binding to CSL, it becomes a transcriptional activator. In combination with Mastermind-like (MAML) proteins, transcriptional induction of Hairy/Enhancer of Split genes occurs, for example Hey, Hes and Notch1 itself. Elevated Notch signaling has also been found to increase transcription of HER2 (17) and Cyclin D1 (18), among others.
Dysregulated Notch signaling has been observed in many human cancers, including endometrial cancer (19), colon cancer (20) and lung cancer (21). Recently it has been shown that Notch signaling is activated in human breast cancer, with the accumulation of Notch1 intracellular domain in tissue (22). Notch signaling in breast cancer has also been shown to activate Akt (23) and survivin (24), which may be involved in mediating chemoresistance. Elevated Notch ligands have been shown to correlate with poor overall survival in patients with breast cancer (25). Notch signaling has previously been shown to play a role in stem cell maintenance (26-27), and may also contribute to the maintenance of the CSC phenotype (28-31), with the strongest evidence for a role in CSCs being in breast cancer. Gamma-secretase inhibition has been shown to prevent the formation of secondary mammospheres from cell lines and primary patient samples (32). A role for CSC in metastasis has been proposed but remains incompletely defined (33-35). Similarly, a role for the Notch pathway in tumor metastasis has been proposed (36-39). Nam et al. reported that a MDA-MB-435 carcinoma cell line selected for metastatic growth in the brain exhibited up-regulation of the Notch pathway as compared to the parental cell line, and that the commercial γ-secretase inhibitor DAPT and RNA interference-mediated knockdown of Notch1 inhibited tumor cell migration and invasion in vitro (40).
In this study, we used an experimental model of brain metastasis of breast cancer to assess the role of the Notch pathway in brain metastases of breast cancer in vivo. Using two different experimental strategies, Notch signaling inhibition significantly prevented the colonization of brain metastatic MDA-MB-231 (231-BR) human breast cancer cells in the brain. We also determined the relationship of the ‘stem-like’ phenotype (CD44hi/CD24lo) to both brain metastasis and Notch signaling inhibition. The data nominate Notch as a potential therapeutic target for inhibition of brain metastases.
MATERIALS AND METHODS
Cell Culture
MDA-MB-231 (231) human breast carcinoma cells (American Type Culture Collection, Manassas, VA) and the brain metastatic variant 231-BR cells (41) were maintained in Dulbecco’s-minimal essential media (DMEM) (Invitrogen, Burlington, ON, Canada), supplemented with 10% fetal bovine serum (FBS) (Sigma Aldrich, Mississauga, ON, Canada).
Cell Surface Marker Analysis
231, 231-BR, 231-BR-shNotch1, 231-BR-shScrambled cells and cells treated with 5μM DAPT ((N-(N-(3,5-Difluorophenacetyl)-L-alanyl)-S-phenylglycine t-butyl ester; Sigma Aldrich) or DMSO (dimethyl sulfoxide; Sigma) (1 × 105) were incubated with fluorescently-conjugated antibodies, including anti-CD24 (clone ML5; BD Biosciences, Mississauga, ON, Canada) conjugated to phycoerytherin (PE); and anti-CD44 (clone IM7; BD Biosciences) conjugated to fluorescein isothiocyanate (FITC). Fluorescently-conjugated IgG isotype controls (BD Biosciences) were used as negative controls. Cells were analyzed using a Beckman Coulter EPICS XL-MCL flow cytometer (Beckman Coulter, Mississauga, ON, Canada). Data are illustrated as percentage of cells with a CD44hi/CD24lo phenotype (bottom right quadrant) ± standard error.
Fluorescence Activated Cell Sorting (FACS)
231-BR cells were labeled with fluorescent antibodies (CD44-FITC + CD24-PE). Cell subsets were isolated using a four-color analysis protocol on a FACS Vantage/Diva cell sorter (BD Biosciences), into CD44hiCD24lo (defined as ‘stem like’) and CD44loCD24hi (‘non-stem like’) subsets (top and bottom 5 %). Cell viability was assessed by plating efficiency post-sorting. Following FACS isolation, cells were used immediately for in vivo assays. Sorting was conducted at the Robarts Research Institute Flow Cytometry facility, University of Western Ontario.
Analysis of ALDH Activity
To assess ALDH activity of the different cell lines, the Aldefluor® assay kit (StemCell Technologies, Vancouver, BC, Canada) was used. The basis for this assay is that uncharged ALDH substrate (BODIPY-aminoacetaldehyde (BAAA)) is taken up by living cells via passive diffusion. Once inside the cell, BAAA is converted into negatively-charged BODIPY-aminoacetate (BAA-) by intracellular ALDH. BAA- is then retained inside the cell, causing the cell to become highly fluorescent. Only cells with an intact cell membrane can retain BAA-, so only viable cells can be identified (13). The Aldefluor® assay was performed essentially as described previously (10-11). Briefly, 231-BR cells were harvested, placed in Aldefluor® assay buffer (2 × 106 cells/ml), and incubated with the Aldefluor® substrate for 45 minutes at 37°C to allow substrate conversion. As a negative control for all experiments, an aliquot of Aldefluor-stained cells was immediately quenched with 1.5-mM diethylaminobenzaldehyde (DEAB), a specific ALDH1 inhibitor. Cells were analyzed using the green fluorescence channel (FL1) on a Beckman Coulter EPICS XL-MCL flow cytometer.
In Vivo Brain Metastasis Assays
All in vivo work was carried out using 6-7 week old female athymic nude nu/nu mice (Charles River Laboratories, Wilmington, MA, USA). Animal procedures were conducted in accordance with the recommendations of the Canadian Council on Animal Care, under a protocol approved by the University of Western Ontario Council on Animal Care. Cells were grown to ~75% confluency in 75 cm2 flasks. Following cell sorting, sorted populations (CD44hiCD24lo and CD44loCD24hi 231-BR cells) were resuspended in sterile Hank’s buffered salt solution (HBSS, Invitrogen) at 1.75 × 105 cells/0.1ml. Mice were anaesthetized with isofluorane/O2 and cells were injected into the left ventricle, as described previously (42). Similarly, 231-BR cells transfected with shNotch1 or shScrambled were injected intracardiac at 1.75 × 105 cells/mouse. For γ-secretase inhibition study, we used a commercially available γ-secretase inhibitor, DAPT (Sigma Aldrich). Following injection of 1.75 × 105 231-BR cells, tumors were allowed to form for 14 days. Mice were treated intraperitoneally with 8 mg/kg DAPT (or vehicle: 5 % ethanol in corn oil), beginning treatment on Day 14 using a 3 day on, 4 day off schedule up to Day 28. Intermittent administration of DAPT has previously been shown to reduce the gastrointestinal toxicity observed with γ-secretase inhibition which can result from expansion of goblet cells (43-44). Mice were euthanized on Day 28 and the whole brains were removed from the skull and assessed for metastatic growth. Brains were fixed in formalin, embedded in paraffin and sectioned (10 μm sections). Sections were stained using standard hematoxylin and eosin (H&E) staining. The number of metastases per section was determined by light microscopy, by two independent blinded investigators. Metastases were binned into micrometastases and macrometastases using 50 μm2 as a cut-off, as described previously (42).
RNA Isolation and Real Time PCR
RNA from cell lines was isolated using the RNeasy Mini kit (Qiagen, Mississauga, ON, Canada). All RNA was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen). Primers were obtained for Notch1-4, Hey1 and 18S (SABiosciences, Frederick, MD, USA). The optimized amplification protocol consisted of an initial denaturation step of 95 °C for 10 min followed by 36 amplification cycles at 95 °C for 10 s, annealing at 58 °C for 5 s, and elongation at 72 °C for 10 s. A constant temperature ramp of 20 °C/s was used throughout each of the steps. Measurements of fluorescence were taken at the end of the annealing phase for each cycle. The PCR products were melted by increasing the temperature to 95 °C (0.1 °C/s). Finally, the samples were cooled to 40 °C. Real time PCR was conducted using the CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Mississauga, ON, Canada). As a negative control, cDNA was replaced with deionized water. To quantify gene expression, the internal control transcript 18S rRNA was used as a reference. Data were analyzed using the Pfaffl method (45) and is displayed as fold changes relative to control cells.
Immunoblotting
Protein was extracted from cell lines using RIPA (radioimmunoprecipitation assay) buffer containing protease inhibitors (EDTA-free-Protease inhibitor cocktail; Roche, Mississauga, ON, Canada). Protein concentrations were measured using the Nanodrop spectrophotometer (Nanodrop 2000, Thermo Scientific, Rockford, IL, USA). 100 μg of protein were separated on SDS-PAGE (sodium dodecyl sulfate – polyacrylamide gel electrophoresis) and transferred to PVDF (polyvinylidene fluoride) membranes. Membranes were blocked in 5 % low fat milk solution in TBS-T (Tris buffered saline with 0.05 % Triton X100) for one hour at room temperature, and then incubated overnight with primary antibody (1:1000 cleaved Notch1: Cell Signaling Danvers, MA, USA; 1: 10,000 GAPDH (Glyceraldehyde 3-phosphate dehydrogenase): Sigma). Following 3 washes for 10 min in TBS-T, the membrane was incubated with 1:1000 HRP (horseradish peroxidase)-conjugated anti-rabbbit Ig secondary antibody (Sigma) for 2 hours at room temperature. Antibody binding was detected using enhanced chemiluminescence (Thermo Scientific) and visualization using ChemiDoc XRS+ System (Bio-Rad Laboratories).
Transfection of 231-BR cells with Notch1 shRNA
Cells were transiently transfected with a pool of 4 pre-designed Notch1 short hairpin RNA (shRNA) vectors or a scrambled vector (SureSilencing, SABiosciences), according to the manufacturer’s instructions (for sequences, see Supplementary Table 1). 231-BR cells were transfected using the Lonza nucleofection technology (Lonza, Shawinigan, QC, Canada). Briefly, 1 ×106 231-BR cells were resuspended in Cell Line Nucleofector Kit V (Lonza), mixed with 2 μg cDNA and pulsed with the program X-13, as suggested by the manufacturer. Immediately after nucleofection, cells were transferred into wells containing pre-warmed (37 °C) culture medium. Silencing of Notch1 expression was confirmed by real-time PCR.
Cell Proliferation Assay
231-BR, scrambled shRNA and Notch1 shRNA-transfected cells were plated at a density of 5 × 104 cells per well in 10 mm dishes in DMEM plus 10 % FBS (n = 3/timepoint). Every 24 hours, for 3 days, triplicate cultures were trypsinized and counted by hemocytometer.
Cell Invasion Assay
231-BR, scrambled shRNA and Notch1 shRNA-transfected cells (2.5 × 104 cells/well), in DMEM containing 10 % FBS, were seeded in the upper compartment of Matrigel™-coated inserts (24-well plate, 8 μM pore size; BD Biocoat, BD Biosciences). The lower chamber was filled with DMEM with 25 % FBS. After 24 hours of incubation at 37 °C, non-migrated cells in the upper chamber were removed from the upper surface of the filters using a phosphate buffered saline (PBS)-soaked cotton swab. This was followed by fixation in 1 % glutaraldehyde and staining with 0.1 % crystal violet. Cells fixed on the lower face of the Matrigel™ chambers were counted under a light microscope. Five high powered fields were counted for each well and mean numbers of invaded cells per field were counted. All experiments were carried out in duplicate, using two independent assays.
Data Analysis
In vivo experiments with FACS-sorted cells were performed three times, with data pooled from all three (Control, unsorted cells: n = 7, CD44hi/CD24lo cells: n = 5, CD44lo/CD24hi cells: n = 10). shNotch1 in vivo experiments were conducted once (Control, untreated: n = 7, 231-BR-shScrambled injections: n = 9, 231-BR-shNotch1 injections: n = 7). DAPT treatments were conducted once (Untreated mice: n = 10, Vehicle-treated mice: n = 9, 8 mg/kg DAPT-treated mice: n = 10). Statistical analysis was performed using SPSS statistics version 15.0 (SPSS Inc., Chicago, IL). Differences between means were determined using the Kruskal Wallis (for in vivo data), ANOVA or Student’s paired t-test (for in vitro data) tests. Data are presented as the mean ± SEM.
Analysis of a Published Microarray Dataset
We interrogated a comprehensive gene expression microarray data set of 295 primary breast tumors (obtained from Rosetta Inpharmatics, Seattle, WA, USA) prepared by van de Vijver and colleagues, who used it to identify gene expression signatures predictive of outcome. The detailed clinical characteristics of these tumours have been previously published (46). Univariate relationships between Notch mRNA log ratio data and patient outcome were assessed using the Log Rank test (RR; relative risk, CI; confidence interval). A RR of 1 indicates that the risk of relapsing or dying is the same in both groups, while a RR of >1 indicates an increased risk of relapse or death. Confidence intervals were calculated for the RR, which was then tested for statistical significance using the Wald test. Data were assigned arbitrary cut-off points of 25th, 50th and 75th percentiles, with the 75th percentile yielding the most interesting results. Data are presented using Kaplan Meier curves. P values of ≤0.05 were regarded as being statistically significant. As above, data was analyzed using SPSS statistics version 15.0 (SPSS Inc., Chicago, IL).
RESULTS
Characterization of CSC properties of 231-BR cells
MDA-MB-231 parental breast cancer cells have been shown to have little or no metastatic growth to the brain in vivo (41). The 231-BR cell line model used in this study has been selected in vivo for increased brain metastatic ability (40). Previous work reported that parental MDA-MB-231 cells had approximately 80 % cells with a CD44hi/CD24lo phenotype (13). The proportion of CD44hi/CD24lo cells in brain metastatic 231-BR cells was compared with parental 231 cells by flow cytometry. We found that 231 cells were 92.12 % CD44hi/CD24lo and the 231-BR cells were 90.68 % CD44hi/CD24lo (Figure 1A). Previous work also showed that a distinct subgroup of 231 parental cells displayed increased ALDH activity, another marker of potential CSC function (13). However, upon analysis of 231-BR cells, a uniform, whole population increase in ALDH activity was observed (Figure 1B), which did not reach statistical significance and was therefore not suitable, in this investigation, for use as an additional enrichment factor.
Figure 1.
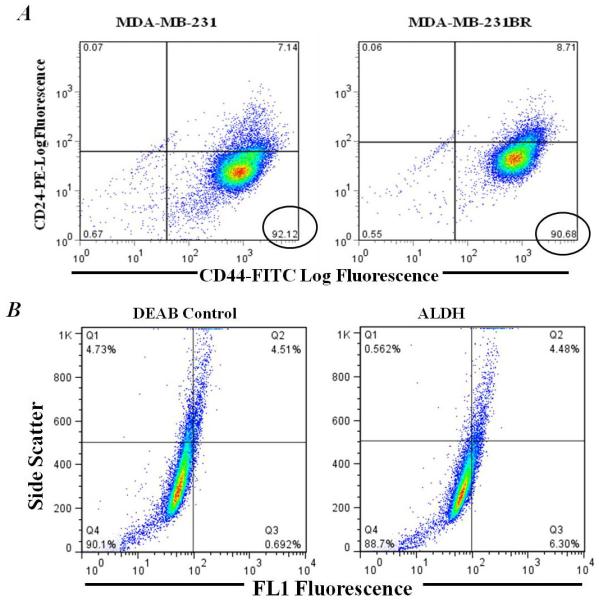
A, Flow cytometry analysis of CD44/CD24 cell surface markers. Antibodies used included an anti-CD44 antibody (clone IM7) conjugated to fluorescein isothiocyanate (FITC), an anti-CD24 antibody (clone ML5) conjugated to phycoerytherin (PE), (appropriate FITC and PE conjugated IgG isotype controls were also included, data not shown). Left hand panel shows representative dot plots of CD44-FITC versus CD24-PE expression for MDA-MB-231 parental cells. The right hand panel shows representative dot plot of CD44-FITC versus CD24-PE expression for 231-BR cells. Cells which fell in the lower right hand quadrant (percentages circled) we considered to express the CSC phenotype of interest (CD44hi/CD24lo). B, Flow cytometry analysis of ALDH activity in 231-BR cells. Cells were analyzed using the Aldefluor® assay kit (StemCell Technologies), according to the manufacturer’s instructions. As a negative control an aliquot of Aldefluor-stained cells was immediately quenched with 1.5 mM DEAB (diethylaminobenzaldehyde), a specific ALDH1 inhibitor, was included as a negative control. Cells were analyzed using the green fluorescence channel (FL1) on a BD FACSCalibur cytometer. The left panel shows a representative dotplot of cells treated with DEAB, the ALDH inhibitor. The right panel shows a representative dotplot of ALDH activity.
To continue our investigation into the CSC phenotype in brain metastases, FACS was used to isolate subsets of cells from 231-BR cells using CD44hi/CD24lo expression as the primary sort criteria. The resulting cell subsets were designated as CD44hi/CD24lo (stem-like, referred to as CSC) and CD44lo/CD24hi (non-stem-like, referred to as non-CSC). CD44hi/CD24lo cells (average of 99.49 % population purity following cell sorting) and CD44lo/CD24hi (average of 95.4 % purity) sorted cells. The sorted cells, as well as unsorted 231-BR cells (which are 90.68 % CD44hi/CD24lo), were injected by the intracardiac route into nude mice, as described previously (42), and allowed to form tumors for 28 days (Table 1). On day 28, mice were sacrificed and brains were removed, formalin fixed and paraffin embedded. The incidence and numbers of micro- and macrometastases were determined by histology (data presented as an average of 3 sections/mouse), by 2 independent observers using an ocular micrometer. Representative H&E stained images can be seen in Supplementary Figure 1. 231-BR cells with a CD44lo/CD24hi non-CSC phenotype formed a mean of 44.5 ± 9.5 micrometastases and 16.2 ± 3.6 macrometastases per histologic section. This represented a 25 % and 45 % reduction, respectively, compared to brain metastasis formation by 231-BR cells with a CD44hi/CD24lo CSC phenotype, which produced 59.4 ± 17.8 micrometastases and 29.9 ± 10.2 macrometastases per section. The formation of both micrometastases and macrometastases was statistically significantly different in the two sorted populations (micrometastases: p < 0.0001; macrometastases: p = 0.02, Kruskal Wallis test). Thus, cells sorted for a reduced stem-like phenotype had a reduced ability to form brain metastases. The number of metastases formed in the CD44lo/CD24hi experimental group is likely due to the reduced purity of the population obtained following cell sorting or the uniform expression of ALDH observed in the entire cell population (Figure 1B). In addition, it is important to note that the phenotype of CD44hi/CD24lo is a dynamic one and the population tends to return to baseline at approximately 7 days post-sorting (data not shown).
Table 1. In vivo analysis of the effect of sorting for CD44hi/CD24lo and CD44lo/CD24hi on the formation of brain metastases compared to unsorted 231-BR cells*.
| Group | Incidence | Mean Micrometastases (Median: Range) | Mean Macrometastases (Median: Range) |
|---|---|---|---|
| Unsorted 231-BR | 7/11 | 73.3 (70.5: 44-135) | 27.7 (20: 2-48) |
| CD44hi/CD24lo | 5/9 | 59.4 (65: 28-119) | 29.9 (30: 8-54) |
| CD44lo/CD24hi | 10/10 | 44.5 (50: 8-79)* | 16.2 (17: 1-29)** |
Data shown were pooled from three independent experiments. 1.75 × 105 cells (sorted and unsorted) were injected in nude mice via the intracardiac route. Mice were sacrificed 28 days later and brains were removed for histological analysis. Numbers shown are average values from 3 sections per mouse (median value and range also displayed). CD44lo/CD24hi formed fewer micrometastases and macrometastases when compared to CD44hi/CD24lo or unsorted 231-BR cells. Data was analyzed using Kruskal Wallis test
p < 0.05
p < 0.001.
Role of Notch pathway in breast cancer aggressiveness and the CSC phenotype of 231-BR cells
Due to the reported role of Notch signaling in the maintenance of the CSC phenotype (28-30), the role of Notch signaling in the 231-BR model was studied. The levels of Notch 1-4 mRNA in 231 and 231-BR cells was determined by RT-PCR. Both 231 and 231-BR cell lines expressed Notch1, Notch2, Notch3 and Notch4 mRNA at similar levels, relative to 18S rRNA (Figure 2A), with no statistical differences observed. To extend our investigation, we analyzed a published database from the Netherlands Cancer Institute (295 consecutive primary breast cancer patients) (46). We found that elevated levels (>75th percentile) of Notch1 mRNA were associated with poor overall survival in patients with primary breast carcinoma (Figure 2B), confirming a potential role for elevated Notch1 in breast cancer aggressiveness. No significant relationships were observed with Notch2, Notch3 or Notch4 in this dataset. Based on these observations, we pursued Notch1 as a target for further study. Levels of cleaved Notch1 protein (NICD, Notch 1 intracellular domain, cleaved by γ-secretase) were increased in 231-BR cells compared to 231 parental cells (Figure 2C), as determined by Western blotting. The data indicate that the Notch pathway is expressed, and that intracellular cleavage of Notch 1 is elevated in 231-BR cells.
Figure 2.

A, Notch 1-4 mRNA was measured in MDA-MB-231 parental cells (231) and 231-BR cells. There were no significant differences in mRNA expression. B, Elevated levels of Notch 1 (>75th percentile) were associated with a significantly shorter overall. Univariate relationships between Notch mRNA log ratio data, from a publicly available database of 295 consecutive breast cancer patients, and patient outcome were assessed using the Log Rank test. Confidence intervals were calculated for the relative risk, which was then tested for statistical significance using the Wald test. Data was assigned arbitrary cut-off points of 25th, 50th and 75th percentiles, with the 75th percentile yielding the most interesting results. Data is presented using Kaplan Meier curves. No significant relationships were observed with Notch 2, Notch 3 or Notch 4. RR; relative risk, CI; confidence interval. C, Cleaved Notch1IC protein was elevated in 231-BR cells compared with parental MDA-MB-231 cells.
We then asked whether inhibition of the Notch pathway would alter the CSC phenotype of 231-BR cells. A commercial inhibitor of γ-secretase, DAPT, was utilized. To test the activity of DAPT on the 231-BR cell line, cells were cultured in 5 μM DMSO +/− 5 μM DAPT for 24 hr, and the mRNA levels of Notch pathway members were quantified by RT-PCR. Reductions in Notch 1-4 mRNA levels (Figure 3A) were observed in response to DAPT treatment, with reductions in Notch 1, 3 and 4 reaching statistical significance.
Figure 3.

Effect of γ-secretase inhibition using DAPT (Sigma) on in vitro cellular behavior. A, DAPT treatment reduced the expression of Notch 1, Notch 2, Notch 3 and Notch 4 mRNA, p < 0.01, ANOVA (*p<0.05, **p<0.001, Student’s paired t-test DAPT compared to DMSO). B, Cancer stem cells markers, CD44 and CD24 were determined in 231-BR, DMSO-treated and 5 μM DAPT-treated cells. DAPT reduced the percentage of CD44hi/CD24lo cells, compared to IgG controls, by approximately 15 % compared to untreated or DMSO-treated control cells (p = 0.001, ANOVA; p < 0.05 DAPT compared to DMSO control, Student’s paired t-test). C, DAPT treatment resulted in a significant reduction in cellular growth at 24 hour and 48 hours (* p = 0.023, p < 0.001, respectively, ANOVA).
To ask if Notch inhibition influenced the stem cell phenotype of 231-BR cells in vitro, cells were cultured in DMSO +/− DAPT and the CD44/CD24 phenotypes quantified. Flow cytometry analysis revealed that the percentage of 231-BR cells with a CD44hi/CD24lo CSC phenotype was reduced by ~15 % (from ~92 % to ~75 %) in cells treated with 5 μM DAPT for 24 hours compared to DMSO-treated or untreated cells (p = 0.001, ANOVA) (Figure 3B). This was accompanied by a parallel increase in CD44hi/CD24hi markers from ~7 % to ~24 %). The effect of DAPT on 231-BR proliferation in vitro was assessed using a growth curve assay (Figure 3C). While vehicle had no significant effect on 231-BR proliferation, 5 μM DAPT reduced proliferation by 54.2 % and 45.1 % at 24 and 48 hours of culture (p = 0.023, p < 0.0001 respectively; ANOVA). In addition, treatment of 231-BR cells with 5 μM DAPT in vitro resulted in a significant reduction in mRNA levels of downstream transcriptional target Hey1 (Supplementary Figure 2A). The data indicate that DAPT, a Notch inhibitor, reduced the stem cell phenotype of 231-BR cells in vitro with a resulting anti-proliferative effect.
Effect of Notch inhibition on 231-BR brain metastasis in vivo
Extending our findings from the in vitro setting, we examined the effect of γ-secretase inhibition, using DAPT, in vivo. 231-BR cells were injected into the left cardiac ventricle of immunodeficient mice, and mice were randomized to vehicle or 8 mg/kg (3 days on, 4 days off schedule) DAPT beginning on day 14 post-injection. On day 28, mice were sacrificed and brains were removed, formalin fixed and paraffin embedded. Micro and macrometastases were counted by two independent observers on three H&E stained sections per brain, using an ocular micrometer (Table 2), as previously described (42). Mice treated with DAPT developed a mean of 26.1 ± 5.9 macrometastases per section, 25 % lower than vehicle treated (34.9 ± 2.4 per section) or untreated (34.6 ± 2.4 per section) mice (p = 0.011, Kruskal Wallis test). A similar reduction in micrometastases was observed for DAPT treated mice, with 81.1 ± 17.7 per section, a 24 % reduction from 106.1 ± 9.5 per section in the vehicle control treated mice (p = 0.046, Kruskal Wallis test). Representative H&E stained sections can be seen in Supplementary Figure 3. These data indicate that inhibition of the Notch pathway using a γ-secretase inhibitor may be a potential therapeutic avenue for treatment of brain metastases from breast cancer.
Table 2. The effect of γ-secretase inhibition, using DAPT, and Notch1 silencing by shRNA, on the formation of brain metastases from 231-BR cells in vivo*.
| Group | Incidence | Mean Micrometastases (Median: Range) | Mean Macrometastases (Median: Range) |
|---|---|---|---|
| γ-secretase inhibition | |||
| Untreated | 10/12 | 103.2 (101.5: 70-129) | 34.6 (34: 22-52) |
| Vehicle Control | 9/12 | 106.1 (97.5: 65-166) | 34.9 (35.5:21-48) |
| 8 mg/kg DAPT | 10/12 | 81.1 (85: 8-180)* | 26.1 (20.5: 2-60)* |
| Notch1 silencing | |||
| 231-BR | 7/8 | 91.7 (80: 22-173) | 32.0 (30: 4-62) |
| 231-BR-shScrambled | 9/11 | 82.7 (84: 35-130) | 23.6 (25: 9-38) |
| 231-BR-shNotch1 | 7/12 | 17.4 (15: 3-49)** | 5.8 (5: 1-18)** |
1.75 × 105 cells were injected in nude mice via the intracardiac route. γ-secretase inhibitor experiment: Metastases were allowed to form for 14 days prior to initiation of treatment. DAPT was administered i.p. on a 3 day-on, 4-day off schedule, up to day 28 (total of 6 treatments). On day 28, mice were sacrificed and brains were removed for histological analysis. Numbers shown are an average of 3 sections per mouse (median value and range also displayed). Mice treated with DAPT developed significantly fewer metastases compared to vehicle-treated (5 % ethanol in corn oil) or untreated mice. Data was analyzed using Kruskal Wallis test
p < 0.05; Notch silencing experiment: 1.75 × 105 cells (231-BR, 231-BR-shScrambled and 231-BR-shNotch1) were injected in nude mice via the intracardiac route. Mice were sacrificed 28 days later and brains were removed for histological analysis. Cells transfected with shNotch1 formed significantly fewer micrometastases and macrometastases compared with untransfected cells or cells transfected with a scrambled sequence, when injected in vivo. Data was analyzed using Kruskal Wallis test
p < 0.001
To confirm and extend these trends, Notch1 silencing via shRNA was used to investigate the role of Notch1 in multiple facets of 231-BR cellular behavior. Notch1 shRNA reduced the expression of Notch1 mRNA, as determined by quantitative real time PCR, in a specific manner (no reduction was observed for Notch2, Notch3 or Notch4 mRNA levels) as shown on Figure 4A, and downstream transcriptional target Hey1 (Supplementary Figure 2B). Notch1 knockdown resulted in a reduction in expression of CSC marker phenotype, from ~90 % to ~70 % of cells in the population exhibiting a CD44hi/CD24lo expression pattern (with a concomitant change in the CD44hi/CD24hi from ~8.5 % to ~27 %), suggesting a potential role for Notch1 in the maintenance of the CSC phenotype in 231-BR cells (Figure 4B). In vitro, Notch1 shRNA resulted in a significant reduction in cellular growth at 24 hour, 48 hour and 72 hour time points (p = 0.033, p = 0.002, p = 0.009, respectively; ANOVA) (Figure 4C). To ascertain in vitro aspects of the metastatic process, invasion assays were performed using serum gradient as an attractant. A ~60% reduction in the ability of 231-BR cells to invade Matrigel™ was observed with Notch knockdown (p < 0.001, ANOVA) (Figure 4D).
Figure 4.

Effect of Notch1 silencing by shRNA on in vitro cellular behavior. A, Notch1 shRNA resulted in knockdown of Notch1 in a specific manner. No significant knockdown of Notch2, Notch3 or Notch4 was observed (*p<0.05, **p<0.001, Student’s paired t-test shNotch1 compared to shScrambled). B, CSC markers, CD44 and CD24 were determined in 231-BR, 231-BR-shScrambled and 231-BR-shNotch1 cells. Notch1 knockdown reduced the percentage of CD44hi/CD24lo cells, compared to 231-BR and shScrambled control cells, by approximately 20 % (p < 0.05, shNotch1 compared to shScrambled; Student’s paired t-test). C, Notch1 silencing resulted in a significant reduction in cellular growth at 24 hour, 48 hour and 72 hours (p = 0.033, p = 0.002, p = 0.009, respectively, ANOVA). D, Notch1 knockdown significantly reduced the ability of 231-BR cells to invade through Matrigel™, using 24-well transwell chambers (p = 0.001, ANOVA and p < 0.05 shNotch1 compared to shScrambled, Student’s paired t-test). Error bars: ± SEM.
The effect of Notch1 knockdown on formation of brain metastases was assessed by injecting nu/nu mice with 1.75 × 105 cells intracardiac, either 231-BR, 231-BR transiently transfected with scrambled shRNA or 231-BR transiently transfected with shNotch1. Metastases were allowed to colonize the brain for 28 days. At this time, brains were removed, formalin-fixed and paraffin-embedded. Micro and macrometastases were counted by two independent observers on H&E stained sections (Table 2: representative stained sections can be seen in Supplementary Figure 4). Cells transfected with shNotch1 formed an average of 5.8 ± 1.0 macrometastases per brain section, a 74 % reduction as compared to cells expressing a scrambled shRNA (23.6 ± 1.5 per section) or untransfected 231-BR cells (32.6 ± 3.9 per section) (p < 0.001; Kruskal Wallis test). A similar reduction was observed in micrometastases using shRNA to Notch 1. Notch 1 shRNA transfected 231-BR cells produced a mean of 17.4 ± 2.9 micrometastases per section, a 79 % decreased as compared to scrambled shRNA transfected 231-BR cells (82.7 ± 5.1 per section) or untransfected 231-BR cells (91.7 ± 9.9 per section) (p < 0.001; Kruskal Wallis test). These results extend the findings observed using γ-secretase inhibition, indicating a role for Notch signaling in the formation of brain metastases in vivo
DISCUSSION
Brain metastasis from breast cancer has become the focus of recent investigations due to the inability of many current targeted therapies to effectively treat these tumors. Patients with brain metastases have very poor prognosis (2). Here we used an experimental model of breast cancer metastasis to brain, coupled with in vitro studies, to clarify mechanisms that can regulate brain metastasis. We have shown that a reduction in the proportion of cancer stem cell surface markers CD44hi/CD24lo can give rise to the formation of significantly fewer brain metastases from breast cancer. This finding is in agreement with previous work by Croker et al., who showed that breast cancer cells with a CD44hi/CD24lo phenotype (albeit in addition to ALDH as a further tool for selection of the stem cell pool) displayed enhanced cell growth, colony formation, adhesion and invasion in vitro and enhanced tumorigenicity and metastases formation in multiple organs in NOD/SCID-IL2Rγ null mice (13). In the results reported here, the CD44lo/CD24high subpopulation retained a considerable amount of brain metastatic activity however, suggesting that additional markers of “stemness” may be needed. In previous studies, CD44hi/CD24lo cell subsets grown as mammospheres have been shown to be radioresistant, at DNA and cellular levels, compared to cells grown as a monolayer and the proportion of these breast cancer initiating cells increased following short courses of fractionated irradiation (47). It will be of interest to determine the radiosensitivity of these subpopulations as brain metastases are treated by both large doses (stereotactic radiation) and fractionated smaller doses (whole brain radiotherapy) of cranial irradiation.
The data presented herein provide a strong case for preclinical consideration of Notch signaling as a therapeutic target for brain metastases of breast cancer. Increased levels of cleaved Notch1 were detected in 231-BR cells, compared to parental 231 breast cancer cells, in agreement with results shown by Nam et al. who isolated a brain-metastatic derivative of the MDA-MB-435 cells, Br4 (40). The authors showed activation of the Notch pathway in the Br4 cells, including elevated levels of Jag2, Hes1 and Hey1 mRNA (40). Proliferative and migratory properties of these cells in vitro were significantly reduced upon treatment with DAPT or Notch1 RNAi (40). In addition, we found that inhibition of Notch1 in vitro resulted in decreased cell proliferation and invasion, similar to results found by Purow et al and Rose et al (48-49) and reduced expression of Notch 1-4 mRNA. The effect of gamma secretase inhibition on Notch mRNA expression appears to be very cell-type and context dependent. In a recent publication, Wang et al. showed that DAPT reduced Notch mRNA levels in the ovarian cancer cell line A2870, in a dose dependent manner (50). The authors treated ovarian cancer cells with varying doses of DAPT and found a reduction in Notch1 mRNA, as determined by real time PCR (data normalized to beta-actin). In addition, the authors showed a significant reduction of Notch1 mRNA expression after 6 hours, which was sustained for up to 72 hours (50). The reason for this is unclear, though the regulation of Notch transcription remains largely unknown. Some studies have shown that DAPT treatment results in decreased NFΚB nuclear translocation, reducing its activity (following LPS stimulation) (51). Reduction in NFΚB has in turn been shown to reduce Notch1 mRNA transcript levels in melanocytes (52). A reduction in Notch1 mRNA levels in response to DAPT treatment was also observed, but this did not reach statistical significance (52).
Bos and colleagues identified a 243 gene signature in a genome wide expression analysis of parental CN34 and MDA-MB-231 breast cancer cells compared to derivatives that formed brain metastasis (53). Notch ligands, Jagged 1 and Jagged 2, were both identified as being differentially regulated in brain metastasis. This suggests, in agreement with the present study, that Notch signaling has a potential role in the formation of brain metastases from breast cancer. Similarly, Min et al. elucidated a panel of differentially regulated genes that may mediate breast cancer metastasis to the lung (54). Jagged 1 was also overexpressed in this study, which analyzed MDA-MB-231 and CM2 breast cancer cells selected for lung metastatic capability. However, in a gene panel of bone metastasis from breast cancer, Jagged 1 (or any Notch receptor or ligand) was not among the genes identified as potentially predicting metastasis to this site (55). It is clear that Notch signaling appears to be involved in many facets of tumor progression and may play a role in mediating metastasis not only to the brain, but also to additional local and distant sites.
Two approaches were used to demonstrate that Notch signaling inhibition was causally linked to brain metastatic colonization in the 231-BR in vivo model system. First, administration of DAPT, beginning 14 days post-injection, resulted in a ~25 % reduction in the formation of both micrometastases and macrometastases. Knockdown of Notch 1 using shRNA resulted in a more complete inhibition of brain metastasis, 74-79 %. The reasons for this discrepancy in efficacy may be numerous. First, using the shRNA approach, Notch 1 signaling was inhibited from the time of tumor cell injection into the circulation, while DAPT was only administered at a later time, when multiple micrometastases and a few macrometastases are generally visible. Though our transfections were transient, Notch1 was silenced for 7 days prior to Notch1 levels returning to baseline. As our stem-like population was also a dynamic one, we felt that this was an appropriate model for comparison.
It is possible that Notch signaling inhibition is more efficacious on single tumor cells or small micrometastases, and that clinical approaches should be aimed at prevention of brain metastases rather than their shrinkage, in line with data reported for vorinostat (56). Second, the brain permeability of DAPT is not known. Previous work using the 231-BR model system has established that brain metastases have heterogeneous levels of blood-brain barrier permeability (57), which could expose some lesions with suboptimal drug levels. In agreement with our study, a recent investigation showed that inhibition of Notch1 in primary breast tumor samples and cell lines reduced tumor formation in vivo and reduced stem cell activity (ESA+CD44+CD24lo phenotype), though they found that inhibition of Notch4 elicited a greater effect than inhibition of Notch1 in their models (58).
Taken together, our data indicate that Notch signaling plays an important role in the formation of brain metastases from breast cancer, in part due to its role in maintaining the pool of CD44hi/CD24lo putative cancer ‘stem-like’ cells. It remains to be determined whether the effects observed vis-à-vis cancer stem cells and Notch inhibition are cell type, context and/or methodology-dependent. Also, rational drug combinations using brain-permeable therapies and/or radiation will be a topic of further investigation. Currently, a clinical trial is underway in patients with advanced breast cancer using a Notch inhibitor (γ-secretase inhibitor), MK0752 (www.clinicaltrials.gov: NTC00106145). Results are anticipated in 2011. Further elucidation of the characteristics of these metastases may enable the development of novel therapeutic strategies for brain metastases from breast cancer.
Supplementary Material
ACKNOWLEDGMENTS
Grant support: This research was supported by grant #W81XWH-06-2-0033 US Department of Defense Breast Cancer Research Program (to AFC, PJF and PSS). PMM was the recipient of a Postdoctoral Fellowship from the Translational Breast Cancer Research Training Program at the London Regional Cancer Program. ALA is supported by research funding from the Ontario Institute of Cancer Research (#08NOV-230) and a Canadian Institutes of Health Research New Investigator Award. AFC is Canada Research Chair in Oncology, supported by the Canada Research Chairs Program.
The authors would like to thank Dr. Kristin Chadwick at the Robarts Research Institute, University of Western Ontario, for valuable advice and for conducting cell sort experiments. Dr. Dwayne Jackson (Dept. Medical Biophysics, University of Western Ontario) for use of molecular facilities and Alysha Croker (Dept. of Anatomy & Cell Biology, University of Western Ontario) and Dr. Ben Hedley (London Laboratory Services Group, Hematology Research, London, ON) for help with flow cytometric analyses.
ABBREVIATIONS
- ALDH
Aldehyde dehydrogenase
- CSC
cancer stem cell
- DAPT
N-[N-(3,5-Difluorophenacetyl]-L-alanyl)-S-phenylglycine t-butyl ester; γ-secretase inhibitor
- DEAB
diethylaminobenzaldehyde
- RIPA
radioimmunoprecipitation assay
REFERENCES
- 1.Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS. Breast cancer metastasis to the central nervous system. Am J Pathol. 2005;167:913–920. doi: 10.1016/S0002-9440(10)61180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmieri D, Smith QR, Lockman PR, et al. Brain metastases of breast cancer. Breast Dis. 2006;26:139–147. doi: 10.3233/bd-2007-26112. [DOI] [PubMed] [Google Scholar]
- 3.Gaedcke J, Traub F, Milde S, Wilkens L, Stan A, Ostertag H, Christgen M, von Wasielewski R, Kreipe HH. Predominance of the basal type and HER-2/neu type in brain metastasis from breast cancer. Mod Pathol. 2007;20:864–870. doi: 10.1038/modpathol.3800830. [DOI] [PubMed] [Google Scholar]
- 4.Lower EE, Drosick DR, Blau R, Brennan L, Danneman W, Hawley DK. Increased rate of brain metastasis with trastuzumab therapy not associated with impaired survival. Clin Breast Cancer. 2003;4:114–119. doi: 10.3816/cbc.2003.n.016. [DOI] [PubMed] [Google Scholar]
- 5.Lin NU, Bellon JR, Winer EP. CNS metastases in breast cancer. J Clin Oncol. 2004;22:3608–3617. doi: 10.1200/JCO.2004.01.175. [DOI] [PubMed] [Google Scholar]
- 6.Lin NU, Winer EP. Brain metastases: the HER2 paradigm. Clin Cancer Res. 2007;13:1648–1655. doi: 10.1158/1078-0432.CCR-06-2478. [DOI] [PubMed] [Google Scholar]
- 7.Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer. 2011;11:352–63. doi: 10.1038/nrc3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke MF. Oncogenes, self-renewal and cancer. Pathol Biol (Paris) 2006;54:109–111. doi: 10.1016/j.patbio.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. PNAS USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hess DA, Meyerrose TE, Wirthlin L, et al. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood. 2004;104:1648–1655. doi: 10.1182/blood-2004-02-0448. [DOI] [PubMed] [Google Scholar]
- 11.Hess DA, Wirthlin L, Craft TP, et al. Selection based on CD133 and high aldehyde dehydrogenase activity isolates long-term reconstituting human hematopoietic stem cells. Blood. 2006;107:2162–2169. doi: 10.1182/blood-2005-06-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croker AK, Goodale D, Chu J, et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009;13:2236–2252. doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann D, de Strooper B, Serneels L, et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 15.Brou C, Logeat F, Gupta N, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5:207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 16.Ray WJ, Yao M, Mumm J, et al. Cell surface presenilin-1 participates in the gamma-secretase-like proteolysis of Notch. J Biol Chem. 1999;274:36801–36807. doi: 10.1074/jbc.274.51.36801. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Fischer WH, Gill GN. Regulation of the ERBB-2 promoter by RBPJkappa and NOTCH. J Biol Chem. 1997;272:14110–14114. doi: 10.1074/jbc.272.22.14110. [DOI] [PubMed] [Google Scholar]
- 18.Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic) Mol Cell Biol. 2001;21:5925–5934. doi: 10.1128/MCB.21.17.5925-5934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki T, Aoki D, Susumu N, Udagawa Y, Nozawa S. Imbalanced expression of TAN-1 and human Notch4 in endometrial cancers. Int J Oncol. 2000;17:1131–1139. doi: 10.3892/ijo.17.6.1131. [DOI] [PubMed] [Google Scholar]
- 20.Veenendaal LM, Kranenburg O, Smakman N, Klomp A, Borel Rinkes IH, van Diest PJ. Differential Notch and TGFbeta signaling in primary colorectal tumors and their corresponding metastases. Cell Oncol. 2008;30:1–11. doi: 10.1155/2008/839076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dang TP, Gazdar AF, Virmani AK, et al. Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J Natl Cancer Inst. 2000;92:1355–1357. doi: 10.1093/jnci/92.16.1355. [DOI] [PubMed] [Google Scholar]
- 22.Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–1525. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- 23.Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66:4715–24. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
- 24.Lee CW, Simin K, Liu Q, et al. A functional Notch-survivin gene signature in basal breast cancer. Breast Cancer Res. 2008;10 doi: 10.1186/bcr2200. [Epub] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickson BC, Mulligan AM, Zhang H, et al. High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol. 2007;20:685–693. doi: 10.1038/modpathol.3800785. [DOI] [PubMed] [Google Scholar]
- 26.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6:R605–615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouras T, Pal B, Vaillant F, et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell. 2008;3:429–441. doi: 10.1016/j.stem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Bolos V, Blanco M, Medina V, Aparicio G, Diaz-Prado S, Grande E. Notch signalling in cancer stem cells. Clin Transl Oncol. 2009;11:11–19. doi: 10.1007/s12094-009-0305-2. [DOI] [PubMed] [Google Scholar]
- 29.Pannuti A, Foreman K, Rizzo P, et al. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010;16:3141–3152. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kakarala M, Wicha MS. Cancer stem cells: implications for cancer treatment and prevention. Cancer J. 2007;13:271–275. doi: 10.1097/PPO.0b013e318156da4e. [DOI] [PubMed] [Google Scholar]
- 31.Farnie G, Clarke RB. Mammary stem cells and breast cancer--role of Notch signalling. Stem Cell Rev. 2007;3:169–175. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 32.Grudzien P, Lo S, Albain KS, et al. Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010;30:3853–67. [PubMed] [Google Scholar]
- 33.Monteiro J, Fodde R. Cancer stemness and metastasis: Therapeutic consequences and perspectives. Eur J Cancer. 2010;46:1198–1203. doi: 10.1016/j.ejca.2010.02.030. [DOI] [PubMed] [Google Scholar]
- 34.Charafe-Jauffret E, Ginestier C, Flora I, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoogen C. v. d., Horst G. v. d., Cheung H, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010;70:5163–5173. doi: 10.1158/0008-5472.CAN-09-3806. [DOI] [PubMed] [Google Scholar]
- 36.Hughes D. How the NOTCH pathway contributes to the ability of osteosarcoma cells to metastasize. Cancer Treat. Res. 2010;152:479–496. doi: 10.1007/978-1-4419-0284-9_28. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Imanaka N, Chen J, Griffin J. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer. 2010;102:351–360. doi: 10.1038/sj.bjc.6605486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafeez BB, Adhami V, Asim M, et al. Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomittant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clin. Cancer Res. 2009;15:452–459. doi: 10.1158/1078-0432.CCR-08-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu X, Feng F, Wang Y, et al. Blockade of Notch signaling in tumor-bearing mice may lead to tumor regression, progression or metastasis, depending on tumor cell types. Neoplasia. 2009;11:32–38. doi: 10.1593/neo.81008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nam D, Jeon H, Kim S, et al. Activation of Notch signaling in a xenograft model of brain metastasis. Clin. Cancer Res. 2008;14:4059–4066. doi: 10.1158/1078-0432.CCR-07-4039. [DOI] [PubMed] [Google Scholar]
- 41.Yoneda T, Williams PJ, Hiraga T, Niewolna M, Nishimura R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J Bone Miner Res. 2001;16:1486–1495. doi: 10.1359/jbmr.2001.16.8.1486. [DOI] [PubMed] [Google Scholar]
- 42.Gril B, Palmieri D, Bronder JL, et al. Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst. 2008;100:1092–1103. doi: 10.1093/jnci/djn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sjolund J, Johansson M, Manna S, et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest. 2008;118:217–228. doi: 10.1172/JCI32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 45.Pfaffl MW. A new mathematical model for relative quantification in real time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 47.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 48.Purow BW, Haque RM, Noel MW, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65:2353–2363. doi: 10.1158/0008-5472.CAN-04-1890. [DOI] [PubMed] [Google Scholar]
- 49.Rose SL, Kunnimalaiyaan M, Drenzek J, Seiler N. Notch 1 signaling is active in ovarian cancer. Gynecol Oncol. 2010;117:130–133. doi: 10.1016/j.ygyno.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Wang M, Wu L, Wang L, Xin X. Downregulation of Notch1 by gamma secretase inhibition contributes to cell growth inhibition and apoptosis in ovarian cancer cells A2780. Biochem Biophys Res Commun. 2010;393:144–149. doi: 10.1016/j.bbrc.2010.01.103. [DOI] [PubMed] [Google Scholar]
- 51.Monsalve E, Ruiz-Garcia A, Baladron V, et al. Notch1 upregulates LPS-induced macrophage activation by increasing NF-kappaB activity. Eur J Immunol. 2009;39:2556–2570. doi: 10.1002/eji.200838722. [DOI] [PubMed] [Google Scholar]
- 52.Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ, Powell MB. Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest. 2008;118:3660–3670. doi: 10.1172/JCI36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smid M, Wang Y, Klijn JG, et al. Genes associated with breast cancer metastatic to bone. J Clin Oncol. 2006;24:2261–7. doi: 10.1200/JCO.2005.03.8802. [DOI] [PubMed] [Google Scholar]
- 56.Palmieri D, Lockman P, Thomas F, et al. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer. Clin. Cancer Res. 2009;15:6148–6157. doi: 10.1158/1078-0432.CCR-09-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lockman PR, Mittapalli RK, Taskar KS, et al. Heterogeneous Blood-Tumor Barrier Permeability Determines Drug Efficacy in Experimental Brain Metastases of Breast Cancer. Clin. Cancer Res. 2010;16:5664–78. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harrison H, Farnie G, Howell SJ, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–18. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.