

Abstract
Introduction. The FSAP-Marburg I polymorphism (1704G > A), which reduces FSAP activity, is associated with late complications of carotid stenosis in humans. Therefore, this study examines the influence of the Marburg I polymorphism and the closely linked Marburg II polymorphism (1280G > C) on various cardiovascular outcomes in two large independent study populations. Methods. The two Marburg polymorphisms in the HABP2 gene encoding FSAP were genotyped in a large population of elderly patients at risk for vascular disease (the PROSPER-study, n = 5804) and in a study population treated with a percutaneous coronary intervention (the GENDER-study, n = 3104). Results. In the PROSPER study, the Marburg I polymorphism was associated with an increased risk of clinical stroke (HR: 1.60, 95% CI: 1.13–2.28) and all-cause mortality (HR: 1.33, 95% CI: 1.04–1.71). In the GENDER study carriers of this variant seemed at lower risk of developing restenosis (HR: 0.59, 95% CI: 0.34–1.01). The Marburg II polymorphism showed similar but weaker results. Conclusion. The increase in stroke risk in Marburg I carriers could be due to differential effects on smooth muscle cells and on matrix metalloproteinases, thereby influencing plaque stability. The possible protective effect on restenosis could be the result of reduced activation of zymogens, which are involved in hemostasis and matrix remodeling.
1. Introduction
Factor seven activating protease (FSAP) is a plasma serine protease known to activate factor VII (FVII) [1] and prourokinase (pro-uPA) [2]. Despite these actions, it is unclear if endogenous FSAP has a relevant role in hemostasis. The Marburg I (1704G > A) polymorphism in the HABP2 gene encoding FSAP, which leads to an amino acid change in the protease domain of this protein, may lead to a pro-thrombotic phenotype when it is associated with reduced activation of pro-uPA, but unchanged activation of FVII [3]. Although its possible association with venous thrombosis remains controversial [4–6], the Marburg I variant is a risk factor for coronary heart disease [7] and late complications of carotid stenosis [8]. The Marburg variant is associated with advanced atherogenesis (a lumen narrowing of >40%) but not with early atherogenesis (occurrence of new plaques) [8].
Furthermore, FSAP is identified as a potent inhibitor of smooth muscle cell proliferation and migration [9], specifically through its ability to cleave platelet derived growth factor-BB (PDGF-BB). The FSAP Marburg I variant, which also has reduced proteolytic activity towards PDGF-BB, is associated with a reduced capability to suppress neointima formation in an animal model [10]. This might be another mechanism by which the Marburg I polymorphism could play a role in carotid stenosis and many other aspects of cardiovascular disease [11].
Therefore, we investigated the influence of this polymorphism on clinical stroke, coronary events, vascular mortality, and all-cause mortality in a large population of elderly patients at risk for vascular disease (the PROSPER study, n = 5804) and on clinical restenosis after a percutaneous coronary intervention (PCI) (the GENDER study, n = 3104). Although not associated with altered enzymatic activity, we also investigated the Marburg II variant (1280G > C), a closely linked polymorphism that leads to an amino acid change in the protease domain of FSAP.
2. Methods
2.1. Study Design and Followup of the PROSPER Study
The protocol of PROSPER has been described in more detail elsewhere [12]. PROSPER is a prospective multicenter randomized placebo-controlled trial to assess whether treatment with pravastatin diminishes the risk of major vascular events in elderly individuals. Between December 1997 and May 1999, we screened and enrolled subjects in Scotland (Glasgow), Ireland (Cork), and the Netherlands (Leiden). Men and women aged 70–82 years were recruited if they had preexisting vascular disease or increased risk of such disease because of smoking, hypertension, or diabetes. A total of 5804 subjects were randomly assigned to pravastatin or placebo. In this genetic substudy, we evaluated the predefined endpoints all-cause mortality, vascular mortality, and the secondary endpoints fatal or nonfatal coronary events and fatal or nonfatal clinical stroke. Median followup was 3.3 (interquartile range 0.5) years and 604 (10.4%) patients died during the study [13].
2.2. Study Design and Followup of the GENDER Study
The present study sample has been described previously [14]. In brief, the GENetic DEterminants of Restenosis project (GENDER) was a multicenter followup study designed to study the association between various gene polymorphisms and clinical restenosis. The overall inclusion period of the GENDER study lasted from March 1999 until June 2001. A total of 3104 patients eligible for inclusion in the GENDER study were treated successfully for stable angina, non-ST-elevation acute coronary syndromes or silent ischemia by PCI in 4 of the 13 referral centers for interventional cardiology in the Netherlands. Patients treated for acute ST elevation myocardial infarction were excluded. Experienced operators, using a radial or femoral approach, performed standard angioplasty with and without stent placement. During the study, no drug-eluting stents were used. Followup lasted for at least 9 months, except when a coronary event occurred. The primary endpoint was clinical restenosis, defined as target vessel revascularization (TVR), either by PCI or coronary artery bypass grafting (CABG). Subjects with asymptomatic restenosis were not included and routine angiography was not obtained. Median followup duration was 9.6 (interquartile range 3.9) months and 304 (9.8%) patients underwent TVR during followup.
For both studies, all endpoints were adjudicated by independent clinical events committees. The protocols meet the criteria of the Declaration of Helsinki and were approved by the Medical Ethics Committees of each participating institution. Written informed consent was obtained from all participating patients.
2.3. Genotyping
Blood was collected in EDTA tubes at baseline and genomic DNA was extracted following standard procedures. The Marburg I (1704G > A) and II (1280G > C) polymorphisms were determined using the Sequenom Massarray genotyping platform (rs7080536 and rs11575688 resp.). A multiplex assay was designed using assay designer software (Sequenom Inc.). As quality controls, 5–10% of the samples were genotyped in duplicate; no inconsistencies were observed. Cluster plots were made of the signals from the low and the high mass allele. Two independent researchers carried out scoring. Disagreements or vaguely positioned dots produced by Genotyper 4.0 (Sequenom Inc.) were left out of the results.
2.4. Replication
We performed in silico replication in the genome-wide association study on stroke from the Rotterdam Study [15]. Genotyping was performed using the Illumina 550 K Beadchip in 5763 study participants with subsequent imputation to 2.5 million HapMap II SNPs. Marburg I polymorphism was not present nor were any proxies of the Marburg I polymorphism with r2 > 0.5. Marburg II polymorphism was present in the imputed dataset with an imputation quality >0.8. The association between this SNP and incident stroke was assessed in 367 incident ischemic stroke cases.
2.5. Statistical Analysis
Allele frequencies were determined by gene counting. The Chi-squared test was used to test the consistency of the genotype frequencies at the SNP locus with Hardy-Weinberg equilibrium. Hazard ratios (HR) with 95% confidence intervals (CI) were calculated using a Cox proportional hazards model. All analyses were adjusted for sex and age. The analyses with PROSPER data were additionally adjusted for pravastatin use and country. In the GENDER study, polymorphisms were included in a multivariable model containing clinical and procedural risk factors for restenosis, such as diabetes, stenting, and total occlusion. The replication was assessed with a logistic regression model adjusted for sex and age. The SPSS software (version 17.0.1, SPSS Inc, Chicago, ILL) was used for all statistical analyses.
3. Results
3.1. The PROSPER Study
Table 1 presents the patient characteristics and minor allele frequencies. Genotyping success rates were 98% and 97% for the Marburg I and II polymorphisms, respectively, and there were no significant deviations from Hardy-Weinberg equilibrium.
Table 1.
Baseline characteristics of the PROSPER and the GENDER study in patients with Marburg I genotype.
| PROSPER | GENDER | |
|---|---|---|
| n = 5697 | ||
| Continuous variates (mean, SD) | ||
| Age (years) | 75.3 (3.4) | 62.1 (10.7) |
| Body mass index (kg/m2) | 26.8 (4.2) | 27.0 (3.9) |
| Categorical variates (n, %) | ||
| Male sex | 2752 (48) | 2110 (71) |
| Current smoker | 1529 (27) | 730 (25) |
| History of diabetes | 602 (11) | 434 (15) |
| History of hypertension | 3530 (62) | 1189 (40) |
| History of myocardial infarction | 764 (13) | 1176 (40) |
| History of stable angina | 1529 (27) | 1977 (67) |
| Genotype, minor allele frequency (%) | ||
| Marburg I G/A | 5 | 4 |
| Marburg II G/C* | 2 | 2 |
All data are presented in number (%) unless otherwise stated.
*In PROSPER and GENDER measured in 5655 and 2959 participants, respectively.
Using a Cox proportional hazards model, which included the variables sex, age, pravastatin use, and country, a significant association was found between the Marburg I polymorphism and clinical stroke. Figure 1 shows that the combined group of heterozygotes (n = 518) and homozygotes (n = 17) was at increased risk for clinical stroke (HR: 1.6, 95% CI: 1.13–2.28, P = .009) when compared to the wild type group (n = 5162). Also, all-cause mortality was significantly higher in patients carrying one or two copies of this variant (HR: 1.33, 95% CI: 1.04–1.71, P = .025). The increased mortality was mainly a result of an increase in vascular mortality (HR: 1.37, 95% CI: 0.96–1.97, P = .082), whereas vascular mortality was mainly determined by death from stroke. The Marburg I polymorphism did not seem to influence the risk for coronary events (HR: 0.98, 95% CI: 0.75–1.29). Additional adjustment for traditional risk factors (hypertension, diabetes, smoking, and cholesterol levels) did not change the results (HR: 1.63, 95% CI: 1.14–2.31, P = .007).
Figure 1.
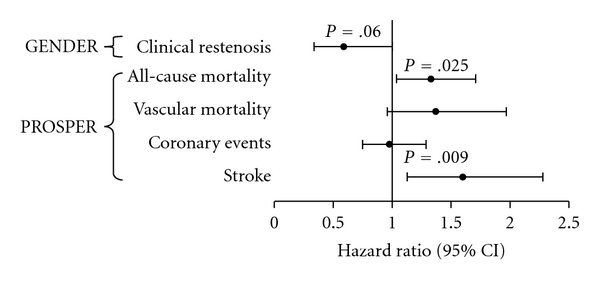
Marburg I hazard ratios for vascular endpoints in GENDER and PROSPER. The Marburg I (G534E) polymorphism is associated with an increased risk for stroke and mortality in the PROSPER study, whereas it tends to reduce the risk for clinical restenosis in the GENDER study.
Due to its location in the same coding sequence and its proximity to the Marburg I polymorphism we also present data of the Marburg II polymorphism which show significant effects on mortality (Figure 2). The similar trends observed for this polymorphism are probably a result of the high linkage disequilibrium between the Marburg variants. The linkage disequilibrium coefficient was 0.79, and 62% of the Marburg II carriers were also genotyped with the Marburg I allele. Due to a lower allele frequency of the Marburg II polymorphism, there were only 234 heterozygotes and 4 homozygotes with the variant allele. The large confidence intervals indeed indicate that these results are less reliable than the Marburg I findings.
Figure 2.
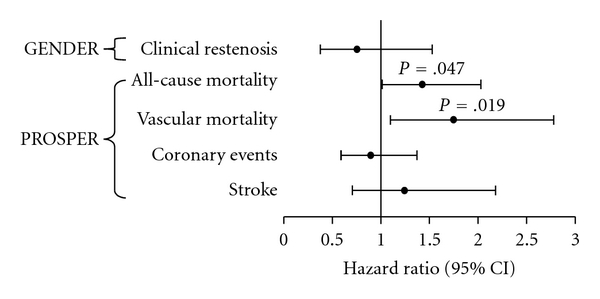
Marburg II hazard ratios for vascular endpoints in GENDER and PROSPER. As the Marburg II polymorphism does not lead to altered enzymatic activity, the similar trends that were observed for this variant are probably a result of the high linkage disequilibrium between the Marburg variants. Despite high confidence intervals, a significant effect was observed on mortality.
Finally, we performed an analysis comparing carriers of both alleles of Marburg I and Marburg II with carriers of the wild-type alleles in relation to clinical stroke. A hazard ratio of 1.60 (95% CI: 0.87–2.94) was found for the risk of clinical stroke for subjects carrying both variant alleles; however, this was not significant due to low numbers.
3.2. The GENDER Study
Table 1 presents the patient characteristics and minor allele frequencies. Genotyping success rates were 95% for both polymorphisms, and there were no significant deviations from Hardy-Weinberg equilibrium.
In contrast with the results from the PROSPER study, the Marburg I polymorphism (208 heterozygotes and 5 homozygotes for the variant allele) tended to reduce the risk for clinical restenosis in the GENDER study (Figure 1), (HR: 0.59, 95% CI: 0.34–1.02, P = .061). The protective effect was borderline significant. Carriers of the Marburg II variant had a slightly and nonsignificantly reduced risk of TVR (Figure 2). In the GENDER population, 67 patients were carriers of both Marburg I and II risk alleles, accounting for 63% of all Marburg II carriers (105 heterozygotes and 1 homozygote).
3.3. Replication
The Marburg II polymorphism showed an increased risk for clinical stroke in 5763 subjects of the Rotterdam Study. Carriers of the variant allele have a 1.4 times increased risk of clinical stroke compared to carriers of the wild-type allele (β: 0.38, SE: 0.23, P = .05).
4. Discussion
Our data suggest that the Marburg I polymorphism, which leads to less FSAP activity, increases stroke risk and mortality. The Marburg I associated increase in mortality was mainly due to an increased risk of fatal stroke. Surprisingly, carriers of this variant seemed at lower risk of developing restenosis. The effect of the Marburg II polymorphism on clinical stroke was replicated in the Rotterdam Study cohort.
Although FSAP was originally identified as a potential activator of factor VII and prourokinase (pro-uPA), its role in hemostasis remains unclear. Endogenous FSAP, of which intravascular levels are low, has not been clearly shown to influence blood coagulation or fibrinolysis [11]. Moreover, the Marburg I polymorphism does not seem to influence the risk for venous thrombosis [4, 16]. However, FSAP cleaves PDGF-BB and has been shown to inhibit vascular smooth muscle cell proliferation and migration in vitro [9] and in vivo [10]. The activity of FSAP in Marburg carriers is low and could therefore be a risk factor for atherosclerosis and restenosis, processes which are known to be determined by vascular smooth muscle cell proliferation.
A possible role for FSAP in human atherosclerosis was suggested by a study showing an association of the Marburg I polymorphism with advanced atherogenesis in carotid arteries [8]. Despite the low number of carriers among cases (n = 8) and controls (n = 2), their findings suggest a role for Marburg I in carotid plaque formation. Another study, investigating the effect of Marburg I on coronary heart disease, found no significant effect in the whole population, but observed an interactive effect on risk between the Marburg I variant and elevated levels of cholesterol and triglyceride [7]. The primary endpoint in that study was a composite of myocardial infarction and the need for a PCI.
In agreement with these studies, we also found no association of Marburg I with myocardial infarction in the whole population of patients taking part in the PROSPER study, whereas we did find a significant association with stroke, which is known to be related to carotid plaque formation. The observed increase in stroke risk in Marburg I carriers could be a consequence of hyperproliferation of smooth muscle cells, due to a reduced proteolytic activity of Marburg I-FSAP towards PDGF-BB. Unfortunately, we have no measurements of carotid atherosclerosis within the PROSPER study on which to draw more definite conclusions.
The possible protective effect of Marburg I on clinical restenosis in patients treated for stable angina pectoris is difficult to explain. Despite some similarities between atherosclerosis and restenosis, such as the involvement of inflammation and smooth muscle cell proliferation, there are important mechanistic differences between these processes. In contrast with atherosclerosis, which develops partly in response to elevated lipoprotein levels and cigarette smoke, the restenotic process is not particularly sensitive to circulating lipids and smokers even seem to have a reduced risk for restenosis [14]. It is therefore not unlikely that a genetic risk factor would have opposite effects on stroke and clinical restenosis after PCI.
However, as opposed to WT-FSAP, Marburg I-FSAP was less effective in preventing neointima formation in an animal model for restenosis [10]. Based on these findings, the Marburg I polymorphism was expected to increase the risk for restenosis in humans. Although the protective effect observed in the GENDER study was (borderline) not significant, there was a strong trend towards protection. The contradicting results not only could relate to the subjects (mice versus humans) or the intervention site (femoral artery versus coronary arteries), but could also be the result of differences in the exact location and concentration of the Marburg I-FSAP-protein, which was locally applied in high concentrations to injured arteries in the mouse model. Furthermore, FSAP plays a role in many different processes known to be important in vascular remodeling. FSAP has recently been shown to activate the matrix metalloproteinases MMP2 and MMP9 (gelatinases) [17], which are important in matrix remodeling. Further research is needed to elucidate the precise role of FSAP in vascular remodeling and the pathogenesis of stroke.
The Marburg II polymorphism, also located in the protease domain of FSAP, has no known functional implications and is not associated with altered catalytic activity.
Due to its low allele frequency (2%), the Marburg II findings are less reliable. The similar, but much weaker, trends that were observed for this polymorphism could be a result of the linkage disequilibrium with the Marburg I variant. Large confidence intervals indicate that the observed significant association with vascular mortality, which is slightly stronger than the effect of Marburg I on vascular mortality, probably occurred by chance.
There were 263 stroke endpoints within the PROSPER population during the 3.3-year followup. Unfortunately, we do not have data on the separate ischemic and hemorrhagic strokes. In the present study, both types of stroke were combined into one clinical endpoint. Because we know from previous studies in elderly populations that approximately 80% of all strokes is attributable to ischemic events [18, 19], the association between the Marburg I polymorphism and clinical stroke is probably driven by an association between the polymorphism and ischemic stroke. If there is no association between the polymorphism and hemorrhagic stroke, then the association we found is an underestimation of the true relative risk for ischemic stroke.
In conclusion, we have demonstrated that carriers of the Marburg I polymorphism are at increased risk for clinical stroke and stroke-related mortality. Furthermore, a strong trend towards a reduced risk for clinical restenosis was observed in Marburg I carriers. As Marburg I-FSAP has reduced proteolytic activity towards PDGF-BB, the increase in stroke risk could be a consequence of hyperproliferation of smooth muscle cells.
The PROSPER Study Group
Scotland—J. Shepherd, C. J. Packard, A. Gaw, (University Department of Pathological Biochemistry, North Glasgow), P. W. Macfarlane, D. J. Stott, (Division of Cardiovascular and Medical Sciences, University of Glasgow), I. Ford, (Robertson Centre for Biostatistics, University of Glasgow); Ireland—M. B. Murphy, B. M. Buckley, (Department of Pharmacology and Therapeutics, University College Cork), I. J. Perry, (Department of Epidemiology and Public Health, University College Cork), M. Hyland, C. Twomey, (Department of Geriatric Medicine, Cork University Hospital), B. J. Sweeney (Department of Neurology, Cork University Hospital); The Netherlands—S. Trompet, G. J. Blauw, A. J. M. de Craen, R. G. J. Westendorp, (Section of Gerontology and Geriatrics, Leiden University Medical Center), E. L. E. M. Bollen, (Department of Neurology, Leiden University Medical Center), J. W. Jukema, (Department of Cardiology, Leiden University Medical Center).
Acknowledgments
The authors thank Dennis Kremer and Eka Suchiman, from the Molecular Epidemiology Section of the Leiden University Medical Center, for their expert assistance with the Sequenom Massarray genotyping platform. This study was supported by Grant 99.210 from the Netherlands Heart Foundation, a grant from the Interuniversity Cardiology Institute of the Netherlands (ICIN), a grant from the Center for Medical Systems Biology (CMSB), a center of excellence approved by the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research (NWO), and by the seventh framework program of the European commission (Grant 223004). The funders had no role in study design, data collection and analysis, decision to publish, or the preparation of the manuscript. Stella Trompet and Douwe Pons are contributed equally to this article.
Abbreviations
- PROSPER:
Prospective Study of Pravastatin in the Elderly at Risk
- GENDER project:
Genetic Determinants of Restenosis project
- HR:
Hazard ratio
- OR:
Odds ratio
- CI:
Confidence interval
- PCI:
Percutaneous coronary intervention
- CABG:
Coronary artery bypass grafting
- TVR:
Target vessel revascularization.
References
- 1.Römisch J, Feußner A, Vermöhlen S, Stöhr HA. A protease isolated from human plasma activating factor vii independent of tissue factor. Blood Coagulation and Fibrinolysis. 1999;10(8):471–479. [PubMed] [Google Scholar]
- 2.Römisch J, Vermöhlen S, Feussner A, Stöhr HA. The FVII activating protease cleaves single-chain plasminogen activators. Haemostasis. 2000;29(5):292–299. doi: 10.1159/000022515. [DOI] [PubMed] [Google Scholar]
- 3.Roemisch J, Feussner A, Nerlich C, Stoehr HA, Weimer T. The frequent Marburg I polymorphism impairs the pro-urokinase activating potency of the factor VII activating protease (FSAP) Blood Coagulation and Fibrinolysis. 2002;13(5):433–441. doi: 10.1097/00001721-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Gulesserian T, Hron G, Endler G, Eichinger S, Wagner O, Kyrle PA. Marburg I polymorphism for factor VII-activating protease and risk of recurrent venous thromboembolism. Thrombosis and Haemostasis. 2006;95(1):65–67. [PubMed] [Google Scholar]
- 5.Hoppe B, Tolou F, Radtke H, Kiesewetter H, Dörner T, Salama A. Marburg I polymorphism of factor VII-activating protease is associated with idiopathic venous thromboembolism. Blood. 2005;105(4):1549–1551. doi: 10.1182/blood-2004-08-3328. [DOI] [PubMed] [Google Scholar]
- 6.Hoppe B, Tolou F, Döner T, Kiesewetter H, Salama A. Gene polymorphisms implicated in influencing susceptibility to venous and arterial thromboembolism: frequency distribution in a healthy German population. Thrombosis and Haemostasis. 2006;96(4):465–470. [PubMed] [Google Scholar]
- 7.Ireland H, Miller GJ, Webb KE, Cooper JA, Humphries SE. The factor VII activating protease G511E (Marburg) variant and cardiovascular risk. Thrombosis and Haemostasis. 2004;92(5):986–992. doi: 10.1160/TH04-05-0275. [DOI] [PubMed] [Google Scholar]
- 8.Willeit J, Kiechl S, Weimer T, et al. Marburg I polymorphism of factor VII-activating protease: a prominent risk predictor of carotid stenosis. Circulation. 2003;107(5):667–670. doi: 10.1161/01.cir.0000055189.18831.b1. [DOI] [PubMed] [Google Scholar]
- 9.Kannemeier C, Al-Fakhri N, Preissner KT, Kanse SM. Factor VII-activating protease (FSAP) inhibits growth factor-mediated cell proliferation and migration of vascular smooth muscle cells. The FASEB Journal. 2004;18(6):728–730. doi: 10.1096/fj.03-0898fje. [DOI] [PubMed] [Google Scholar]
- 10.Sedding D, Daniel JM, Muhl L, et al. The G534E polymorphism of the gene encoding the factor VII-activating protease is associated with cardiovascular risk due to increased neointima formation. Journal of Experimental Medicine. 2006;203(13):2801–2807. doi: 10.1084/jem.20052546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanse SM, Parahuleva M, Muhl L, Kemkes-Matthes B, Sedding D, Preissner KT. Factor VII-activating protease (FSAP): vascular functions and role in atherosclerosis. Thrombosis and Haemostasis. 2008;99(2):286–289. doi: 10.1160/TH07-10-0640. [DOI] [PubMed] [Google Scholar]
- 12.Shepherd J, Blauw GJ, Murphy MB, et al. The design of a prospective study of pravastatin in the elderly at risk (PROSPER) American Journal of Cardiology. 1999;84(10):1192–1197. doi: 10.1016/s0002-9149(99)00533-0. [DOI] [PubMed] [Google Scholar]
- 13.Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360(9346):1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 14.Agema WRP, Monraats PS, Zwinderman AH, et al. Current PTCA practice and clinical outcomes in the Netherlands: the real world in the pre-drug-eluting stent era. European Heart Journal. 2004;25(13):1163–1170. doi: 10.1016/j.ehj.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Ikram MA, Seshadri S, Bis JC, et al. Genomewide association studies of stroke. New England Journal of Medicine. 2009;360(17):1718–1728. doi: 10.1056/NEJMoa0900094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Minkelen R, De Visser MCH, Vos HL, et al. The Marburg I polymorphism of factor VII-activating protease is not associated with venous thrombosis. Blood. 2005;105(12):4898–4899. doi: 10.1182/blood-2005-02-0576. [DOI] [PubMed] [Google Scholar]
- 17.Parahuleva MS, Kanse SM, Parviz B, et al. Factor Seven Activating Protease (FSAP) expression in human monocytes and accumulation in unstable coronary atherosclerotic plaques. Atherosclerosis. 2008;196(1):164–171. doi: 10.1016/j.atherosclerosis.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 18.Melcon CM, Melcon MO. Prevalence of stroke in an Argentine community. Neuroepidemiology. 2006;27(2):81–88. doi: 10.1159/000094978. [DOI] [PubMed] [Google Scholar]
- 19.Sagui E, M’Baye PS, Dubecq C, et al. Ischemic and hemorrhagic strokes in Dakar, Senegal: a hospital-based study. Stroke. 2005;36(9):1844–1847. doi: 10.1161/01.STR.0000177864.08516.47. [DOI] [PubMed] [Google Scholar]