

Abstract
Radiation therapy remains a promising modality for curative treatment of localized prostate cancer, but dose-limiting toxicities significantly limit its effectiveness. Agents that enhance efficacy at lower radiation doses might have considerable value in increasing tumor control without compromising organ function. Here, we tested the hypothesis that the PARP inhibitor ABT-888 (veliparib) can enhance the response of prostate cancer cells and tumors to ionizing radiation (IR). Following exposure of DU-145 and PC-3 prostate cancer cell lines to the combination of 10μM ABT-888 and 6 Gy, we observed similar persistence between both cell lines of DNA damage foci and in vitro radiosensitization. We have previously observed that persistent DNA damage foci formed after ABT-888 plus IR efficiently promote accelerated cell senescence, but only PC-3 cells displayed the expected senescent response of G2/M arrest, induction of p21 and β-galactosidase expression, and accumulation as large, flat cells. In turn, combining ABT-888 with 6 Gy resulted in delayed tumor regrowth compared with either agent alone only in PC-3 xenograft tumors while DU-145 tumors continued to grow. By 7 days after treatment with ABT-888 plus IR, PC-3 tumors contained abundant senescent cells displaying persistent DNA damage foci, but no evidence of senescence was noted in the DU-145 tumors. That equivalent radiosensitization by ABT-888 plus IR in vitro failed to predict comparable results with tumors in vivo suggests that the efficacy of PARP inhibitors may partially depend on a competent senescence response to accumulated DNA damage.
Keywords: Prostate cancer, PARP, radiation, senescence, PTEN
INTRODUCTION
Current treatment options for localized prostate cancer are radiation therapy or surgery. Both have shown similar survival outcomes in low- and intermediate-risk patients. Contemporary radiotherapy approaches such as intensity-modulated radiation therapy (IMRT) have permitted increased delivery of radiation to the prostate while sparing adjacent organs, reducing the potential for acute and chronic toxicity. However, proctitis, cystitis and erectile dysfunction remain significant complications of high dose radiotherapy. In turn, local failure after radiotherapy remains 20–35% in intermediate and high risk patients (1, 2), leading to increased metastasis and lower survival. Hormone therapy has proven value when combined with localized radiotherapy in intermediate- and high-risk prostate cancer patients, but carries its own set of morbidities, including increased cardiovascular and thromboembolic risk (3, 4). Novel agents with more attractive side effect profiles that can be combined with radiotherapy to improve local control in high risk patients and/or permit a dose reduction in lower risk patients would be of great value.
An emerging strategy to improve efficacy at lower IR doses is the use of radiosensitizers to target recognition and repair of DNA damage (5). Poly(ADP-ribose) polymerases (PARP) are a family of enzymes that use NAD+ as a substrate to polymerize PAR onto their cellular targets (6). The PARP1 and PARP2 isozymes are activated by DNA damage and participate in repair of single-strand breaks (SSBs) by activating XRCC1 and base-excision repair (BER), and double-strand breaks (DSBs) likely through influence on both the homologous recombination (HR) and non-homologous end joining (NHEJ) mechanisms. After a DSB, PARP is rapidly recruited and triggers poly-ADP ribosylation of PARP itself, histones and other mediator proteins to stimulate chromatin loosening and DNA repair. PARP has long been considered a promising therapeutic target, and several small-molecule PARP1 and PARP2 inhibitors are currently in preclinical and clinical trials, alone or in combination with DNA-damaging agents (7, 8). It has been observed that PARP is activated by ionizing radiation (IR) and chemotherapy agents, and this has provided the rationale to examine the combined effects of PARP inhibitors and genotoxic therapy in tumor models and in clinical trials (9–11). Recent results have established an ability of PARP inhibitors to target cancers of specific genotypes via “synthetic lethality” (12–15), wherein PARP inhibition exposes the deficiency in tolerance for DNA damage created by defects in a DNA repair pathway such as HR by inhibiting the compensatory pathway, NHEJ. A specific example is the sensitization of BRCA mutant cancer cells to PARP inhibition, causing selective tumor cytotoxicity (16, 17). BRCA1 and BRCA2 mutations are not considered a major cause of familial or sporadic prostate cancer. However, a number of other mutations that decrease HR repair responses can also sensitize cells to PARP inhibitors, including defects in the inositide phosphatase PTEN, a gene commonly inactivated in prostate cancer (18). Downregulation of the HR pathway under hypoxic conditions can also lead to sensitization to PARP inhibition, an effect dubbed “contextual synthetic lethality” (19). Cells deficient in DNA DSB repair have been shown to be sensitized by PARP inhibitors to DNA damaging agents (20). Perhaps mediating all these effects, PARP may be continuously recruited to persistent damage in HR-defective cells, resulting in its modification by poly(ADP-ribose) and partial inactivation (21).
Nonetheless, the mechanisms by which PARP inhibitors mediate their beneficial effects in vivo remain poorly defined. Our previous work demonstrated the combination of IR and the PARP inhibitor ABT-888 (veliparib; 2-[(R)-2-methylpyrrolidin- 2-yl]-1H-benzimidazole-4-carboxamide) increased breast cancer cell senescence in vitro and in vivo, implicating persistent DNA damage as a mechanism (22). Senescence is an important tumor suppressive mechanism (23–25). The cellular equivalent of aging, replicative senescence is a DNA damage checkpoint response to telomere erosion mediated by activation of the p53/p21 and/or p16/ARF/Rb pathways and characterized by irreversible cell cycle arrest rather than cell death (26–28). Stress-induced premature senescence or accelerated senescence is an analogous persistent cell cycle arrest in cells with otherwise unlimited proliferative capacity, due to oncogene activation, oxidative stress, excessive mitogenic signals, chromatin perturbation or accumulation of unrepaired DNA damage (25, 29–31). Current data suggest that the modified chromatin foci at sites of persistent DNA breaks serve a role in signaling to promote senescent arrest (31, 32). The defects in the p53 and/or Rb tumor suppressor pathways common in cancer may allow tumor cells to maintain genomic instability and tolerate persistent DNA damage, by blocking senescent signaling while promoting cell proliferation and survival.
γH2AX and 53BP1 localization to IR induced foci (IRIF) can serve as proxies for unrepaired DSBs and the DNA damage response (33, 34). Herein, by exploiting green fluorescent protein (GFP) fused to the chromatin-binding domain of 53BP1 as a live-cell reporter, and fluorescent immunocytochemistry for IRIF markers γH2AX and endogenous 53BP1, we monitored the effects of PARP inhibition on irradiated prostate cancer cells and tumor xenografts. We hypothesized that inhibition of PARP using ABT-888 would enhance the antitumor effects of radiation in human prostate cancer cells in vitro and in experimental prostate cancer tumors in mice. Surprisingly, although PARP inhibition mediated radiosensitivity in both tumor cell lines in vitro, only PC-3 exhibited significant tumor regression in vivo. Our results suggest that in vitro assays of radiosensitivity may not predict in vivo efficacy of PARP inhibitors with radiation and that induction of senescence may be an important mechanism of PARP induced radiosensitivity in vivo.
MATERIALS AND METHODS
Cell cultures and constructs
Two human androgen-unresponsive prostate cancer cell lines were used: PC-3, which is PTEN-negative, p53-null and DU-145, PTEN wild type, p53 mutated (35). Both cell lines were purchased from American Type Culture Collection (ATCC). They have been authenticated by short tandem repeat (STR) analysis at Johns Hopkins University on February of 2011, using the Identifiler kit (Applied Biosystems), and compared to known profiles (ATCC STR profile database and NCI-60 cell line panel, ref. 36).
To examine the effect of PARP inhibition on IRIF persistence in living cells, we exploited our previously described IRIF reporter consisting of GFP fused to the 53BP1 IRIF binding domain, expressed under tetracycline-inducible control (GFP-IBD, ref. 22). GFP-IBD cloned into the pLVX-Tight-Puro vector (Clontech) was transfected along with pLVX-Tet-On Advanced (Vector) into the PC-3 cell line, using FuGENE HD transfection reagent (Roche). Following G418 and puromycin selection, cells cultured in DMEM/F12 medium (Invitrogen) with 10% Tet system-approved fetal bovine serum (Clontech) were induced with 1 μg/ml doxycycline and sorted to establish a PC-3 GFP-IBD cell line.
Immunofluorescence
Antibodies used were rabbit anti-phospho-H2AX Ser139 diluted 1:500 (γH2AX, Cell Signaling Technology) and rabbit anti-53BP1 diluted 1:500 (Novus Biologicals), detected by Texas Red anti-rabbit IgG diluted 1:1000 (Vector Laboratories). Cells were grown in cover slips and after treatment they were rinsed with PBS and fixed with 4% paraformaldehyde for 30 minutes at 4°C. Permeabilization was done with 0.3% Triton X-100 and blocking with 5% bovine serum albumin in PBST for 30 minutes. Incubation with primary antibodies was performed at room temperature for 1 hour. After washing with PBS, incubation with secondary antibodies was done at room temperature for 1 hour in dark. Cover slips were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted using ProLong Gold anti-fade reagent (Invitrogen). Harvested tumors were fixed in 10% formalin for 48 h and embedded in paraffin. 4 μm sections were deparaffinized and then stained using the same protocol. Images were captured on a Zeiss Axiovert 200M epifluorescence microscope with a 63X PlanApo 1.4 NA objective using a Hamamatsu ORCA ER digital camera and Improvision OpenLab software.
Clonogenic assay
Five hundred cells were seeded to form colonies in p100 plates and treated the next day with 6 Gy alone, or with 10 μM of ABT-888 followed by 6 Gy 30 minutes later. When sufficiently large colonies with at least 50 cells were visible (after 1 week for PC-3 and 2 weeks for DU-145 cells) the plates were fixed with methanol and stained with crystal violet. Colonies with more than 50 cells were counted.
Cell death and cell cycle analysis
For cell death analysis, cells were collected at 48 h after treatment and stained with propidium iodide (PI). For cell cycle analysis, cells were fixed in ice cold methanol while shaking to avoid agglutination and resuspended in PBS plus RNAse and PI. Stained cells were analyzed in an LSR-II flow cytometer (Becton Dickinson). Data were analyzed on FlowJo (Becton Dickinson) using the Cell Cycle platform and calculating G1, S and G2/M fractions by fit to the Watson Pragmatic model.
qPCR gene expression analysis
Total RNA was isolated using Trizol (Invitrogen) and quantified using the QuBit platform (Invitrogen). 850 ng total RNA was subjected to DNAse I digestion using Amplification Grade DNAseI (Invitrogen) following the manufacturer’s protocol. 550 ng DNAsed RNA was subjected to cDNA synthesis in a 20 μl reaction volume using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and subsequently diluted 1:10 for use in qRT-PCR. qRT-PCR was performed on the ABI7900HT in a 384-well plate in a 5 μl reaction volume containing 2.5 μl 2x Power SYBR master Mix (Applied Biosystems), 0.5 μl of 10 μM primer mixture and 2 μl of the diluted cDNA. GAPDH was used as the endogenous control and fold change calculations were made using the comparative ct method. Primer sequences were as follows:
CDKN1A(p21)-f: GCGAGGCCGGGATGAGTTGG
CDKN1A(p21)-r: CAGCCGGCGTTTGGAGTGGT
GAPDH-f: CTCTGCTCCTCCTGTTCGAC
GAPDH-r: GTTAAAAGCAGCCCTGGTGA
Senescence-associated β galactosidase (SA β-Gal) staining
The SA β-Gal assay was performed as described before (37). Images were captured on a Zeiss Axiovert 200M and Zeiss Axiocam color digital camera controlled by OpenLab software with a 20X objective.
Xenograft tumors
Female athymic nude mice underwent s.c. injection of 1 × 107 PC-3 or DU-145 cells in 100 μl of PBS. Once tumors grew to 100 mm3, mice received a dose of 0 or 6 Gy and no ABT-888 or 25 mg/kg of ABT-888 in water twice daily by oral gavage 48 hours before IR and for 48 hours after IR.
RESULTS
PARP inhibition induces DSB foci in PC-3 cells without additional genotoxic therapy
To study the molecular effects of treatment with the PARP inhibitor ABT-888 and radiation in prostate cancer cell lines, we first investigated their effects on formation of DNA damage foci in PC-3 and DU-145 cells. We initially monitored the effect of ABT-888 alone in PC-3 cells expressing GFP-IBD (PC-3 GFP-IBD). Unirradiated PC-3 cells displayed pan-nuclear GFP-IBD, with absent or sporadic nuclear foci. Addition of 10 μM ABT-888 to the culture media caused the GFP-IBD reporter to relocalize to nuclear foci, noted at 2 hours and that did not resolve after 24 hours. These foci have been shown to represent accumulation of unrepaired endogenous DNA damage (38) and formed in the absence of genotoxic exposure (Fig 1a). Formation of DNA damage foci in PC-3 cells was confirmed with immunofluorescence for γH2AX and endogenous 53BP1 (Fig 1b). In comparison, DU-145 cells did not show increased GFP-IBD nuclear foci after treatment with 10 μM ABT-888 alone and immunofluorescence revealed only pan-nuclear fluorescence or sporadic foci for the same markers (Fig 1b).
Figure 1.
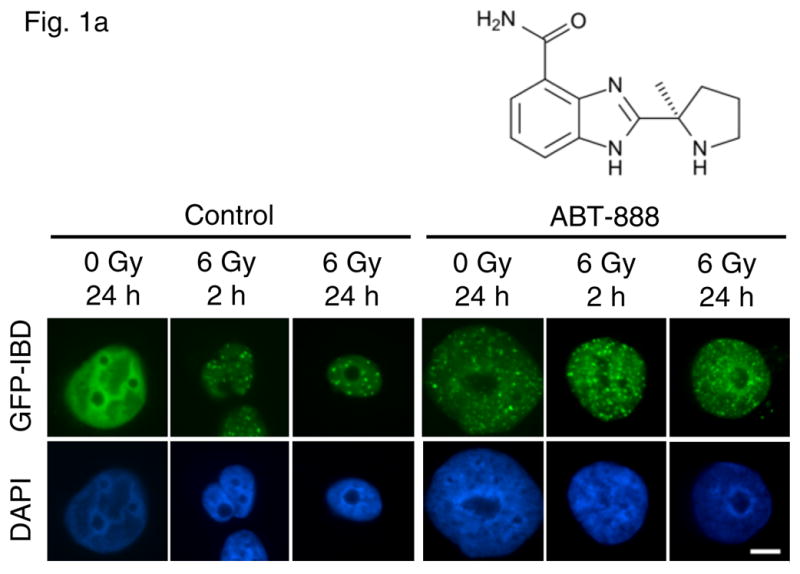

(a) PC-3 GFP-IBD cells show pan-nuclear fluorescence before IR treatment but display ionizing radiation induced foci (IRIF) at 2 h after irradiation that partially resolve by 24 h. The PARP inhibitor ABT-888 induced DNA damage foci on its own. Addition of ABT-888 to IR markedly increased IRIF persistence at 24 h. Bar, 10 μm. (b) Immunofluorescence staining of PC-3 and DU-145 cells after treatment with ABT-888 ± IR. Both cells lines display few γH2AX and 53BP1 foci without ABT-888 or IR treatment. 6 Gy induces similar numbers of γH2AX and 53BP1 foci in each cell line at 2 h, which partially resolve by 24 h. As observed with GFP-IBD, ABT-888 alone induced both γH2AX and 53BP1 foci in PC-3 cells, with no similar effect observed with DU-145 cells. The addition of ABT-888 to IR increased γH2AX and 53BP1 foci persistence at 24 h in both cell lines. Bar, 10 μm.
PARP inhibition increases persistence of IR-induced DNA damage foci in both PC-3 and DU-145 cells
Following treatment with ABT-888 alone, we explored its effect in combination with IR. Nuclear GFP-IBD foci formed rapidly in PC-3 cells after 6 Gy and were prominent at 2 h but then diminished over the next 24 h (Fig 1a). Combining IR and ABT-888 markedly slowed the resolution of GFP-IBD foci over 24 h (Fig 1a). Immunofluorescence staining for γH2AX and endogenous 53BP1 in PC-3 cells after 6 Gy alone or combined with ABT-888 revealed a similar pattern and kinetics of nuclear foci (Fig 1b). ABT-888 induced persistent foci, IR alone induced nuclear foci within 2 h that mostly resolved after 24 h, and the addition of ABT-888 to 6 Gy of IR prevented the resolution of foci after 24 h. As observed in PC-3 cells, DU-145 cells treated with 6 Gy displayed γH2AX and 53BP1 foci at 2 h that partially resolved by 24 h. Similarly, the foci persisted when 6 Gy was combined with 10 μM ABT-888. We conclude that DU-145 cells are not sensitive to induction of DNA damage foci by PARP inhibition alone but they are able to form foci after IR and demonstrate foci persistence after combined treatment with 6 Gy and ABT-888.
PARP inhibition enhances the effect of IR treatment in PC-3 and DU-145 prostate cancer cell lines in vitro
In order to determine the effect on cell survival, PC-3 and DU-145 cells were analyzed by clonogenic assays after treatment with ABT-888 and IR, alone or in combination. 10 μM ABT-888 alone induced a significant inhibition in colony formation in PC-3 cells (100 ± 5% for control versus 77 ± 6% for ABT-888; P = 0.006, t test), but no significant effect was observed in DU-145 cells (100 ± 11% for control versus 90 ± 10% for ABT-888; P = 0.37, t test, Fig. 2a). However, 10 μM ABT-888 combined with IR reduced colony formation in both cell lines, compared to IR alone. In PC-3 cells significant differences were noticeable from 1 Gy (89 ± 10% for IR alone versus 44 ± 3% for IR + ABT-888; P = 0.002, t test), with similar fold effects at each IR dose up to 6 Gy (Fig. 2b). In DU-145 cells, the survival fractions began to differ from 2 Gy (58 ± 4% for IR alone versus 47 ± 4% for IR + ABT-888; P = 0.038, t test, Fig. 2b).
Figure 2.


(a) Effect of ABT-888 treatment alone on colony formation in PC-3 and DU-145 cell lines. Significant growth inhibition was observed only in PC-3 cells. (b) Clonogenic survival of PC-3 and DU-145 cells after treatment with IR doses from 0 to 6 Gy, with or without 10 μM ABT-888. Although the PARP inhibitor sensitizes both cell lines to radiation, the effect is more significant in the PC-3 cells. Bars, SD.
We explored the effect of PARP inhibition and IR on cell cycle kinetics 48 h after treatment using permeabilization, propidium iodide staining and flow cytometry. Consistent with the formation of IRIF after addition of ABT-888 alone, PC-3 cells shifted toward G2/M DNA content, while DU-145 cells displayed no appreciable change in cell cycle distribution (Fig. 3). IR treatment increased the proportion of cells with G2/M DNA content in each cell line. This effect was enhanced by ABT-888, with PC-3 cells displaying a greater G2/M shift than DU-145 cells.
Figure 3.

Cell cycle analysis of PC-3 and DU-145 cells after treatment with ABT-888 and IR alone or in combination. Propidium iodide flow cytometry was performed 48 h after treatment with ABT-888 ± IR and cell cycle statistics were modeled with FlowJo. 48 h incubation in 10 μM ABT-888 increased the G2/M fraction in PC-3 cells but had no effect on DU-145 cells. Irradiation with 6 Gy decreased S phase and increased the G2/M fraction in both cell lines. This effect was enhanced by ABT-888 in each. The combined effect of ABT-888 and IR on PC-3 cells also markedly depleted G1 phase cells from the population, suggesting a G2 cell cycle arrest.
PARP inhibition induces senescence in combination with IR in PC-3 cells
The persistence of DNA damage foci and G2/M shift led us to investigate cellular consequences of treatment with ABT-888 and IR. No differences were noted in cell death at short times in either cell line after any treatment combination via analysis of propidium iodide uptake by unpermeabilized cells; the percentage of cells with PI uptake ranged from 2.5% to 6.8% across all groups in both cell lines. When cells remain viable and display persistent DNA damage, one potential outcome is the induction of accelerated senescence. Thus, we evaluated the treated cells for characteristic markers of senescence including a large and flattened cell morphology, accumulation of SA-βGal and p21 overexpression. ABT-888 or IR treatment alone did not induce significant SA-βGal activity or p21 expression in either cell line. However, we observed markers of accelerated senescence in PC-3 cells by 4 d after treatment with ABT-888 and IR, including an enlarged flat morphology and positive staining for SA-βGal (Fig 4a). There was also a five-fold increase in p21 gene expression as determined by PCR (Fig 4b), and increased protein expression detected by immunofluorescence (Fig 4c). In DU-145 cells, only isolated cells demonstrated SA-βGal activity, and no increase in p21 was detected (Figs. 4a–c). We did not observe overexpression of other senescence markers, including p16 and p27, in either cell line.
Figure 4.



Accelerated senescence in DU-145 and PC-3 cells induced by ABT-888 and IR alone or in combination. (a) Analysis of SA-βGal activity 7 d after treatment showed that 6 Gy with or without 10 μM ABT-888 increased SA-βGal activity in PC-3 cells. IR alone had less effect on DU-145 cells, which showed some increase in SA-βGal staining with IR + ABT-888. Bar, 20 μm. (b) qPCR analysis of p21 expression demonstrated a significant increase only in PC-3 cells 48 h after 6 Gy with 10 μM ABT-888. Bars, SD. (c) Immunofluorescence analysis at 72 h after treatment showed significant accumulation of p21 protein only in PC-3 cells treated with both IR and ABT-888. Bar, 10 μm.
Effect of ABT-888 and IR on prostate tumors in vivo
To investigate the effects of PARP inhibition and IR alone and in combination in tumor xenografts, PC-3 and DU-145 cells were injected into nude mice to form tumors. Mice were treated with ABT-888 and/or IR as described above. Tumors were harvested 4 d after treatment, and tissue sections were evaluated for γH2AX and 53BP1 foci. The findings in tumor tissues mirrored the results in vitro, showing DNA damage foci induction by ABT-888 alone in PC-3 xenografts, but not in DU-145 cells. In irradiated PC-3 and DU-145 tumors, the addition of ABT-888 caused foci persistence 4 d after treatment compared to IR alone (Fig. 5a). Additional tumors were harvested at 7 d after treatment and frozen sections were analyzed for senescence markers. In PC-3 tumors, numerous cells stained positive for SA-βGal (Fig 5b) and PCR demonstrated increased expression of p21 (not shown) after combined treatment. In DU-145 tumors, there were only isolated cells with detectable SA-βGal activity (Fig 5b) and no induction of p21 was apparent (not shown).
Figure 5.


Foci persistence and accelerated senescence in PC-3 and DU-145 tumors treated with ABT-888 and IR alone or in combination. (a) Immunofluorescence staining in PC-3 and DU-145 tissue sections from tumors harvested 4 d after treatment with ABT-888 ± IR. Both cell lines display small numbers of γH2AX and endogenous 53BP1 nuclear foci 4 d after 6 Gy alone. ABT-888 increased the number of persistent foci in PC-3 but had a less marked effect on DU-145 tumors. Bar, 10 μm. (b) Tumors excised at 7 d after treatment, sectioned and analyzed for SA-βGal display increased activity in PC-3 tumors after treatment with IR + ABT-888. DU-145 tumors show only isolated cells with SA-βGal staining. Bar, 20 μm.
We also analyzed tumor growth in mice after treatment with 6 Gy and ABT-888 alone or in combination. ABT-888 had a moderate effect slowing tumor growth in PC-3 tumors (mean V/V0 at 15 d, 13 in the ABT-888 group vs. 10 in controls, Fig 6a). In combination with 6 Gy, the effect of PARP inhibition was more robust, and it significantly delayed regrowth in PC-3 tumors (mean V/V0 at 32 d, 12 in IR group vs. 4.5 in combination group, P = 0.07, t test). DU-145 tumor growth was not slowed by ABT-888 treatment, alone or combined with IR. In fact, we note some protective effect of ABT-888 on DU-145 tumor growth after 6 Gy (Fig 6b).
Figure 6.


PC-3 and DU-145 tumor growth kinetics after treatment with ABT-888 and IR alone or in combination. Tumor volume was followed over time in PC-3 (a) and DU-145 (b) xenografts in animals treated with ABT-888 followed by IR. PC-3 tumors showed minimal sensitivity to ABT-888 treatment alone but significantly delay in tumor regrowth after 6 Gy with the addition of ABT-888. DU-145 tumor growth was not significantly slowed by the 6 Gy dose and addition of ABT-888 appeared to have a slight protective effect.
DISCUSSION
PARP inhibitors are a class of highly promising targeted therapy agents that are showing benefits alone and in combination with genotoxic therapy in a wide range of cancers in preclinical models and clinical trials (7, 8). Much of the focus has been on the “synthetic lethality” mechanism (12–15) that has brought these agents to the forefront in treatment of BRCA mutant and triple negative breast cancers. Trials in prostate cancer patients remain at an early stage and have yet to demonstrate clear benefits. Among the few papers examining PARP inhibitors in prostate cancer cells, prominent examples have focused on the potential for exploiting synthetic lethality. These studies have examined PARP inhibition as a sensitizer to defects in HR resulting from loss of Rad51 expression due to PTEN deficiency (PC-3, ref. 18) or induced by hypoxia (DU-145, ref. 39). Other studies have examined the effects of combining a PARP inhibitor with temozolomide on prostate cancer cells (40). In prostate cancer, PARP inhibitors are most likely to find their use in combination with conventional treatments such as radiation therapy. Perhaps most significant to in vivo studies, although the molecular targets of PARP inhibitors are known and a mechanism of action appears to be established, how PARP inhibition synergizes with genotoxic agents and how it mediates its growth inhibitory effects on tumors remains poorly defined.
When IR treatment was added to PARP inhibition in vitro, we observed radiosensitization in both the PC-3 and DU-145 cell lines, manifested by a decrease in colony formation, increased IRIF persistence and induction of cell cycle arrest. The in vitro effects were greater in PC-3 cells compared to DU-145, and PC-3 cells developed hallmarks of senescence after combined treatment. The effectiveness of the combination was confirmed in vivo in PC-3 tumor xenografts, delaying tumor growth compared to IR and inducing a senescent phenotype. However, no improvement in the effect of IR was observed in DU-145 xenografts. We hypothesize that senescence in PC-3 cells can partially explain the difference in the response in vivo, since no significant senescence was observed in DU-145 cells. DU-145 cells have been found to be more radioresistant in vitro to IR treatment, whereas PC-3 tumors have been reported to have a higher hypoxic fraction compared to DU-145 tumors (52% vs. 7%, respectively), which could potentially increase the radioresistance of PC-3 in vivo (41). However, ABT-888 has also been reported to sensitize hypoxic cancer cells to a level similar to oxic radiosensitivity (39); this mechanism might overcome the potential radioresistance from a higher hypoxic fraction in PC-3 tumors.
Consistent with prior work from Mendes-Pereira et al. (18), we found that treatment with ABT-888 alone had some efficacy in vitro in PC-3 prostate cancer cells, which are defective in PTEN. It has been described that PTEN deficiency causes a homologous recombination (HR) defect in human tumor cells, making them a therapeutic target of PARP inhibitors (18, 42, 43). PTEN, besides inactivating the P13-K/AKT pathway, has a nuclear function controlling chromosomal integrity and regulating the expression of Rad51, which reduces the incidence of spontaneous double strand breaks (44). PARP inhibition alone induced DNA damage foci, inhibited cell proliferation and promoted G2/M cycle arrest in PC-3 cells. A similar trend in PC-3 tumor xenografts was observed with a moderate suppression of tumor growth. ABT-888 alone did not exert any of the above effects in DU-145 cells in vitro at a similar drug concentration. These results support the current model that the efficacy of PARP inhibitors in HR deficient BRCA1-, BRCA2-, or PTEN-negative cancer cells is the result of accumulation of unrepaired endogenous DNA damage. It could be hypothesized that if the efficacy of PARP inhibitors in PTEN-deficient prostate tumors translates into a significant clinical benefit, in selected cases they could be used as monotherapy, allowing adequate tumor control while preventing the complications associated with radiation therapy and surgery.
Our results are consistent with other reports that have shown increased efficacy of IR with the addition of PARP inhibitors (9, 11), and further implicate IRIF persistence as a determinant of the increase in accelerated senescence (31, 32). Senescence can result from several inducers, including accumulation of unrepaired DNA damage. Therapy-induced senescence is increasingly being reported as an alternative mode of cell death in addition to apoptosis and necrosis and is proposed to contribute to tumor control following treatment with cytotoxic agents (29, 45). Along with prior studies (11, 39), our results in PC-3 cells and tumors suggest that PARP inhibitors can be an effective radiosensitizing strategy. Additionally, consistent with our previous work (22), accelerated senescence may be a factor in the therapeutic response of some human tumors to IR combined with PARP inhibition.
Liu et al. (39) previously observed that DU-145 cells treated with IR and ABT-888 displayed increased toxicity without undergoing apoptosis. In our experiments, ABT-888 sensitized DU-145 cells to IR as measured by clonogenic assay and induction of DNA damage foci, without inducing senescence or apoptosis, suggesting a mitotic death. This p53 mutant cell line also has a nonsense mutation in the Rb gene (35), impairing both the p53/p21 and p16/ARF/Rb senescence pathways (32). We ascribe the lack of benefit of ABT-888 on the DU-145 tumors to their distinct response to persistent DNA damage. We also acknowledge that there may be other unidentified host or tumor factors that may impinge upon the effectiveness of ABT-888 in DU-145 tumors. A favorable interpretation of our data is that a key determinant of the efficacy of PARP inhibitors in vivo may be their ability to drive cells toward senescent arrest. Insofar as senescent cells induced by genotoxic treatment can persist in tissue for weeks or months and can alter the microenvironment via paracrine signaling (46, 47), they may be able to limit the recovery of other tumor cells. These results also highlight the importance of tailoring therapy to each tumor.
We conclude that PARP inhibitors have therapeutic potential in specific types of prostate cancer in combination with radiation therapy, and even as monotherapy in DNA repair defective tumors. Induction of accelerated senescence is a novel therapeutic approach, and deserves consideration in clinical trials. Despite its increasing recognition as a potential alternative for tumor control, there is still a lack of reliable senescence-inducing agents and this area remains an open field for further research. PARP inhibitors are strong candidates for this purpose, though potentially limited to specific tumor types.
Acknowledgments
Financial support: Ludwig Center for Metastasis Research, NCI SPORE in Prostate Cancer P50 CA090386 (RRW, SJK), NIH R21 CA138365 (SJK, RRW), NIH R01 GM60443 (SJK), Foglia Family Foundation, The Francis L. Lederer Foundation, Center for Radiation Therapy, American Society of Clinical Oncology Translational Research Professorship (EEV).
Abbreviations List
- PARP
Poly(ADP-ribose) polymerase
- IR
ionizing radiation
- DSB/SSB
double strand breaks, single strand breaks
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- HR
homologous recombination
- NHEJ
non-homologous end joining
- 53BP1
p53 binding protein 1
- IRIF
ionizing radiation induced foci
- GFP
green fluorescent protein
- H2AX
Histone 2A, member X
- PI
propidium iodide
- SA β-Gal
senescence-associated beta galactosidase
- PCR
polymerase chain reaction
Footnotes
Conflicts of interest: None of the authors has potential conflicts of interest.
References
- 1.Zelefsky MJ, Chan H, Hunt M, Yamada Y, Shippy AM, Amols H. Long-term outcome of high dose intensity modulated radiation therapy for patients with clinically localized prostate cancer. J Urol. 2006;176:1415–9. doi: 10.1016/j.juro.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Bill-Axelson A, Holmberg L, Ruutu M, Haggman M, Andersson SO, Bratell S, et al. Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med. 2005;352:1977–84. doi: 10.1056/NEJMoa043739. [DOI] [PubMed] [Google Scholar]
- 3.Van Hemelrijck M, Garmo H, Holmberg L, Ingelsson E, Bratt O, Bill-Axelson A, et al. Absolute and relative risk of cardiovascular disease in men with prostate cancer: results from the Population-Based PCBaSe Sweden. J Clin Oncol. 2010;28:3448–56. doi: 10.1200/JCO.2010.29.1567. [DOI] [PubMed] [Google Scholar]
- 4.Van Hemelrijck M, Adolfsson J, Garmo H, Bill-Axelson A, Bratt O, Ingelsson E, et al. Risk of thromboembolic diseases in men with prostate cancer: results from the population-based PCBaSe Sweden. Lancet Oncol. 2010;11:450–8. doi: 10.1016/S1470-2045(10)70038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ljungman M. Targeting the DNA damage response in cancer. Chem Rev. 2009;109:2929–50. doi: 10.1021/cr900047g. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–28. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 7.Rodon J, Iniesta MD, Papadopoulos K. Development of PARP inhibitors in oncology. Expert Opin Investig Drugs. 2009;18:31–43. doi: 10.1517/13543780802525324. [DOI] [PubMed] [Google Scholar]
- 8.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 10.Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14:7917–23. doi: 10.1158/1078-0432.CCR-08-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell C, Mikropoulos C, Kaye SB, Nutting CM, Bhide SA, Newbold K, et al. Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. Cancer Treat Rev. 2010;36:566–75. doi: 10.1016/j.ctrv.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–90. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 13.Moeller BJ, Pasqualini R, Arap W. Targeting cancer-specific synthetic lethality in double-strand DNA break repair. Cell Cycle. 2009;8:1872–6. doi: 10.4161/cc.8.12.8743. [DOI] [PubMed] [Google Scholar]
- 14.Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci. 2010;31:372–80. doi: 10.1016/j.tips.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Chalmers AJ, Lakshman M, Chan N, Bristow RG. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin Radiat Oncol. 2010;20:274–81. doi: 10.1016/j.semradonc.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 17.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 18.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, Bhogal N, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010;70:8045–54. doi: 10.1158/0008-5472.CAN-10-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9:1775–87. doi: 10.1158/1535-7163.MCT-09-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70:5389–98. doi: 10.1158/0008-5472.CAN-09-4716. [DOI] [PubMed] [Google Scholar]
- 22.Efimova EV, Mauceri HJ, Golden DW, Labay E, Bindokas VP, Darga TE, et al. Poly(ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res. 2010;70:6277–82. doi: 10.1158/0008-5472.CAN-09-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 24.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 25.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 26.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23:2919–33. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 27.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–22. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Campisi J. Cellular senescence: putting the paradoxes in perspective. Curr Opin Genet Dev. 2010 doi: 10.1016/j.gde.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–15. [PubMed] [Google Scholar]
- 30.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 31.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–11. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 34.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubin SJ, Hallahan DE, Ashman CR, Brachman DG, Beckett MA, et al. Two prostate carcinoma cell lines demonstrate abnormalities in tumor suppressor genes. J Surg Oncol. 1991;46:31–6. doi: 10.1002/jso.2930460108. [DOI] [PubMed] [Google Scholar]
- 36.Lorenzi PL, Reinhold WC, Varma S, Hutchinson AA, Pommier Y, Chanock ST, et al. DNA fingerprinting of the NCI-60 cell line panel. Mol Cancer Ther. 2009;8:713–24. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhogal N, Jalali F, Bristow RG. Microscopic imaging of DNA repair foci in irradiated normal tissues. Int J Radiat Biol. 2009;85:732–46. doi: 10.1080/09553000902785791. [DOI] [PubMed] [Google Scholar]
- 39.Liu SK, Coackley C, Krause M, Jalali F, Chan N, Bristow RG. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother Oncol. 2008;88:258–68. doi: 10.1016/j.radonc.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 40.Palma JP, Wang YC, Rodriguez LE, Montgomery D, Ellis PA, Bukofzer G, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15:7277–90. doi: 10.1158/1078-0432.CCR-09-1245. [DOI] [PubMed] [Google Scholar]
- 41.Leith JT, Quaranto L, Padfield G, Michelson S, Hercbergs A. Radiobiological studies of PC-3 and DU-145 human prostate cancer cells: x-ray sensitivity in vitro and hypoxic fractions of xenografted tumors in vivo. Int J Radiat Oncol Biol Phys. 1993;25:283–7. doi: 10.1016/0360-3016(93)90350-5. [DOI] [PubMed] [Google Scholar]
- 42.McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70:5457–64. doi: 10.1158/0008-5472.CAN-09-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2:53ra75. doi: 10.1126/scitranslmed.3001538. [DOI] [PubMed] [Google Scholar]
- 44.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 45.Lleonart ME, Artero-Castro A, Kondoh H. Senescence induction; a possible cancer therapy. Mol Cancer. 2009;8:3. doi: 10.1186/1476-4598-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 47.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doom R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]