

Abstract
His272 (7.43) in the seventh transmembrane domain (TM7) of the human A3 adenosine receptor (AR) interacts with the 3′ position of nucleosides, based on selective affinity enhancement at a H272E mutant A3 AR (neoceptor) of 3′-ureido, but not 3′-OH, adenosine analogues. Here, mutation of the analogous H278 of the human A1 AR to Ala, Asp, Glu, or Leu enhanced the affinity of novel 2′- and 3′-ureido adenosine analogues, such as 10 (N6-cyclopentyl-3′-ureido-3′-deoxyadenosine), by >100-fold, while decreasing the affinity or potency of adenosine and other 3′-OH adenosine analogues. His278 mutant receptors produced a similar enhancement regardless of the charge character of the substituted residue, implicating steric rather than electrostatic factors in the gain of function, a hypothesis supported by rhodopsin-based molecular modeling. It was also demonstrated that this interaction was orientationally specific; i.e., mutations at the neighboring Thr277 did not enhance the affinity for a series of 2′- and 3′-ureido nucleosides. Additionally, H-bonding groups placed on substituents at the N6 or 5′ position demonstrated no enhancement in the mutant receptors. These reengineered human A1 ARs revealed orthogonality similar to that of the A3 but not the A2A AR, in which mutation of the corresponding residue, His278, to Asp did not enhance nucleoside affinity. Functionally, the H278D A1 AR was detectable only in a measure of membrane potential and not in calcium mobilization. This neoceptor approach should be useful for the validation of molecular modeling and the dissection of promiscuous GPCR signaling.
The engineering of G protein-coupled receptors (GPCRs)1 (1–3) and other receptor proteins (4) has been reported. Here we focus on adenosine receptors (ARs), which are a family of four GPCR subtypes, termed A1, A2A, A2B, and A3. The structural basis of ligand recognition at the ARs has been studied by mutagenesis and molecular modeling based on homology to the light-sensing transducing protein rhodopsin (5–10). Resulting hypotheses for ligand binding have permitted the introduction of complementary structural changes in reengineered receptor proteins, e.g., neoceptors, and in tailored small molecule ligands, e.g., neoligands (11). This technique has already been used to probe the putative binding sites of two subtypes of ARs, A2A and A3, which were successfully reengineered for orthogonal activation by neoligands (12–14), i.e., nucleosides derived from the structures of known agonists but modified to avoid activation of the native receptors. The selective enhancement of affinity of these structurally matched pairs may be based on novel ionic, van der Waals, or H-bonds. This neoceptor approach is useful in verifying the accuracy of homology modeling of a given GPCR and dissecting its signaling pathways. Ultimately, this approach, which combines classical medicinal chemistry and receptor engineering, may be useful in gene therapy, following tissue-specific delivery of the neoceptor to a target site.
The A1 AR is biologically important in regulating the heart rhythm, in inhibiting neurotransmitter release in the central nervous system, tubuloglomerular feedback in the kidney, protection of cardiac and skeletal muscle, and in other systems (15–20). A1 AR agonists, antagonists, and allosteric modulators are under clinical development for a variety of conditions, including cardiac arrhythmias, renal damage, and pain (16, 20, 21). Thus, modulation of this receptor has therapeutic potential; however, past attempts to introduce selective agonists have encountered problems of side effects. Therefore, we extended the neoceptor approach to the A1 AR following the expectation that there are structure–function parallels within the AR family (8, 10).
With the aim of reengineering the human (h) A1 AR, site-directed mutagenesis and rhodopsin-based homology modeling of the A1 AR have helped to identify crucial amino acids for agonist and antagonist recognition within the putative binding pocket of the A1 AR as shown in Figure 1A (6–10, 22, 23). This led to the identification of two residues in transmembrane helical domain 7 (TM7), Thr277 and His278, which were predicted by molecular modeling to be in the proximity of the 3′ position on the ribose ring of adenosine-based agonists. This His residue (7.43) is conserved among all ARs, and support for it being a critical recognition element has come from diverse approaches (5, 11, 22, 24). For example, His (7.43) is important for agonist but not antagonist binding for the A1,A2A, and A3 ARs. By mutating this residue, i.e., substituting it with a negatively charged residue, such as Glu, which was done with the A3 AR neoceptors (14), we expected the standard adenosine agonists to undergo a decrease in affinity, while those (neoligands) modified at the 3′ position in a complementary fashion were not.
FIGURE 1.
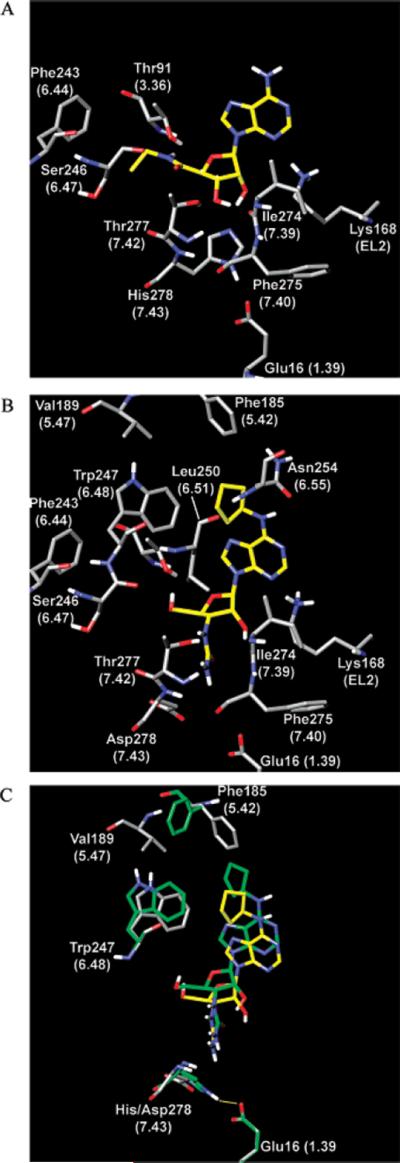
(A) Binding mode of NECA at the wild-type A1 adenosine receptor obtained after MCMM calculations. The receptor model was initially generated by homology to bovine rhodopsin and was energetically refined in stages, both before and after ligand docking. The carbon atoms of NECA are colored yellow. (B) Binding mode of compound 10 at the H278D mutant A1 adenosine receptor obtained after MCMM calculations. Several residues appear to be involved in H-bonding with the ligand: Thr91 (TM3), Asn254 (TM6), Ile274, Thr277, and His278 (TM7), and Lys168 (EL2). In addition, several hydrophobic residues are in the direct proximity of the N6 position of the ligand: Phe185, Trp247, and Leu250. (C) Superimposition of the models obtained for compound 10 at the wild-type (green) and H278D mutant (residues colored by atom type, with the carbon atoms of the ligand colored yellow) A1 adenosine receptor. The H-bond between His278 and Glu16 observed in the model is shown as a yellow line. All atomic coordinates of Val189 were identical in these two models. For this reason, only one “copy” of Val189 is shown.
By comparing the binding affinity among variously modified nucleoside analogues, we have identified pairs of a reengineered A1 AR and a strategically modified nucleoside that display orthogonal affinity enhancement. These results are shown to support the molecular modeling and proposed ligand docking of this receptor subtype. Unlike the neoceptors derived from both the A3 AR (12, 14) and the A2A AR (13), the present set of A1 AR neoceptors does not preserve all of the second-messenger coupling pathways associated with the native receptor (15). This finding promises to be useful in the dissection of promiscuous GPCR signaling (25).
EXPERIMENTAL PROCEDURES
Biological Materials
The pcDNA3.1 vector containing a hemagglutinin A-tagged wild-type human A1 AR was obtained from the University of Missouri Rolla cDNA Resource Center (Rolla, MO). Oligonucleotides used for the mutagenesis were synthesized by MWG Biotech (High Point, NC). Adenosine deaminase was obtained from Worthington Biochemical Corp. (Lakewood, NJ). CGS15943 {N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine}, NECA (5′-N-ethylcarboxamidoadenosine), CCPA (2-chloro-N6-cyclopentyladenosine), and DPCPX (8-cyclopentyl-1,3-dipropylxanthine) were obtained from Sigma (St. Louis, MO). [3H]DPCPX (116 Ci/mmol) was purchased from Amersham Bioscience (Buckinghamshire, U.K.). All other compounds, reagents, or solutions were obtained from standard commercial sources and were of analytical grade.
Cell Culture
Human embryonic kidney (HEK-293) cells and Chinese hamster ovary (CHO) cells were maintained at 37 °C with 5% CO2 in a 1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 medium, supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine.
Site-directed mutagenesis
was carried out using the Stratagene (La Jolla, CA) QuickChange site-directed mutagenesis kit. Mutations were confirmed by DNA sequencing using MWG Biotech.
Transient Transfection of HEK-293 and CHO Cells with Wild-Type and Mutant A1 ARs
HEK-293 and CHO cells were transiently transfected with cDNA using 1 mg/mL Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Briefly, 10 μg of cDNA was combined with 625 μL of OptiMEM, and 30 μL of Lipofectamine 2000 was combined with 625 μL of OptiMEM, for each 150 mm plate of 85–90% confluent HEK-293 cells. After 5 min, the two solutions were mixed and incubated at room temperature for 30 min. Meanwhile, the medium for the cells was changed to a 1:1 DMEM/Ham's F12 mixture with 10% FBS. The cDNA and Lipofectamine mixture was then added to the HEK-293 cells, and they were allowed to incubate at 37 °C with 5% CO2 for 48 h.
Membrane Preparation
After transfection for 48 h, cells were harvested in Dulbecco's phosphate-buffered saline and centrifuged twice at 2000 rpm for 5 min. The resulting pellet was homogenized with a Polytron homogenizer in 50 mM Tris-HCl buffer (pH 7.4) containing 10 mM MgCl2 and then centrifuged at 20 000 rpm and 4 °C for 20 min. The pellet was resuspended and homogenized in the Tris·HCl buffer with the addition of 3 units/mL adenosine deaminase and incubated at 37 °C for 30 min; 1 mL aliquots were stored at −80 °C. The protein concentration was measured using the method of Bradford (27).
Radioligand Binding Assays
For competitive binding studies, cell membranes containing 15 μg (HEK-293hA1-wt), 10 μg (HEK-293hA1-T277A), or 35–40 μg (HEK-293hA1-H278 mutant receptors) of protein were incubated for 1 h at 25 °C in 50 mM Tris-HCl buffer (pH 7.4) containing 10 mM MgCl2, with 0.4–0.6 nM [3H]DPCPX, and increasing concentrations of ligand, in a total volume of 200 μL. For saturation experiments, 10 μg (HEK-293hA1-wt and HEK-293hA1-T277A) or 15–20 μg (HEK-293hA1-mutant receptors) of membrane were incubated for 1 h at 25 °C in 50 mM Tris-HCl with 10 mM MgCl2 buffer (pH 7.4) with six different concentrations of [3H]DPCPX (0–9 nM). Nonspecific binding was assessed using 10 μM CGS15943. Incubations ended with filtration through What-man GF/B filters using a Brandel harvester and cold 50 mM Tris-HCl buffer. Radioactivity was counted using liquid scintillation. IC50 values were converted to Ki values as described previously (28).
Cyclic AMP Accumulation Assay
As described previously (46), CHO cells stably expressing the wild-type human A2B AR were treated with an agonist for 30 min and then lysed. Cyclic AMP was assayed as reported previously (47).
Calcium Assay
Intracellular calcium mobilization by stimulation of the wild-type and mutant human A1 ARs was assessed using a fluorescence imaging plate reader (Flex Station, Molecular Devices, CA). Briefly, CHO cells, transiently expressing either the wild-type or mutant human A1 AR, were split into 96-well plates after growing for 24 h. CHO cells stably expressing human A1 ARs were grown overnight in 100 μL of medium in 96-well flat bottom plates at 37 °C in 5% CO2 or until approximately 90% confluency. The calcium or membrane potential assay kit (Molecular Devices) was used as directed without washing cells, and with probenecid added to the loading dye at a final concentration of 2.5 mM to increase the extent of dye retention. Cells were loaded with 50 μL of dye with probenecid in each well and incubated for 60 min at room temperature. The compound plate was prepared using dilutions of various compounds in Hanks buffer. Samples were run in duplicate using a Molecular Devices Flexstation I at room temperature. Cell fluorescence (excitation at 485 and 535 nm and emission at 525 and 565 nm for the calcium assay and the membrane potential assay, respectively) was monitored following exposure to a test compound. Increases in the level of intracellular calcium or membrane hyperpolarization are reported as the maximum fluorescence value after exposure minus the basal fluorescence value before exposure (normalized relative fluorescent units, RFU%).
Conformational Search Analysis
Methods used for molecular modeling of ARs have been described previously (29–34). The ligand binding modes were studied with the Monte Carlo multiple-minimum (MCMM) conformational search analysis using MacroModel (35). The MCMM calculations were performed for a ligand and all residues located within 5 Å, using a shell of residues located within 2 Å. The following parameters were used: MMFFs force field, water used as an implicit solvent, a maximum of 1000 iterations of the Polak-Ribier Conjugate Gradient minimization method with a convergence threshold of 0.05 kJ mol−1 Å−1, 100 conformational search steps, and an energy window for saving structures of 1000 kJ/mol.
To study the binding mode of compound 10 in the wild-type and H278D mutant A1 receptor, the same initial atomic coordinates of the ligand and receptor residues were used, but in the model of the mutated receptor, the side chain of His278 was replaced with the side chain of Asp using the Mutate Residue option of MacroModel.
RESULTS
Design and Chemical Synthesis of Nucleoside Analogues
The nucleosides selected for probing the mutant A1 ARs were closely related to known AR agonists, such as NECA (adenosine 5′-N-ethyluronamide), a potent nonselective AR agonist. The 5′ position mainly consists of either the ribose-like CH2OH (1–21, Figure 2A) or the NECA-like 5′-uronamide (22–27, Figure 2B). Compounds 2, 5, 6, 13, 14, 23, and 26 were prepared as described previously (12, 14). Compound 7 was prepared as shown in Scheme 1. The new 3′-urea derivatives 3, 10, 16, and 18 were prepared as shown in Scheme 2 by the reported approach (14). Novel 2′-amino and 2′-urea derivatives 19–21 were prepared as shown in Scheme 3 using a reagent introduced by Katritzky et al. to effect a one-step conversion of the amino to urea derivatives (36). In the process of synthesizing the 2′-urea derivative 20, the N-acylurea derivate 21 was also isolated as a stable side product.
FIGURE 2.

Known and novel nucleoside analogues used to probe the putative binding site of the wild-type and mutant human A1 ARs. 9-Riboside (A) and NECA-type (5′-uronamide) derivatives (B) are shown.
Scheme 1.

Procedure for the Synthesis of Urea Derivative 7 from the Corresponding Primary Amine 6 (26)a
aReagents and conditions: (a) benzotriazole-1-carboxamide (36), DMF, room temperature.
Scheme 2.

Procedure for the Synthesis of 3′-Urea Derivatives of Adenosinea
aReagents and conditions: (a) NH4OH, 1,4-dioxane, room temperature, 24 h, or 2-phenylethylamine, Et3N, EtOH, 50 °C, 18 h, 2-methylbenzylamine, Et3N, EtOH, 50 °C, 18 h, or cyclopentylamine, Et3N, EtOH, 50 °C, 18 h, then NaOMe, MeOH, room temperature, 2 h; (b) TBSCl, imidazole, DMF, room temperature, 24 h; (c) Ph3P, NH4OH/H2O, THF, room temperature, 18 h; (d) chloroacetyl isocyanate, DMF, 0 °C, 3 h; (e) NaOMe, MeOH, room temperature, 18 h; (f) TBAF, THF, room temperature, 4 h.
Scheme 3.

Procedure for the Synthesis of 2′-Urea Derivatives of Adenosinea
aReagents and conditions: (a) ethyl trifluoroacetate, diisopropylethylamine, DMF, room temperature; (b) 3-iodobenzylbromide, DMF, 40 °C; (c) NH4OH, MeOH, 90 °C; (d) benzotriazole-1-carboxamide, DMF, room temperature.
Initially, the native agonist, adenosine 1, was modified at the 3′ position with amino 2 or ureido 3 groups. Other compounds were modified with such groups at the N6 (6 and 7) or 5′ position (23). A urea group in compounds 3, 5, 7, 10, 13, 14, 16, 18, 26, and 27 was selected as a functional change that could act complementarily with the introduction of Asp or Glu residues in the receptor through the formation of stabilizing H-bonds between the ligand and anionic residues in the mutant receptor. From previous results, the 3′-ureido moiety was known to result in inactivity in wild-type ARs, possibly due to unfavorable steric interactions (37). This approach was proven to be successful in the design of neoligands matched to neoceptors of the human A3 AR (14). Similarly, an amino group was introduced into nucleoside analogues 2, 6, and 23 and acted as a possible stabilizing counterion to anionic residues introduced into the mutant A1 ARs (12). An amino group is useful for comparison to the larger urea group, since steric factors, as well as electrostatics and H-bonding, may influence which modification will prove to be more beneficial in selective affinity enhancement at the mutant A1 ARs.
Substitution at the N6 and C2 positions of adenosine derivatives is known to enhance the potency and selectivity at ARs and the stability of the nucleoside analogues (16). These well-characterized modifications were then combined with additional amino or ureido derivatization to generate a library of neoligands. N6-Methyladenosine 4, a relatively weak human A1 AR agonist (38), was modified at the 3′ position with a ureido group in 5. The N6-methyl group was extended to a 2-aminoethyl moiety in 6 or a 2-ureidoethyl group in 7.
The potent, selective A1 AR agonist, CPA 8, was transformed into the corresponding 3′-ureido analogue in 10. MRS541 11 and MRS542 12 are a partial agonist and an antagonist, respectively, at the human A3 AR, and they are also fairly potent at the A1 AR (39). Their corresponding 3′-ureido analogues, 13 and 14, were prepared. The 3′-ureido analogue 13 was reported as a neoligand for the H278E and H278D mutant A3 ARs (14) and was therefore applied to analogous A1 AR mutations. Two other 5′-hydroxyl-3′-ureido analogues, the N6-(2-methylbenzyl) 16 and the N6-(2-phenylethyl) 18 derivatives, were prepared and evaluated, because the parent analogues 15 and 17 potently activate the ARs nonselectively (13, 40). Compound 15 (metrifudil) has already been evaluated in human trials (41).
The potent, nonselective AR agonist, NECA 22, a 5′-uronamide, was appended on the N-ethyl moiety with an amino group in 23. Compound 23 was identified previously as a suitable neoligand when paired with the A2A AR neoceptors (13). Other 5′-uronamide analogues, 24 and 25, are currently undergoing human clinical trials and exhibited favorable in vivo pharmacokinetics (42, 43). The A3 AR-selective agonists, 24 and 25, also activate the A1 AR at higher concentrations. Therefore, the related 3′-ureido derivatives, 26 and 27, were prepared for investigation of activation of the mutant A1 ARs.
Mutagenesis of the hA1 AR
Targeted amino acid replacements in the hA1 AR were selected to facilitate an enhanced interaction with the ribose moiety based upon its putative binding mode as illustrated by rhodopsin-based homology modeling (Figure 1A). Specifically, Thr277 (7.42) was mutated to both Ala and Glu, while His278 (7.43) was mutated to Ala, Asp, Glu, and Leu. Both of these residues in TM7 were predicted to be involved in coordination of the ribose moiety in the putative A1 AR binding site (8, 10, 44). Thr277 was predicted by modeling to interact with both the 5′-OH and 3′-OH groups, and His278 was situated close to the 2′-OH and 3′-OH groups (10). The mutations to the charged residues, Asp and Glu, versus the neutral residues, Leu and Ala, were selected for investigation of what features of the ligand–receptor interaction, i.e., electrostatic interactions, hydrogen bonding, or steric factors, will prove to be more beneficial in selective affinity enhancement at the mutant A1 ARs. For binding and functional analysis, the wild-type or mutant A1 ARs were expressed in either CHO or HEK-293 cells.
Pharmacological Analysis of Nucleoside Analogues at hA1 AR Constructs Using Saturation Analysis
Since the TM7 mutations of the A1 AR occur in a region predicted to coordinate the ribose moiety of the nucleoside ligands, modification of the selected amino acid residues would be expected to be more detrimental to binding of agonist than antagonist. Attempted saturation analysis of the mutant receptors with the agonist [3H]CCPA (9, 1–5 nM) did not produce measurable specific binding (data not shown), making it impractical to use this radioligand. Therefore, subsequently radioligand saturation and competition analysis at the wild-type and mutant A1 ARs were conducted using the antagonist [3H]DPCPX, which retained high affinity (7).
Saturation binding using [3H]DPCPX provided a Kd value of 2.37 ± 0.20 nM with a Bmax value of 10.5 ± 1.2 pmol/mg of protein, for the wild-type A1 AR transiently expressed in HEK-293 cells (Table 1). These values are similar to those previously reported (45). In the case of the mutant A1 ARs, the antagonist Kd values varied only slightly from that of the wild-type receptor, thus allowing the use of [3H]DPCPX in competitive binding studies. However, unlike the relative uniformity in the Kd values, the Bmax for the mutant receptors was on average ~3-fold lower than that of the wild-type A1 AR, with the exception of the T277A mutant receptor.
Table 1.
Saturation Analysis of [3H]DPCPX at the Wild-Type and Mutant Human A1ARs Transiently Expressed in HEK-293 Cella
| A1 AR construct | Kd (nM) | Bmax (pmol/mg) |
|---|---|---|
| wild-type | 2.37 ± 0.20 | 10.5 ± 1.2 |
| T277A | 1.12 ± 0.09 | 14.7 ± 3.8 |
| T277E | 5.14 ± 0.74 | 6.0 ± 0.2 |
| H278A | 4.72 ± 0.60 | 3.0 ± 0.8 |
| H278D | 3.11 ± 0.34 | 2.2 ± 0.4 |
| H278E | 4.96 ± 0.93 | 2.7 ± 0.6 |
| H278L | 3.94 ± 0.10 | 3.1 ± 0. |
Structures are given in Figure 2. Values are expressed as the mean ± sem from four to eight experiments, each performed in duplicate.
Pharmacological Analysis of Nucleoside Analogues of Wild-Type ARs Stably Expressed in CHO Cells
Competitive radioligand binding studies were performed using the agonist radioligands [3H]CCPA, [3H]CGS21680, and [125I]I-AB-MECA in membranes of CHO cells stably expressing wild-type human A1, A2A, and A3 ARs, respectively (7, 46). Ki values for the binding of nucleoside analogues to the wild-type ARs are given in Table 2. At the A2B AR, a functional assay, i.e., stimulation of adenylate cyclase in CHO cells stably expressing the wild-type human A2B AR, was used (47).
Table 2.
Binding Affinity or Functional Potency of Nucleoside Derivatives at Wild-Type Human ARs
| compound (ref) | A1a,bKi (nM ± sem) or % inhibition | A2Aa,bKi (nM ± sem) or % inhibition | A2Bc EC50 (nM ± sem) or % activity | A3a,bKi (nM ± sem) or % inhibition |
|---|---|---|---|---|
| 3 | <10% | <10% | <10% | 17% |
| 5 (14) | <10% | <10% | <10% | <10% |
| 6 | 47.5 ± 18.4 | 22% | 33% | 3000 |
| 7 | 43.4 ± 13.0 | 26% | 48% | 252 |
| 8, CPA (40) | 1.8 ± 0.5 | 820 ± 220 | <10% | 70 ± 10 |
| 9, CCPA (16) | 0.83 | 2270 | 39% | 38 ± 6 |
| 10 | <10% | 13% | <10% | <10% |
| 11 (MR541) (39) | 7.4 ± 1.7 | 135 ± 22 | 58% | 5.8 ± 0.4 |
| 12 (MR542) (39) | 16.8 ± 2.2 | 197 ± 34 | 16% | 1.8 ± 0.1 |
| 13 (14) | <10% | <10% | <10% | <10% |
| 14 (14) | 34% | <10% | <10% | 21% |
| 16 | <10% | <10% | <10% | <10% |
| 18 | <10% | 13% | 15% | 25% |
| 19 | 314 ± 26 | 38% | 20% | 78.7 ± 4.9 |
| 20 | 36% | 20% | <10% | 34% |
| 21 | 23% | 15% | <10% | 53% |
| 22, NECA (40) | 6.8 ± 2.0 | 2.2 ± 0.6 | 140 ± 20 | 16.0 ± 5.0 |
| 23 (13) | 250 ± 50 | 3100 ± 400 | 15600 ± 3000 | <100000 |
| 25 (40) | 1240 ± 320 | 5400 ± 2500 | NDd | 1.4 ± 0.3 |
| 26 (37) | <10% | <10% | <10% | <10% |
| 27 | <10% | <10% | <10% | 24% |
Experiments were performed using cells stably expressing human adenosine receptors (ARs). For A1, A2B, and A3 ARs, CHO cells were used, and for the A2A AR, HEK-293 cells were used. Binding was carried out using 1 nM [3H]CCPA, 10 nM [3H]CGS21680, and 0.5 nM [125I]AB-MECA as radioligands for A1, A2A, and A3 ARs, respectively. Ki values are expressed as the mean ± sem (n = 3–4) and normalized against the nonspecific nucleoside ligand, NECA 22. Ki values for other compounds are reported in refs 40 and 46: 2, 4, 15, 17, and 24.
The percent inhibition of radioligand binding when given in italics is for 10 μM nucleoside, relative to the maximum inhibition by NECA at 10 μM (100%).
The potency at the A2B AR was measured with a cyclic AMP functional assay (47), expressed as EC50 or percent activation at 10 μM, compared with the maximum efficacy of NECA at 10 μM being 100%.
Not determined.
The presence of a 3′-ureido group caused a dramatic reduction in the affinity at the A1 AR. This was observed for the following N6-substituted 9-riboside derivatives (compared to the 3′-OH analogue which generally displayed a Ki value of <100 nM): N6-cyclopentyl 10 (8), N6-(3-iodobenzyl) 13 (11) and 14 (12), N6-(2-methylbenzyl) 16 (15), and N6-(2-phenylethyl) 18 (17) analogues. Less dramatic reductions were evident for the adenosine analogue 3 (1) and its N6-methyl 5 (4) analogue. The 2′-ureido group also caused a reduction in the affinity of 9-riboside analogues at the A1 AR, as evidenced by the N6-(3-iodobenzyl) 20 (11) derivative. The 5′-N-methyluronamide derivatives also exhibited a dramatic loss of affinity upon substitution of the 3′-OH group with a 3′-ureido group, as in N6-(3-iodobenzyl) analogues 26 (24) and 27 (25).
Ligand Binding Properties of the Wild-Type and Mutant hAR Receptors
Competitive radioligand binding studies were performed using the high-affinity antagonist [3H]DPCPX against a range of ligands at the wild-type and mutant hA1 ARs. Initially, previously reported A1 AR agonists and antagonists were surveyed at the wild-type and mutant receptors (Table 3). The wild-type receptor maintained its binding affinity with the agonists NECA (adenosine 5′-N-ethyluronamide, 22) and CCPA 9, and the antagonists DPCPX and CGS15943 with affinities consistent with reported values (48). At the H278 mutant receptors, the binding affinity of CGS15943 was reduced 30–50 fold, while the loss of affinity of DPCPX, CCPA, and NECA was less pronounced. The binding affinity of antagonists was better preserved in the Thr277 than in the His278 mutant receptors. It is important to note that the agonist affinities represent a mixture of high- and low-affinity states, which is expected for the inhibition of an antagonist radioligand by an agonist (44). The Ki values obtained for the nucleoside derivatives more closely resemble those reported for the agonist binding component with a low affinity for the A1 AR. Since it was not feasible to use an agonist radioligand for the mutant A1 ARs, the affinities measured for the nucleoside derivatives are to be compared in a relative manner.
Table 3.
Ki Values for the Inhibition by Nucleoside Derivatives of Binding of Antagonist [3H]DPCPX at Wild-Type and Mutant Human A1 ARs Transiently Expressed in HEK-293 Cellsa
| binding affinity, Ki (nM ± sem), or % inhibition |
|||||||
|---|---|---|---|---|---|---|---|
| compound | wild-type hA1 | H278E | H278D | H278A | H278L | T277E | T277A |
| DPCPXb | 4.5 ± 1.2 | 36 ± 2 | 50 ± 3 | 40 ± 8 | 35 ± 2 | 18.3 ± 1.6 | 3.5 ± 0.9 |
| CGS15943b | 3.4 ± 1.1 | 120 ± 20 | 110 ± 40 | 160 ± 3 | 90 ± 3 | 8.6 ± 0.3 | 4.6 ± 1.5 |
| 1 | 4600 ± 480 | 22% | 15% | 13% | <10% | 13% | <10% |
| 2 | <10% | 17% | <10% | 11% | 12% | <10% | <10% |
| 3 | <10% | 14% | 20% | 20% | 17% | <10% | <10% |
| 5 | <10% | 24% | 16% | 15% | 25% | <10% | <10% |
| 6 | 30% | 14% | <10% | 14% | 17% | <10% | <10% |
| 7 | 17% | 18% | <10% | <10% | 15% | <10% | <10% |
| 9, CCPA | 610 ± 50 | 1200 ± 150 | 1000 ± 170 | 1400 ± 120 | 820 ± 90 | 7000 ± 800 | 11% |
| 10 | 12% | 380 ± 80 | 340 ± 20 | 570 ± 60 | 400 ± 60 | 41% | <10% |
| 13 | 15000 ± 3100 | 560 ± 170 | 450 ± 40 | 570 ± 30 | 550 ± 50 | 48% | <10% |
| 14 | 6200 ± 1900 | 210 ± 60 | 240 ± 30 | 220 ± 30 | 290 ± 50 | 50% | 17% |
| 16 | 13% | 750 ± 150 | 590 ± 20 | 1000 ± 60 | 700 ± 100 | 39% | <10% |
| 18 | 13% | 44% | 48% | 50% | 39% | 22% | <10% |
| 19 | 38% | 530 ± 30 | 410 ± 120 | 834 | NDc | <10% | <10% |
| 20 | <10% | 980 ± 280 | 810 ± 250 | 940 | NDc | <10% | NDc |
| 21 | <10% | 560 ± 80 | 500 ± 120 | 670 | NDc | 11% | NDc |
| 22, NECA | 1600 ± 100 | 9700 ± 900 | 8500 ± 1400 | 8800 ± 700 | 8800 ± 900 | 25% | <10% |
| 23 | <10% | 26% | 13% | 14% | 32% | 12% | <10% |
| 26 | 14% | 1200 ± 400 | 1200 ± 300 | 980 ± 450 | 58% | 10% | <10% |
| 27 | <10% | 220 ± 20 | 350 | NDc | NDc | 16% | NDc |
Structures are given in Figure 2. Experiments were performed using HEK-293 cells transiently expressing (using Lipofectamine 2000) a mutant hA1 AR. Binding was carried out using 0.3–0.5 nM [3H]DPCPX as the radioligand. Ki values are expressed as the mean ± sem (n = 3–6) and normalized against a nonspecific binder, CGS15943. Values in italics are percent inhibition at 10 μM, relative to the maximum inhibition by CGS15943 at 10 μM (100%).
Non-nucleoside AR antagonists.
Not determined.
The endogenous ligand for ARs, adenosine 1, bound to the wild-type A1 AR with a Ki value of 4.60 ± 0.48 μM. However, for the mutant A1 ARs, even at a high concentration of 10 μM, binding of the radioligand [3H]DPCPX was not appreciably inhibited, by 20% at most. On the other hand, the compounds designed in a complementary fashion, intended to produce a gain in selectivity for the mutant receptors (neoceptors), exhibited selectivity in binding at the mutant A1 ARs over the wild-type A1 AR (Table 3), while exhibiting no binding affinity for any of the wild-type ARs (Table 2). Specifically, the N6-(3-iodobenzyl) 3′-ureidoadenosine derivatives 13 and 14, which bound to the wild-type A1 AR in the micromolar range, with Ki values of 15.0 ± 3.1 and 6.2 ± 1.9 μM, respectively, were found to bind in the submicromolar range at all the His278 mutant receptors, while losing affinity at both Thr277 mutant receptors. Similarly, the N6-cyclopentyl and N6-(2-methylbenzyl) derivatives of 3′-ureidoadenosine, 10 and 16, respectively, which were inactive at the wild-type A1 AR and able to inhibit only <15% of the binding of radioligand [3H]-DPCPX, also bound in the submicromolar range at all the His278 mutant receptors, while losing affinity at both Thr277 mutant receptors. However, where these two groups of compounds differ is in their pattern of enhancement of affinity between the His278 mutant and wild-type hA1 ARs. While compounds 13 and 14 exhibited modest 27- and 30-fold enhancements for the H278E mutant receptor, respectively, compounds 10 and 16 exhibited 263- and 133-fold enhancements, respectively. These reported enhancements for compounds 10, 13, 14, and 16 at the H278E mutant can be extended to the other His278 mutant receptors as well, e.g., H278D, H278L, and H278A. That is, the enhancement for these compounds was relatively similar for all the His278 mutant receptors; there was not one mutant that generated a significantly larger improvement than the others.
All four of these derivatives contain a 3′-ureido group and large (cyclopentyl or benzyl) N6 substituent. When the N6 substituent of 3′-ureido derivatives, such as in N6-phenylethyl 18, N6-methyl 5, or unsubstituted N6 3 analogue, was enlarged or truncated, the enhancement at the His278 mutant receptors was lost. Similarly, placing hydrogen acceptor groups at other positions, such as appending a urea moiety at the N6 position in 7 or an amino group at the 3′ position in 2, N6 position in 6, or 5′ position in 23, did not produce an affinity enhancement at any of the hA1 AR mutant receptors. It is also important to note that despite derivatization at the 3′, 5′, N6, or 2 positions, none of the compounds displayed a preference in binding affinity for the Thr277 mutant receptors over the His278 mutant receptors.
The 2′-amino and 2′-ureido groups in compounds 19–21 were also well tolerated by the H278 mutant A1 ARs. In comparison to binding to the native AR subtypes, compounds 20 and 21 displayed orthogonality of binding at the H278 mutant A1 ARs. Derivatization of the 5′ position, as in 3′-ureido derivatives 26 and 27, resulted in an enhanced affinity (Ki ) 1.6 μM) at the H278D mutant receptor.
To probe the structural similarities and differences among three of the four hAR subtypes, binding curves using 3′-urea derivatives 10 and 13 were determined for wild-type and analogous mutant human ARs (Figure 3). The H278D mutation of the human A1 AR and the corresponding mutations of conserved His in TM7 of the A2A AR (H278D) and A3 AR (H272D) were studied. The behavior of both the N6-cyclopentyl 10 and the N6-(3-iodobenzyl) 13 derivatives was similar at the A1 and A3 ARs; i.e., the affinity was selectively enhanced at the Asp mutant receptors in comparison to wild-type receptors. However, there was no affinity enhancement at the A2A ARs, implying a similar binding mode of the nucleosides at the A1 and A3 ARs and a different mode at the A2A AR.
FIGURE 3.

Representative inhibition curves from binding assays for wild-type and mutant human ARs using urea derivatives 10 and 13. The H278D mutation of the human A1 AR and the corresponding mutations of the conserved His in TM7 of the A2A AR (H278D) and A3 AR (H272D) were studied. The mutant receptors were expressed transiently in HEK-293 cells. Antagonist radioligands were used at the A1 ARs ([3H]DPCPX) and the A2A ARs ([3H]ZM241385), and an agonist radioligand was used at the A3 ARs ([125I]I-AB-MECA).
Functional Effects of Nucleosides at Wild-Type and Mutant Receptors
In general, neoceptors are intended to activate the same effector systems as the native receptor (11). However, an examination of mutant A1 ARs in several measures of receptor activation in this study indicated major differences with the wild-type A1 AR. A preliminary functional study of the effect of adding GTP during the binding of CCPA 9 and its 3′-ureido analogue 10 at the H278A and H278D mutant receptors indicated that in both cases there was no pronounced GTP-induced shift (data not shown).
Activation of the wild-type A1 AR has been shown to mobilize intracellular calcium (49). We confirmed that CPA caused a concentration-dependent increase in the intracellular calcium concentration in CHO cells stably expressing the wild-type A1 AR. However, among the agonists that exhibited selectively enhanced affinity for the mutant A1 ARs, none were able to activate calcium transients in CHO cells transiently expressing the H278D mutant receptor (Figure 4).
FIGURE 4.

Stimulation of intracellular calcium levels at the wild-type and H278D mutant hA1 ARs using known agonists CPA 8 and NECA 22 and urea derivatives 10 and 13 (n = 3–4). The receptors were expressed transiently in CHO cells. EC50 values determined at the wild-type hA1 AR are 25.1 ± 6.2 (8) and 56.4 ± 16.7 (22).
Activation of the A1 AR has been shown to hyperpolarize cells (50). When membrane potential changes were measured in the same cells using a fluorescence method, CPA caused hyperpolarization in CHO cells expressing the wild-type A1 AR with an EC50 value of 10.1 ± 2.6 nM (Figure 5). The 5′-CH2OH derivative 13, at a concentration as high as 10 μM, did not have any effect on the H278D mutant. However, the 5′-N-methylamide derivative 26 was shown to be more potent in activating the H278D mutant receptor (3.5 ± 1.6 μM) than at the wild-type A1 AR (>10 μM) or the control (nontransfected cells), although it was only partially efficacious compared with the efficacy of CPA at the wild-type A1 AR. More systematic studies of other signaling pathways will be needed to fully characterize the spectrum of effector mechanisms coupled to the neoceptor-(s).
FIGURE 5.

Membrane hyperpolarization at the wild-type and H278D mutant hA1 AR using known agonist CPA 8 and urea derivative 26. The receptors were expressed transiently in CHO cells.
Modeling the Interaction of Nucleosides with Wild-Type and Mutant AL1 ARs
Modeling was used to predict detailed features of the interaction of the A1 AR constructs with their nucleoside ligands. A recently published molecular model (9) of the human A1 AR, based on the high-resolution structure of bovine rhodopsin (51, 52), was used in the docking of NECA 22. The docked complex was subsequently altered in structure, both the ligand and receptor, to predict nucleoside binding modes at the mutant A1 ARs. An initial complex of the A1 AR with NECA was further refined by energy optimization and MCMM calculations implemented in MacroModel (35). The position of NECA inside the A1 AR obtained after MCMM calculations was found to be similar to its initial position. In particular, Thr91 (3.36) formed a H-bond with the amide NH group of NECA, the OH group of Thr277 (7.42) was found in the proximity of the 3′-OH group and the C=O group of the amide moiety, and His278 (7.43) was located near the 2′- and 3′-OH groups of the ligand. In addition, the backbone oxygen atom of Ile274 (7.39) was found in the proximity of both 2′- and 3′-OH groups. Lys168 (EL2.49) was observed between the 2′-OH group and the N3 nitrogen atom of the adenine ring (Figure 1A).
To predict the binding mode of adenosine at the A1 AR, the 5′-ethylcarboxamide moiety of NECA located inside the binding pocket was replaced with a CH2OH group, and the model was subjected to MCMM calculations. Then, a cyclopentyl ring was attached to the N6-amino group of adenosine, providing an initial position of CPA inside the binding site. This A1 AR–CPA complex was also subjected to MCMM calculations.
The results of conformational searches performed for adenosine and CPA indicated that both ligands were involved in almost the same interactions with the receptor. Moreover, the binding modes obtained for these compounds were very similar to the binding mode obtained for NECA. In particular, the 5′-OH group formed a H-bond with Thr91 (3.36), and the 3′-OH group was H-bonded to Thr277 (7.42) and His278 (7.43). His278 also established a H-bond with the 2′-OH group of the ligands. The N6-amino group was found to be H-bonded with the oxygen atom of the side chain amido group of Asn254 (6.55), and the N6-cyclopentyl ring of CPA was located among the hydrophobic side chains of Phe185 (5.43), Trp247 (6.48), and Leu250 (6.51).
Since adenosine, NECA, and CPA had almost the same position and arrangement inside the binding site of the A1 AR, it seems reasonable to propose that compound 10 could attain a similar position in the receptor. However, an attempted manual docking of 10 (considered as a starting point for MCMM calculations) showed that the 3′-ureido group was in a sterically undesirable position with respect to His278. In general, a more energetically favorable state of an initial docking mode could be obtained through rearrangement of the residues around the ligand or through a change in the ligand position within the receptor. It should be noted that in this model of the A1 AR, the side chain of His278 forms a constraining H-bond with Glu16 (1.39). The same interaction was previously proposed between Glu13 and His278 of the A2A AR and suggested to be important for receptor activation (24). For this reason, it seemed unlikely that the side chain of His278 would significantly shift outwardly and would break the bond with Glu16 to provide more free space for the 3′-ureido group. This hypothesis was confirmed by MCMM calculations performed on 10, demonstrating that the position of the nucleoside shifted from TM7 toward TM5, instead of a His side chain shift. The orientation of the His278 side chain was also changed in the resulting model, but the H-bonding between His278 and Glu16 was not lost. Also, the phenyl ring of Phe185 (5.42) shifted toward Trp247 (6.48) and Val189 (5.47) from its initial position, to accommodate the cyclopentyl ring of 10 during its movement from TM7 to TM5 (Figure 1C). These major rearrangements of the wild-type A1 AR needed to bind compound 10 would constitute an energetically unfavorable process.
In contrast, in the model obtained after MCMM calculations for compound 10 docked in the H278D mutant A1 AR, we did not observe any significant changes in the ligand position or residue side chain orientations (Figure 1B). The following ligand–receptor interactions were observed in the resulting model. The 5′-OH group of compound 10 formed a H-bond with Thr91 (3.36). Lys168 (EL2.49) was observed between the 2′-OH group and the N3 atom of the adenine ring. The 2′-OH group of 10 was H-bonded to the backbone oxygen atom of I274 (7.39). The N6-cyclopentyl ring of 10 had the same position as the N6-cyclopentyl ring of CPA and was located among Phe185 (5.43), Trp247 (6.48), and Leu250 (6.51).
The side chain of Asp278 (7.43) was found to be oriented outwardly from Glu16, and also outwardly from the 3′-ureido group of 10. Thus, in the model, Asp278 was not involved in H-bonding interactions with the ligand. However, the switching of the Asp278 side chain, in comparison to the orientation of H278 in the wild-type receptor, provided free space for the 3′-ureido group of 10 inside the binding pocket. Moreover, in the model that was obtained, both amino groups of the 3′-ureido moiety were involved in H-bonding with Thr277 (7.42), and the terminal NH2 group of this moiety was H-bonded to the backbone oxygen atom of Phe275 (7.40).
The obtained binding mode of compound 10 at the H278D mutated A1 AR, demonstrating the absence of a direct interaction between the ligand and Asp278, is in a good agreement with the experimental data obtained for other mutations, in particular, for H278A and H278L. As follows from Table 3, compound 10 has similar binding affinity at these mutant receptors; however, the side chains of Ala278 or Leu278 cannot establish H-bonds with the 3′-ureido moiety.
DISCUSSION
By implementing an integrated approach of mutagenesis, competitive radioligand binding assays, and functional assays, combined with molecular modeling, we were able to identify matched pairs of neoligands and neoceptors. We tested a range of 2′- and 3′-ureido nucleoside analogues, combining functionalization of the adenine ring at the N6 and 2 positions and the ribose ring at the 5′ position, ultimately identifying tailored ligands that could be used to select for a class of His278 mutant receptors in the hA1 AR. The 2′- and 3′-ureido nucleoside analogues tended to lose the ability to bind to all native subtypes of ARs, achieving an aim of designing neoceptors. A 2′-amino-2′-deoxynucleo-side derivative 19 lost the ability to interact with native A2A and A2B ARs, but not with native A1 and A3 ARs.
The H278 mutant receptors exhibited more than 100-fold affinity enhancement for newly synthesized adenosine derivatives, such as 10 (N6-cyclopentyl-3′-ureido-3′-deoxyadenosine), while showing decreased affinity or potency for adenosine and other corresponding 3′-OH adenosine analogues. The present reengineered human A1 ARs revealed orthogonality similar to that of the A3 but not the A2A AR, in which mutation of the corresponding residue, His278, to Asp did not enhance the affinity of nucleosides derivatized with amine functionality on the ribose moiety (8). The key characteristics of the best compounds for affinity enhancement at the reengineered human A1 ARs are a 2′- or 3′-ureido group and a large (cyclopentyl or benzyl) N6 substituent. The 2 position may be substituted with a Cl group, as in 14.
The lack of affinity enhancement of 2′- and 3′-ureido derivatives at the Thr277 mutant receptors implies a spatial orientation of the ligand, facilitating an interaction with the residue at the position of His278, as predicted. Additionally, H-bonding groups placed on the adenine N6 or ribose 5′ positions demonstrated no enhancement of activity in the mutant receptors. The observation that all of the His278 mutant A1 ARs produced a similar type of effect regardless of the charge character of the substituted side chain implies that steric factors are more important in this gain of function than electrostatic factors, a hypothesis that is supported by the molecular modeling analysis.
It should be noted that the measured affinities of the new compounds, such as 10 and 13, for H278 mutant receptors are not very high, which is a less than ideal basis for accurate modeling. However, the fact that the pattern of interactions of 10 and 13 with the A1 AR is very different from that of the A2A AR and the more than 100-fold difference in affinity between the wild-type and H278 mutant receptors for some compounds, such as 10 and 13, does support a specific ligand–A1 AR pair. Also, it is worth noting that the displacement of an antagonist radioligand (such as [3H]-DPCPX) by a competing ligand that is an agonist, in general, tends to underestimate the affinity of the agonist at the receptor, in relation to its high-affinity binding component. We noted this phenomenon much earlier in a study of the A1 AR, in which the binding of a variety of agonists was probed using an antagonist radioligand (53). The modeling is not suitable for conformationally resolving multiple affinity states of the receptor. Thus, there is likely a high-affinity component of the binding of the nucleoside analogues at the mutant receptors that is not resolved in the Ki values that are obtained.
While the design of the neoceptor–neoligand pairs in the case of A1 AR follows that reported for the A3 AR, the diversity of mutated receptors and the repertoire of ligands in the present study are larger. This allows for a better understanding of the interactions that affect the neoceptor–neoligand affinities and for further elaboration of the approach. In this context, the observation that modification of the interaction between the 3′ substituent of the ligand and the residue at position 278 narrows the spectrum of functional responses could be particularly significant. The H278D A1 AR was functionally responsive only as a measure of membrane potential and not calcium mobilization. This neoceptor approach should be useful for the validation of molecular modeling and the dissection of promiscuous GPCR signaling. The observation of differences in the neoceptor–neoligand characteristics in different AR subtypes should contribute to the pinpointing of their structural and functional distinction. The successful application of the neoceptor–neoligand approach to the different ARs suggests that it may also be applicable to other GPCRs.
The proposed nucleoside docking mode (Figure 1) is in good agreement with previous studies of the ligand–receptor interactions of ARs (8, 9). The available experimental data and results of computational studies suggested that agonists of all four AR subtypes share almost the same general binding region, which includes interactions with Thr3.36, Trp6.48, Asn6.55, Ser/Thr7.42, His7.43, and EL2, as recently reviewed (10). The predicted conformation of docked nucleosides at the A2A AR differs from that of the A1 and A3 ARs in characteristic features, such as the preferred angle of the glycosidic bond (8, 13).
Interestingly, the residues located in the positions mentioned above were also proposed to be important for nucleotide recognition in the P2Y receptor family (54), and their proposed role in agonist coordination is also analogous to mutagenesis and modeling data published for other, nonpurinergic GPCRs. Thus, residue 3.36 is important for a ligand binding in melatonin (55) and dopamine receptors, and residue 6.55 is involved in ligand recognition in dopamine, histamine, and adrenergic receptors (56). Residues located in TM7, namely, 7.39 and 7.43, are involved in ligand binding in acetylcholine, serotonin, dopamine, and adrenergic receptors, as reviewed in refs 52 and 56. Also, it is known that conserved aromatic residue 6.48 (Trp in the case of ARs) is essential for activation of various GPCRs. Therefore, the arrangement of the A1 AR binding site and the binding mode of its agonists proposed in this study are in good relation with current, general mechanisms of ligand–receptor interactions for the rhodopsin family of GPCRs.
CONCLUSIONS
Our neoceptor approach to the human A1 AR revealed orthogonality similar to that of the A3 AR but not that of the A2A AR. This neoceptor approach should be useful for the validation of molecular modeling and the dissection of promiscuous GPCR signaling. The full range of signaling associated with the A1 AR was not preserved in these mutant receptors, and therefore, the future use of the A1 AR neoceptors for selective cytoprotection, which was accomplished with an A3 AR neoceptor, is uncertain. In this study, we have been unable to separate the functions of the key conserved His residue in TM7: to recognize and bind a small nucleoside ligand and to allow that molecule to induce the conformational changes required for activation of the full spectrum of functional effects.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases, and by a grant from the NCRC Program of MOST/KOSEF (R15-2006-020-00000-0). P.B. thanks Gilead Sciences (Foster City, CA) for financial support.
Footnotes
Abbreviations: AR, adenosine receptor; CCPA, 2-chloro-N6-cyclopentyladenosine; CGS15943, N-[9-chloro-2-(2-furanyl)[1,2,4]-triazolo[1,5-c]quinazolin-5-amine]; CHO, Chinese hamster ovary; CPA, N6-cyclopentyladenosine; DMEM, Dulbecco's modified Eagle's medium; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine; FBS, fetal bovine serum; GPCR, G protein-coupled receptor; HEK, human embryonic kidney cells; MCMM, Monte Carlo multiple-minimum; NECA, 5′-N-ethylcarboxamidoadenosine; RFU, relative fluorescent units; sem, standard error of the mean; TM, transmembrane helical domain; Tris, tris(hydroxymethyl)aminomethane.
SUPPORTING INFORMATION AVAILABLE Compound 10 inside the putative binding site of the H278D mutant A1 adenosine receptor and details of chemical synthesis. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Strader CD, Gaffney T, Sugg EE, Candelore MR, Keys R, Patchett AA, Dixon RA. Allele-specific activation of genetically engineered receptors. J. Biol. Chem. 1991;266:5–8. [PubMed] [Google Scholar]
- 2.Redfern CH, Coward P, Degtyarev MY, Lee EK, Kwa AT, Hennighausen L, Bujard H, Fishman GI, Conklin BR. Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat. Biotechnol. 1999;17:165–169. doi: 10.1038/6165. [DOI] [PubMed] [Google Scholar]
- 3.Elling CE, Frimurer TM, Gerlach LO, Jorgensen R, Holst B, Schwartz TW. Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J. Biol. Chem. 2006;281:17337–17346. doi: 10.1074/jbc.M512510200. [DOI] [PubMed] [Google Scholar]
- 4.Koh JT, Biggins JB. Ligand-receptor engineering and its application towards the complementation of genetic disease and target identification. Curr. Top. Med. Chem. 2005;5:413–420. doi: 10.2174/1568026053828420. [DOI] [PubMed] [Google Scholar]
- 5.Kim J, Wess J, van Rhee AM, Shöneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivkees SA, Lasbury ME, Barbhaiya H. Identification of domains of the human A1 adenosine receptor that are important for binding receptor subtype-selective ligands using chimeric A1/A2a adenosine receptors. J. Biol. Chem. 1995;270:20485–20490. doi: 10.1074/jbc.270.35.20485. [DOI] [PubMed] [Google Scholar]
- 7.Barbhaiya H, McClain R, IJzerman A, Rivkees SA. Site-directed mutagenesis of the human A1 adenosine receptor: Influences of acidic and hydroxy residues in the first four transmembrane domains on ligand binding. Mol. Pharmacol. 1996;50:1635–1642. [PubMed] [Google Scholar]
- 8.Kim SK, Gao Z-G, Van Rompaey P, Gross AS, Chen A, Van Calenbergh S, Jacobson KA. Modeling the adenosine receptors: Comparison of the binding domains of A2A agonists and antagonists. J. Med. Chem. 2003;46:4847–4859. doi: 10.1021/jm0300431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanov AA, Palyulin VA, Zefirov NS. Computer aided analysis of the binding modes of the adenosine receptor agonists for all known subtypes of adenosine receptors. J. Mol. Graphics Modell. 2007;25:740–754. doi: 10.1016/j.jmgm.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Costanzi S, Ivanov AA, Tikhononva IG, Jacobson KA. Structure and function of G protein-coupled receptors studied using sequence analysis, molecular modeling, and receptor engineering: Adenosine receptors. Front. Drug Des. Discovery. 2007 in press. [Google Scholar]
- 11.Jacobson KA, Gao ZG, Liang BT. Neoceptors: Reengineering GPCRs to recognize tailored ligands. Trends Pharmacol. Sci. 2007;28:111–116. doi: 10.1016/j.tips.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson KA, Gao ZG, Chen A, Barak D, Kim SA, Lee K, Link A, Van Rompaey P, Van Calenbergh S, Liang BT. Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J. Med. Chem. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson KA, Ohno M, Duong HT, Kim SK, Tchilibon S, Cesnek M, Holy A, Gao ZG. A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chem. Biol. 2005;12:237–247. doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao ZG, Duong HT, Sonina T, Kim SK, Van Rompaey P, Van Calenbergh S, Mamedova L, Kim HO, Kim MJ, Kim AY, Liang BT, Jeong LS, Jacobson KA. Orthogonal activation of the reengineered A3 adenosine receptor (neoceptor) using tailored nucleoside agonists. J. Med. Chem. 2006;49:2689–2702. doi: 10.1021/jm050968b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discovery. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boison D. Adenosine and epilepsy: From therapeutic rationale to new therapeutic strategies. Neuroscientist. 2005;11:25–36. doi: 10.1177/1073858404269112. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Cerniway RJ, Byford AM, Berr SS, French BA, Matherne GP. Cardiac overexpression of A1-adenosine receptor protects intact mice against myocardial infarction. Am. J. Physiol. 2002;282:H949–H955. doi: 10.1152/ajpheart.00741.2001. [DOI] [PubMed] [Google Scholar]
- 19.Zheng J, Zambraski E, Liang BT. Adenosine A1 receptors mediate potent anti-ischemic skeletal muscle protection in a mouse hindlimb model. J. Mol. Cell. Cardiol. 2005;38:64. [Google Scholar]
- 20.Belardinelli L, Shryock JC, Song Y, Wang D, Srinivas M. Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 1995;9:359–365. doi: 10.1096/fasebj.9.5.7896004. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Conklin D, Pan HL, Eisenach JC. Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J. Pharmacol. Exp. Ther. 2003;305:950–955. doi: 10.1124/jpet.102.047951. [DOI] [PubMed] [Google Scholar]
- 22.Olah ME, Ren H, Ostrowski J, Jacobson KA, Stiles GL. Cloning, expression, and characterization of the unique bovine A1 adenosine receptor. Studies on the ligand binding site by site-directed mutagenesis. J. Biol. Chem. 1992;267:10764–10770. [PMC free article] [PubMed] [Google Scholar]
- 23.Townsend-Nicholson A, Schofield PR. A threonine residue in the seventh transmembrane domain of the human A1 adenosine receptor mediates specific agonist binding. J. Biol. Chem. 1994;269:2373–2376. [PubMed] [Google Scholar]
- 24.Gao Z-G, Jiang Q, Jacobson KA, IJzerman AP. Site-directed mutagenesis studies of human A2A adenosine receptors: Involvement of Glu13 and His278 in ligand binding and sodium modulation. Biochem. Pharmacol. 2000;60:661–668. doi: 10.1016/s0006-2952(00)00357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenakin T. New concepts in drug discovery: Collateral efficacy and permissive antagonism. Nat. Rev. Drug Discovery. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 26.Golisade A, Wiesner J, Herforth C, Jomaa H, Link A. Anti-malarial activity of N6-substituted adenosine derivatives. Part I. Bioorg. Med. Chem. 2002;10:769–777. doi: 10.1016/s0968-0896(01)00331-5. [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Stewart JJ. MOPAC: A semiempirical molecular orbital program. J. Comput.-Aided Mol. Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 30.Sybyl Molecular Modeling System. version 6.9 Tripos Inc.; St. Louis, MO: 2006. [Google Scholar]
- 31.Halgren TA. MMFF VII. Characterization of MMFF94, MMFF94s, and Other Widely Available Force Fields for Conformational Energies and for Intermolecular-Interaction Energies and Geometries. J. Comput. Chem. 1999;20:730–748. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 32.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of the Cartesian Equations of Motion for a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1997;23:327–341. [Google Scholar]
- 33.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 34.Klebe G, Mietzner T. A fast and efficient method to generate biologically relevant conformations. J. Comput.-Aided Mol. Des. 1994;8:583–606. doi: 10.1007/BF00123667. [DOI] [PubMed] [Google Scholar]
- 35.Mohamadi FN, Richards GJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. MacroModel: An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990;11:440–467. [Google Scholar]
- 36.Katritzky AR, Kirichenko N, Rogovoy BV. Synthesis of mono- and N,N-disubstituted ureas. ARKIVOC (Gainesville, FL, U.S.) 2003:8–14. [Google Scholar]
- 37.Jeong LS, Kim MJ, Kim HO, Gao ZG, Kim SK, Jacobson KA, Chun MW. Design and synthesis of 3′-ureidoadenosine-5′-uronamides: Effects of the 3′-ureido group on binding to the A3 adenosine receptor. Bioorg. Med. Chem. Lett. 2004;14:4851–4854. doi: 10.1016/j.bmcl.2004.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G. N6-Alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligands. J. Med. Chem. 2002;45:3271–3279. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- 39.Gao Z-G, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J. Med. Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tchilibon S, Kim SK, Gao ZG, Harris BA, Blaustein JB, Gross AS, Duong HT, Melman N, Jacobson KA. Exploring distal regions of the A3 adenosine receptor binding site: Sterically constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg. Med. Chem. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wildbrandt R, Frotscher U, Freyland M, Messerschmidt W, Richter R, Schulte-Lippern M, Zschaege B. Treatment of glomerulonephritis with metrifudil. Preliminary Report. Med. Klin. (Muenchen, Ger.) 1972;67:1138–1140. [PubMed] [Google Scholar]
- 42.van Troostenburg AR, Clark EV, Carey WD, Warrington SJ, Kerns WD, Cohn I, Silverman MH, Bar-Yehuda S, Fong KL, Fishman P. Tolerability, pharmacokinetics and concentration-dependent hemodynamic effects of oral CF101, an A3 adenosine receptor agonist, in healthy young men. Int. J. Clin. Pharmacol. Ther. 2004;42:534–542. doi: 10.5414/cpp42534. [DOI] [PubMed] [Google Scholar]
- 43.van Schaick E, Jacobson KA, Kim HO, IJzerman AP, Danhof M. Hemodynamic effects and histamine release elicited by the selective adenosine A3 receptor agonist 2-Cl-IB-MECA. Eur. J. Pharmacol. 1996;308:311–314. doi: 10.1016/0014-2999(96)00373-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heitman LH, Mulder-Krieger T, Spanjersberg RF, von Frijtag Drabbe Künzel JK, Dalpiaz A, IJzerman AP. Allosteric modulation, thermodynamics and binding to wild-type and mutant (T277A) adenosine A1 receptors of LUF5831, a novel nonadenosine-like agonist. Br. J. Pharmacol. 2006;147:533–541. doi: 10.1038/sj.bjp.0706655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kourounakis A, Visser C, de Groote M, IJzerman AP. Differential effects of the allosteric enhancer (2-amino-4,5-dimethyl-trienyl)[3-(trifluoromethyl)phenyl]methanone (PD81,-723) on agonist and antagonist binding and function at the human wild-type and a mutant (T277A) adenosine A1 receptor. Biochem. Pharmacol. 2001;61:137–144. doi: 10.1016/s0006-2952(00)00536-0. [DOI] [PubMed] [Google Scholar]
- 46.Gao ZG, Mamedova LK, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: Affinity and efficacy at four subtypes of human adenosine receptors. Biochem. Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body-fluid. Anal. Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 48.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes: Characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 49.Ethier MF, Madison JM. Adenosine A1 receptors mediate mobilization of calcium in human bronchial smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2006;35:496–502. doi: 10.1165/rcmb.2005-0290OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Márián T, Rubovszky B, Szentmiklósi AJ, Trón L, Balkay L, Boros I, Gáspár R, Székely A, Kraszna Z. A1 and A2 adenosine receptor activation inversely modulates potassium currents and membrane potential in DDT1 MF-2 smooth muscle cells. Jpn. J. Pharmacol. 2002;89:366–372. doi: 10.1254/jjp.89.366. [DOI] [PubMed] [Google Scholar]
- 51.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 52.Visiers I, Ballesteros JA, Weinstein H. Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol. 2002;343:329–371. doi: 10.1016/s0076-6879(02)43145-x. [DOI] [PubMed] [Google Scholar]
- 53.Jacobson KA, Ukena D, Kirk KL, Daly JW. [3H]Xanthine amine congener of 1,3-dipropyl-8-phenylxanthine: An antagonist radioligand for adenosine receptors. Proc. Natl. Acad. Sci. U.S.A. 1986;83:4089–4093. doi: 10.1073/pnas.83.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ivanov AA, Costanzi S, Jacobson KA. Defining the nucleotide binding sites of P2Y receptors using rhodopsin-based homology modeling. J. Comput.-Aided Mol. Des. 2007;20:417–426. doi: 10.1007/s10822-006-9054-2. [DOI] [PubMed] [Google Scholar]
- 55.Chugunov AO, Farce A, Chavatte P, Efremov RG. Differences in binding sites of two melatonin receptors help to explain their selectivity to some melatonin analogs: A molecular modeling study. J. Biomol. Struct. Dyn. 2006;24:91–107. doi: 10.1080/07391102.2006.10507103. [DOI] [PubMed] [Google Scholar]
- 56.Shi L, Javitch A. The binding site of aminergic G protein-coupled receptors: The transmembrane segments and second extracellular loop. Annu. Rev. Pharmacol. Toxicol. 2002;42:437–467. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.