

Abstract
Infectious salmon anaemia (ISA) is an important infectious disease in Atlantic salmon farming causing recurrent epidemic outbreaks worldwide. The focus of this paper is on tracing the spread of ISA among Norwegian salmon farms. To trace transmission pathways for the ISA virus (ISAV), we use phylogenetic relationships between virus isolates in combination with space–time data on disease occurrences. The rate of ISA infection of salmon farms is modelled stochastically, where seaway distances between farms and genetic distances between ISAV isolates from infected farms play prominent roles. The model was fitted to data covering all cohorts of farmed salmon and the history of all farms with ISA between 2003 and summer 2009. Both seaway and genetic distances were significantly associated with the rate of ISA infection. The fitted model predicts that the risk of infection from a neighbourhood infectious farm decreases with increasing seaway distance between the two farms. Furthermore, for a given infected farm with a given ISAV genotype, the source of infection is significantly more likely to be ISAV of a small genetic distance than of moderate or large genetic distances. Nearly half of the farms with ISA in the investigated period are predicted to have been infected by an infectious farm in their neighbourhood, whereas the remaining half of the infected farms had unknown sources. For many of the neighbourhood infected farms, it was possible to point out one or a few infectious farms as the most probable sources of infection. This makes it possible to map probable infection pathways.
Keywords: infection rate, infection pathway, genetic distance, space–time data
1. Introduction
Knowledge on key infection pathways for infectious diseases greatly enhances the prospects of disease control as it allows targeted surveillance and intervention [1]. Recently, approaches based on mathematical and statistical models have been successfully used to disentangle infection pathways and risk factors in fish and animal farming. Keeling et al. ([2], foot-and mouth-disease; [3], foot-and-mouth-disease; Höhle ([4], swine fever virus), Scheel et al. ([5], ISA) and Aldrin et al. ([6], ISA and other salmon diseases) all model the spread of diseases between production farms based on similar stochastic models where distances between farms play a prominent role.
A different approach to study the spread of diseases is through phylogeographic analysis of genetic data characterizing pathogens. Phylogeographic methods have, for example, been used to trace the global spread of avian influenza over the last decade [7], as well as fine-scale local transmission pathways for the foot-and-mouth disease virus (FMDV) in the UK [8]. In the FMDV study, epidemiological data relating to the timing of infection and infectiousness of farms were integrated with data on genetic relatedness of pathogens isolated from infected farm livestock, to construct the most probable infection pathways (transmission trees) between farms. One result from this study was that the projected most probable infection pathways were between farms that were located in closer proximity than expected by chance [8]. However, distances between farms were not explicitly a part of the FMDV model.
Infectious salmon anaemia (ISA) is an important disease in Atlantic salmon (Salmo salar L.) farming. Infections induce a systemic and lethal condition characterized by severe anaemia and variable organ necrosis. Morbidity and mortality are variable, but in severe cases, cumulative mortality may exceed 90 per cent during a three month period [9]. ISA has had recurrent epidemic outbreaks worldwide and most salmon-producing countries have been affected [9]. Since 2007, a large-scale epidemic of ISA has unfolded in Chilean salmon farming [10], causing losses in the order USD two billion between 2007 and 2009 [11]. ISA is caused by the ISA virus (ISAV) within the family Orthomyxoviridae [12]. The virus has a segmented genome consisting of eight segments [13]. The haemagglutinin–esterase (HE) is encoded by the HE gene, which consists of a highly polymorphic region (HPR) and more conserved 5′ part [14,15]. The 5′ part of the HE gene has been widely used for phylogenetic studies [16–18].
Aldrin et al. [6] used a model where the rate of infection for a susceptible farm depends on (among other factors) the seaway distances from neighbourhood infectious farms, such that the probability of infection depends on the distance to infectious farms. The model is a stochastic continuous time model with location of the farms being fixed in time and can be seen as a spatial survival or event history model. The aim of the present study is to extend this model by including genetic distances between ISAV genotypes from different infected farms. Hence, both physical distances between farms and phylogenetic relationships between ISAV isolates play prominent roles in the model. Including genetic distance between ISAV isolates in the model introduces a source of information that is independent of physical distance. Combined, these variables enable a more accurate reconstruction of the space–time spread of ISA.
We model the rate by which a susceptible farm is infected by ISAV with genotype g. This infection rate is decomposed into two possible infection pathways: (i) infection from neighbourhood infectious farms and (ii) infection via other non-specified pathways, such as well boats transporting live fish or infected smolts. The rate of infection from neighbourhood farms decreases by increasing seaway distance to an infectious farm and by increasing genetic distance between the genotype g under consideration and the genotype g ′ at the infectious farm. The infection rate may also depend on characteristics of the fish populations at the susceptible and infectious salmon farms.
The model was fitted to data on ISA-infected salmon farms from February 2003 until August 2009 and used to predict the probability of neighbourhood infection versus other pathways for all ISA-infected farms. Furthermore, the most probable infection pathway between farms was estimated in an area where neighbourhood infection predominated according to model predictions.
2. Data
The data used to model the spread of ISA in this paper are an updated version of the data used by Aldrin et al. [6], including the period from February 2003 to August 2009. More importantly, genetic information on ISAV isolates is now included. Details on virus detection, sequencing and compilation of the genetic data are given in Lyngstad et al. [19], whereas all data on salmon farms and salmon cohorts are described in Kristoffersen et al. [20].
The present data include all 1201 Norwegian marine salmon farms with a standing stock of Atlantic salmon in any month from February 2003 to August 2009. For each salmon farm, the location and monthly figures of total biomass and the number of fish on a farm are given. Within the data period, each salmon farm normally had several consecutive periods of production of fish populations, interrupted by periods of fallowing (no fish on farm). The fish population within a production period is termed a cohort and the present data consist of 3202 cohorts, where a farm had between one and eight cohorts (see animation 1 in the electronic supplementary material).
Each cohort was classified into one of four categories; (i) autumn–smolt cohorts, (ii) spring–smolt cohorts, (iii) mixed cohorts, and (iv) relocated cohorts (see [20]). Each category contained between 11 and 37 per cent of the cohorts. For autumn-smolt cohorts and spring-smolt cohorts, the median times from when they were stocked until they were removed were 13 and 17 months, respectively. Mixed cohorts consist of fish stocked several times during the production period, but treated as one cohort in our modelling approach, and stay in median 20 months at a farm. Relocated cohorts consist of fish moved from other farms and stay in median nine months at a farm. It was not possible to trace movement of cohorts in the present data.
As a measure of distance between farms, we calculated seaway distances, i.e. for a given pair of sites, the shortest path over sea water. This is motivated by the fact that ISA is an aquatic disease and that spread by passive diffusion in the water current is supported [21]. Hence, over a coastline like the Norwegian, with an abundance of islands and fjords, seaway distance is a more logical approximation to spread in the water current than the Euclidean distance. Seaway distances between all possible combinations of two farms within 100 km of each other were computed using ArcView extension Spatial Analyst (ESRI, Redlands, CA, USA). Distances of more than 100 km were truncated to 100 km. The geographical coordinates of the farms were downloaded from the aquaculture register of the Directorate of Fisheries (http://www.fiskeridir.no/). Details of the distance calculations are given in Kristoffersen et al. [20]. The various salmon farms had between 0 and 27 other salmon farms within a seaway distance of 10 km (median 5 farms).
Data on clinical diagnoses of ISA in fish cohorts were compiled from the Laboratory information system at the National Veterinary Institute in Norway. The recorded month of an ISA diagnosis corresponds to the month when the first samples of fish were received at the National Veterinary Institute, and that shortly afterwards resulted in a clinical diagnosis of ISA on a given salmon farm. ISA is a notifiable disease in Norway. In the data period there were 72 recorded diagnoses of ISA, whereof 63 in 2004 or later (animation 1 in the electronic supplementary material). For convenience, a farm with a clinical diagnosis of ISA in a fish cohort is termed an ISA-infected farm in the following. Details regarding ISA diagnostics and regulations are given in the electronic supplementary material.
The ISAV HE gene was sequenced from all except six of the ISA-infected farms. Sequencing was performed on samples of kidney tissue and mostly from two individual fish (see below) sampled at the same time, and from each ISA-infected farm. We refer here to a specific sequence as a genotype. The genetic distance between two genotypes was calculated based on the 5′ part of the HE gene. We used the Kimura 2 parameter model for nucleotide substitution [22] to calculate the genetic distances. The genetic distance between two sequences takes non-negative integer values 0, 1, 2, … , 1782 according to the Kimura 2 model. When more than one genotype was found in an infected farm, sequences with the least genetic distance between farms were used. Details regarding the ISAV HE gene sequences are given in the electronic supplementary material.
ISAV isolates are assumed to be related if the genetic distance between them is small. Six per cent of the pairs of ISAV isolates from different infected farms had genetic distance equal to 0; 6 per cent equal to 1; 7 per cent equal to 2; and the remaining 81 per cent of pairs had distances between 3 and 384.
3. Methods
The stochastic infectious disease model in this paper is inspired by similar models on FMDV (e.g. [3]). Furthermore, it is a direct extension of the model of Aldrin et al. [6] on ISA and two other fish diseases, also taking into account the genetic distance between ISAV isolated from different infected farms. However, the model of Aldrin et al. [6] included two terms that have been omitted from the main body of the present paper, since their effect on the spread of ISA was estimated to be exactly zero in the preliminary analyses of the data (see the electronic supplementary material).
3.1. The infection rate model
Our statistical model simultaneously includes all salmon farms in Norway. The core of the model is the rate by which a salmon farm is infected by a virus of a given genotype; cf. equation (3.1) below.
The time of an ISA diagnosis is recorded, but the actual time when a salmon farm becomes infected (the infection event) is unknown. There is always a time delay between the infection event and the recording of ISA on a farm. Here, we assume that this time delay is fixed and equal to six months for all infected farms. If this resulted in an infection time prior to the stocking of the cohort, the time of infection was set to the time of stocking. See Scheel et al. [5] for a discussion of the length of the time delay. In addition, we investigate the sensitivity of our results to the choice of time delay by refitting the model using a time delay of one, two, three or nine months.
Further, we assume that a fish cohort on a salmon farm is susceptible to infection from the time the cohort is stocked until it (and the salmon farm) becomes infected or it is removed from the salmon farm. When a cohort (and a salmon farm) becomes infected, it is assumed to be infectious from immediately after the infection event and until the cohort is removed (see fig. 1 in [6]).
Figure 1.

Most probable infection pathways in the cluster of ISA-infected farms in Troms County after 2007. Each infected farm is denoted by a pie chart that represents probabilities of infection attributed to neighbourhood farms (red colour) or to other sources (blue colour), (a) when genetic information was accounted for and (b) when genetic information was ignored. The arrows indicate the most probable infection pathway for each infected farm, and the colours represent the probability of each specific pathway.
Let λgi(t) denote the rate by which salmon farm i is infected by a virus of genotype g at time t. Then, λgi(t) dt is approximately the probability that the salmon farm will be infected by a virus of genotype g in the small time interval from t to t + dt. We assume that the rate is of the following additive-multiplicative form
| 3.1 |
The three multiplicative terms in equation (3.1) are:
— Si (t) is an at-risk indicator that is 1 if salmon farm i is susceptible at time t and 0 otherwise.
— λb (t) is a time-varying rate of infection, common for all salmon farms and independent of space, called the baseline rate. This term is left unspecified, and cancels out in the partial likelihood used for estimation; see §3.2.
— λix (t) is a factor proportional to the susceptibility of salmon farm i, and functionally related to explanatory variables x that characterizes the fish cohort at fish farm i at time t; see §3.1.3.
We assume that conditioned on the history up to time t, disease transmission through a given pathway may occur independently of other pathways. As the infection rates or intensities are small probabilities, the contributions from the different pathways may then be added to a total infection rate. The two additive terms in equation (3.1) represent two possible transmission pathways:
— λgid(t) describes the relative rate of infection at time t from infectious salmon farms in the neighbourhood, and it is related to the seaway distance to infectious farms and the genetic distance between the given genotype g and the genotypes of the viruses isolated from the infectious farms.
— λgio(t) describes the relative rate of infection at time t via other (non-specified) pathways.
The infection pathways and expressions for susceptibility and infectiousness are presented in more detail in the following sections.
3.1.1. Distance
The component representing the relative rate of infection from infectious salmon farms in the neighbourhood can be decomposed into a sum of contributions from each salmon farm j that may infect salmon farm i with a virus of genotype g, denoted by λgijd(t). Specifically, the total relative rate attributed to distance by which salmon farm i is infected by a virus of genotype g takes the form λgid(t) = ∑j≠i λgijd(t). Here, the contribution from salmon farm j is modelled as
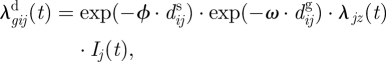 |
3.2 |
where
— dijs is the seaway distance between salmon farms i and j.
— ϕ is a parameter that expresses the effect of the seaway distance on the risk of infection.
— dijg is the genetic distance between genotype g and the genotype g′ of the virus isolated from the infectious salmon farm j. Note that dijg depends on the genotypes g and g′, but for convenience this is not given explicit in the notation.
— ω is a parameter that expresses the effect of the genetic distance on the risk of infection.
— λjz(t) is a factor proportional to the infectiousness of salmon farm j, and functionally related to explanatory variable z that characterizes the fish cohort at salmon farm j at time t. The variable z may, in principle, differ from the variable x used in the susceptibility factor λjx(t), but some variables may also be shared; see §3.1.3.
— Ij(t) is an indicator variable that is 1 if salmon farm j is infectious at time t, and 0 otherwise.
Note that positive values of ϕ and ω mean that there is a reduced risk of infection when seaway distance or genetic distance increases.
3.1.2. Other infection pathways
ISA infection via other possible pathways accounts for other sources of infection, such as well boats, infected smolts or a potential reservoir of non-virulent ISAV that may mutate to virulent ISAV [23]. It can also include infection from other infectious fish farms, but where the infection remained undetected until slaughtering of the cohort. In the present application, the relative rate attributed to infection via other possible pathways is assumed to be constant in time and space, i.e.
| 3.3 |
where the parameter θ expresses the effect of other infection pathways.
3.1.3. Susceptibility and infectiousness
The susceptibility factor λix(t) of equation (3.1) and the infectiousness factor λjz(t) of equation (3.2) can, in principle, vary continuously over time for each salmon farm, for instance, as a function of sea temperature. However, in our application, both factors are assumed to be constant for a fish cohort.
The susceptibility factor λix(t) depends on the category and the size of the cohort, and it takes the form:
| 3.4 |
Here xa(t), xm(t) and xr(t) are indicator variables that are 1 if the cohort is an autumn, a mixed or a relocated cohort, respectively, while all these variables are 0 if the cohort is a spring cohort. Thus, the corresponding β-parameters measure the susceptibility of each type of cohort relative to a spring cohort. Furthermore, xin(t) is the maximum number of fish in cohort i, measured in millions, during the production period. The coefficient βn measures the effect of the number of fish on the cohort susceptibility.
The infectiousness factor λjz(t) depends only on the size of the fish cohort at salmon farm j at time t, and it is given as
| 3.5 |
Here zjn(t) = xjn(t) is the maximum number of fish in cohort j during the production period, which also is included in the susceptibility factor λjz(t), and the coefficient αn expresses the effect of the number of fish on the infectiousness.
3.1.4. Full model
Using equations (3.2)–(3.5), the model in equation (3.1) can now be written as
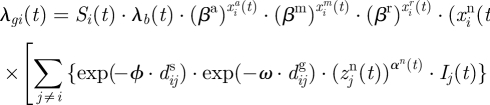 |
3.6 |
3.1.5. Missing genotypes
The genotype is unknown for a few infected farms, with corresponding unknown genetic distances. In these cases, the unknown genetic distances dijg in equation (3.6) are replaced by a parameter δ, which is estimated from the data. Thus, the missing genetic distances are imputed by a common value that gives the best fit to the data. The interpretation of δ is not of particular interest in itself. A short discussion of an alternative strategy for dealing with the missing genotypes is given in the electronic supplementary material.
3.2. Parameter estimation
The unknown parameters ϕ, ω, δ and θ, the four β's and αn are estimated by maximum partial likelihood [3,24,25]. The first four of these are constrained to be non-negative, because negative values of these are meaningless (e.g. with the consequence that the risk of infection could increase by increasing the distance to an infectious farm). The partial likelihood consists of a product of conditional probabilities.
We first consider the situation where ISAV isolated from an infected farm at time t is known to have genotype g. For this situation, we consider the conditional probability that salmon farm i becomes infected by a virus of genotype g at time t, given that one farm is infected by a virus of genotype g at that time. This conditional probability is given by
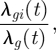 |
3.7 |
where λg(t) = ∑k∈All λgk(t) is the sum over all susceptible salmon farms at time t. Here ‘All’ denotes the list of all salmon farms but remember that, by equation (3.1), λgk(t) = 0 for a salmon farm k that is not susceptible at time t.
Next, we consider the situation where ISAV from an infected farm at time t is of an unknown genotype. Since there are only a finite number of possible genotypes (§2), the rate of infection with ISAV of any genotype is the genotype-specific infection rates summed over all possible genotypes:
| 3.8 |
Here, we consider the conditional probability that salmon farm i becomes infected (by any genotype) at time t, given that one farm gets infected at that time. This conditional probability takes the form:
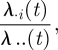 |
3.9 |
where λ .. (t) = ∑k∈All λ·k (t).
The partial likelihood consists of a product of the conditional probabilities given by equations (3.7) and (3.9). Its derivation and exact expression are found in the electronic supplementary material together with some other details regarding estimation.
3.3. Tracing infection routes
For an infected salmon farm i with ISAV genotype g and infection time t, the (model-based) probability that the source of the infection is a specific neighbourhood salmon farm j is given by
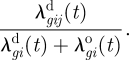 |
3.10 |
Further, the probability that salmon farm i is infected by any of the neighbourhood salmon farms is
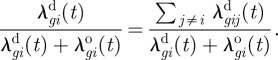 |
3.11 |
Similar expressions can be used for infected farms with unknown genotypes. Thus, for some of the infected farms it may be possible to point out one or a few neighbourhood farms as the probable source of infection. This may be valuable information for understanding the infection process. Taking the average over all infected farms in equation (3.11), we obtain a measure of the overall relative importance of transmission from neighbourhood salmon farms.
4. Results
We estimated the infection rate model using all infected farms recorded in the period 2004 to August 2009 as events in the partial likelihood. There were 63 ISA-infected farms in this period, whereof four had unknown genotypes of ISAV. The infected farms recorded in 2003 were used to initialize the statistical model for those recorded from January 2004 and onwards. The time delay was assumed to be six months. A change from six to one, two, three or nine months gave similar results (see the electronic supplementary material). With a time delay of six months, 10 of the 63 infection times were set equal to the stocking times. This is a consequence of our algorithm and does not necessarily mean that these fish cohorts were infected when they were stocked. However, eight of these 10 were relocated cohorts that had been in the sea at another farm, indicating that they may have been infected before they were re-stocked to a new salmon farm.
Table 1 shows the estimated parameter values with 95% confidence intervals (CI). The ϕ and ω parameters, measuring the effect of the seaway and genetic distances, respectively, were both positive and significant (the CI is well above 0). The estimated value of ϕ was 0.095, implying that the risk of infection from a neighbourhood farm decreases by about 60 per cent (exp(−0.095 · 10) approx. 0.4)) when the seaway distance increases from 0 to 10 km. The estimated value of ω was 1.42, i.e. for a given farm, the probability of infection from a specific source farm decreases by about 75 per cent (exp(−1.42 · 1) approx. 0.25)) when the genetic distance increases from 0 to 1.
Table 1.
Estimated parameters with 95% confidence intervals for the model of equation (3.6) for the rate of ISA infection of salmon farms. From left to right: (i) full model estimated on data from February 2003 to August 2009, (ii) gene effect ω set to 0, estimated on data from February 2003 to August 2009, and (iii) gene effect ω set to 0, estimated on data from February 2003 to 2007 as presented in Aldrin et al. [6]. The time delay is assumed to be six months. Gen. dist., genetic distance; CI, confidence interval; est., estimate; susc., susceptible; inf.; infectious.
| parameters |
full model February 2003–August 2009 95% CI |
ω = 0 February 2003–August 2009 95% CI |
ω = 0 February 2003–2007 95% CI |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| effect of | symbol | est. | lower | upper | est. | lower | upper | est. | lower | upper |
| seaway distance | ϕ | 0.095 | 0.046 | 0.145 | 0.095 | 0.041 | 0.149 | 0.42 | 0.22 | 0.62 |
| genetic distance | ω | 1.42 | 0.40 | 2.45 | — | — | — | — | — | — |
| imputed gen. dist. | δ | 2.88 | 0 | 5.80 | — | — | — | — | — | — |
| other | θ | 0.001 | 0 | 0.002 | 0.018 | 0.002 | 0.034 | 0.003 | 0.001 | 0.006 |
| autumn cohort | βa | 0.44 | 0.17 | 1.14 | 0.35 | 0.14 | 0.90 | 0.24 | 0.05 | 1.14 |
| mixed cohort | βm | 1.12 | 0.52 | 2.39 | 0.81 | 0.39 | 1.66 | 0.80 | 0.35 | 1.85 |
| relocated cohort | βr | 1.30 | 0.65 | 2.62 | 0.90 | 0.47 | 1.75 | 0.64 | 0.27 | 1.53 |
| susc. cohort size | βn | 0.57 | 0.11 | 1.02 | 0.70 | 0.25 | 1.15 | 0.62 | 0.08 | 1.15 |
| inf. cohort size | αn | 2.71 | −0.13 | 5.55 | 1.89 | 0.71 | 3.06 | 2.00 | 0.52 | 3.47 |
| proportion of infection from neighbourhood farms | 43 | 40 | 64 | 47 | 43 | 62 | 22 | 21 | 37 | |
There were no significant effects of the stocking categories of the fish cohorts, because none of the parameters βa, βm or βr were significantly different from 1, which is the reference value for the spring cohort.
Susceptibility (βn) increased significantly by increasing cohort size, whereas infectiousness (αn) was marginally non-significant (p-value 0.06). However, when refitting the model with the stocking category parameters set to 1, the estimate of the infectiousness was almost unchanged, but became slightly significant (p-value 0.04). The refitting had little effect on the estimates of other parameters (not shown).
A direct interpretation of the estimated value of the parameter θ related to infection from other sources is difficult, since it must be seen in relation to the values of ϕ and ω. It is reasonable that θ decreases when genetic distances are included, since the term exp(−ω . dijg) is less than 1 for all genetic distances greater or equal to 1. Hence, it follows that θ has to be smaller to give the same proportion of infection attributed to other sources. It is more useful to study the relative importance of infection from neighbourhood farms as the average of the expression in equation (3.11) over all infected farms. This average implies that the source of infection is attributed to neighbourhood infectious farms for about 43 per cent of the infected farms.
Table 1 also includes parameter estimates of the model in Aldrin et al. [6], which is equal to the model presented here without the gene effect, i.e. setting the parameter ω to 0, for the same data period. Including or excluding the gene effect has little influence on the other parameter estimates, except for θ. The CI for the seaway distance parameter ϕ is slightly tighter when the gene effect is included. However, this does not mean that including the gene distance in the model is unimportant; it is shown below that the genetic information is indeed useful for predictions of the historic pattern of the spread of ISA.
Finally, included in table 1 are the estimates from Aldrin et al. [6] based on data only up to December 2007. The estimate of ϕ was then much higher. This can be explained by a larger uncertainty because fewer infected farms were included. The estimated relative importance of infection originating from neighbourhood farms is considerably higher in the period 2004–2009 than in 2004–2007.
The infection rate model can be used to trace probable infection pathways between neighbourhood farms. Most infected farms were isolated in time and space, but there were also small space–time clusters of infected farms. In addition, since June 2007, there has been a series of diagnosed infections in Troms County (see animation 2 in the electronic supplementary material). The left part of figure 1 shows the most probable infection pathway for each infected farm in this epidemic cluster, for the full model with genetic information. The figure shows that the epidemic started at one farm (in blue), with ISA recorded in June 2007, and was further spread to 16 other farms within 2 years. The ISAV isolates from 15 of the infected farms had identical genotypes (genetic distance between isolates equal to 0). The ISAV isolates from the remaining two infected farms had a genetic distance of 1 to the isolates from the other infected farms and a distance of 2 between them.
For a comparison, the right part of figure 1 shows the most probably infection pathways estimated from the reduced model ignoring the genetic information. The pattern of spread was then more diffuse. Some extra possible infection routes appeared. These had probabilities approximately equal to 0 in the full model owing to large genetic distances. Furthermore, specific infection pathways with probabilities close to 1 in the full model tended to have lower probabilities in the reduced model.
5. Discussion
5.1. Infectious salmon anaemia virus transmission and implications for disease control
In this paper, we present a stochastic spatio-temporal model for the rate of ISA infection among salmon farms, taking into account seaway distances between farms, genetic distances between ISAV isolates from different infected farms and other non-specified infection pathways. The model quantifies the importance of seaway distances between farms and suggests that 43 per cent of the ISA infection events between January 2004 and August 2009 were due to infection from neighbourhood salmon farms. We also demonstrate how the model can be used to trace probable routes of infection between farms in an area where farm to farm infection predominates according to model predictions.
The most informative estimate from the present model is arguably the proportion of infected farms that are attributed to infection from neighbourhood farms. This estimate changed only marginally (from 47% to 43%) between the reduced model where the genetic distance effect was set to 0 and the full model incorporating genetic information. From this, it could be argued that the simpler model ignoring genetic information should be preferred since the inclusion of genetic distances did not contribute on the whole to explain the spread of ISA. However, when considering model estimates for individual ISA outbreaks, the inclusion of genetic information clearly increased the accuracy of the model. The distribution of probability estimates for infection from a neighbourhood farm was shifted towards 0 and 1 for individual infected farms in the full model (figure 2). Some infected farms shift from likely neighbourhood infection to probabilities of nearly 1 for other origins, owing to relatively large genetic distances between virus isolates from proximate infectious farms. These events were probably falsely connected to infection from neighbourhood farms in the model where the genetic distance effect was set to 0. There were also farms for which the probability of infection from a neighbourhood farm increased in the full model. Some of these farms were typically located relatively distant to farms that were probable sources of infection. However, by introducing genetic distance into the relative infection rate, for these farms, the infection rate attributed to seaway distance increased compared with the infection rate from other sources. Hence, we argue that for predicting the source of infection at individual farms, model accuracy is increased with the inclusion of genetic information.
Figure 2.

Salmon farms in Norway with ISA during 2004 to August 2009. Each infected farm is denoted by a pie chart that represents probabilities of infection attributed to neighbourhood farms (red colour) or to other sources (blue colour), (a) when genetic information was accounted for and (b) when genetic information was ignored. Note that locations of farms are displaced in areas with dense clusters of infected farms to avoid overlapping symbols. The histograms show the probability distributions of being infected by neighbourhood farms.
Previous modelling has shown that many of the ISA-infected farms in Norway appear isolated in space and time [5,6]. However, the estimated relative importance of infection from neighbourhood farms was considerably increased in the present prolonged study period. This increase is due to the many infected farms occurring in a small area in the Troms County after 2007. In the core of this area, there was a high degree of genetic similarity between ISAV isolates implying estimates of close to 100 per cent certainty of neighbourhood infection for 16 ISA-infected farms in the area. This accounts for a large amount of the total proportion of infection by neighbourhood farms in the model.
The model was used to calculate the probability that the source of infection for a given infected farm had been a specific neighbourhood farm in the Troms County area (figure 1), when accounting for genetic distance versus not accounting for genetic distance. The two most distant infection pathways in figure 1 appear only in the right panel representing the model ignoring genetic distance effects. Owing to large genetic distances, the estimated probabilities for these pathways were close to 0 (less than 0.005) in the full model. Apart from this difference, the two models predict practically the same pattern of most probable infection pathways between farms. However, a noteworthy difference in the Troms County area is that the probabilities of the most probable infection pathways tend to increase in the full model. This is due to increasing relative infection rates for many of the given infection pathways where virus isolates were genetically similar. From this, we argue that the genetic information increases the accuracy of predictions of the historic pattern of the spread of ISA.
The estimated probabilities of the source of infection being a specific neighbourhood salmon farm are interesting for evaluating the distances over which such transmission may occur. Exemplifying this in the full model are two infection pathways in the Troms County area with seaway distances of more than 20 km and probability predictions of more than 0.9 for such transmission to have occurred. A further example is the southern most infected farm in figure 2. This farm was estimated to have been infected by a farm located about 30 km to the north, with a probability of 0.87. Distances of up to 30 km, however, exceed expected distances over which the disease agent may be infective by passive diffusion [26,27]. One possible explanation is that the larger range transmission events predicted by our model are due to anthropogenic activity resulting in contagious contacts. Through a programme where all ISA-infected farms are followed up by collecting epidemiological data, we have checked for possible contagious contacts between the three pairs of farms where interfarm transmission was predicted over seaway distances extending 20 km. None of these pairs of arms shared ownership or any common source of salmon smolts, but two pairs of arms reported to have shared both the services of a common well boat used for fish transportation and the services of a common team of divers. This emphasizes the importance of high biosecurity standards in all aspects of salmon production and that there are many potential transmission processes for ISAV that could be adsorbed in to these away distance effects in our model by being facilitated by short distance. Epidemiological investigations identified numerous potential infectious contacts between an ISA-affected zone and neighbouring areas of Shetland [28]. Although these did not, in this case, result in spread of infection, ISA risk factors existed there at the scale of tens of kilometres. Even so, an important point to make here is that the risk involved in neighbourhood transmission extends well beyond 5 and 10 km. Distances of 5 and 10 km correspond to the radial extension of restriction zones and observationzones, respectively, that are enforced by the Norwegian Food Safety Authority around ISA-infected farms [29].
Cottam et al. [8] constructed the most probable transmission tree covering the complete history of known farm FMDV infections in the Durham area, UK, in the spring of 2001. This was accomplished by integrating the timing of farm infection events and infectiousness with information from genetic data. There are both similarities and differences in this approach that are interesting to compare with the present approach. An important difference relates to the nature of the pathogens. ISAV, as opposed to FMDV, exists in both low- and high-virulent variants [23]. The prevalence of low virulent ISAV is not known and their possible role as the source of ISA infection for isolated farms is not fully established [19]. Nevertheless, it is informative to also model the part of the ISA disease history for which there is no coherent explanation at present.
Timing of infection events is central both in the FMDV model and in the present ISAV model. Such timing narrows down the number of possible infectious contacts in both models. Owing to more detailed knowledge on the progression of FMDV infection in farm livestock compared with ISAV infection in salmon cohorts, the FMDV model is more finely tuned with respect to timing of events. It incorporates temporal likelihood profiles of infection events as well as farm infectiousness [8]. By comparison, infection events are fixed in time and a salmon cohort is considered infectious from the infection event and until the cohort is removed in the ISAV model. In this aspect, there is room for improvement of the present ISAV model. However, an important addition in the ISAV model, compared with the FMDV model, is that it incorporates the effect of seaway distance to infectious farms. This is a central part of the ISAV model since there is reason to believe that adjacent farms are connected with respect to transmission of fish pathogens through water contact [21,30].
In conclusion, nearly half of the ISA-infected farms in the period from January 2003 to August 2009 are suggested to have been infected by an infectious farm in their neighbourhood, whereas the remaining half were attributed to other, non-specified, sources. The model also suggests that the risk involved in neighbourhood transmission of ISAV extends well beyond the extension of control zones enforced to prevent the spread of ISA. For farms that are predicted to have been infected from neighbourhood farms, it is possible to point out one or a few specific farms as the most probable sources of infection. Genetic sequence information from ISAV isolates contributes to gained insight into the main pathways for the spread of ISA, with implications for the design of targeted control measures against the disease. A key measure to prevent the local spread of ISA is early removal of infectious fish [10,28]. The present study suggests that such a control measure could be more efficient by intensifying ISAV surveillance in fish farms surrounding infected farms, and where the surroundings are extended to 20–30 km surveillance zones.
5.2. Model criticism and future developments
In about half of the infected fish cohorts, the genotypes of the ISA viruses were not the same for the viruses sequenced from two different fish. This may be due to errors in genotyping, mutations that occurred after a farm was infected, or that a farm has been infected more than once. The last possibility is not covered by the spatio-temporal model in equation (3.1), which only considers the occurrence of a single infection for each fish cohort. Our solution to the problem has been to use the least genetic distance between infected farms when fitting the model. A motivation for this choice was given in §2. Our choice is further supported by the finding that for the epidemic cluster in Troms County, a single genotype was found in 15 of the 17 infected cohorts (with the genotypes of the remaining two farms only having a genetic distance 1 from this genotype), while the other genotype varied from cohort to cohort. Nevertheless, we acknowledge that further methodological work is needed to obtain a complete satisfactory handling of cohorts with more than one genotype of the ISAV.
Other aspects of the present spatio-temporal model should be investigated further in future research. One obvious improvement would be to replace the seaway distances between salmon farms by a distance measure that takes into account information on the local sea current conditions, implying that the distance between two farms could depend on the directions (i.e. dij ≠ dji). For the moment, such information is only available for limited areas of the Norwegian coast, but will probably be available for whole of Norway within a few years. The importance of hydrodynamics has been demonstrated by Gustafson et al. [21] for ISA and by Viljugrein et al. [30] for pancreas disease.
Other parametric forms of the model could also be investigated. For instance, one could include an interaction term between the seaway distance and the genetic distance, or maybe more important, one could use other functional forms than the exponential for the decay of seaway and genetic distance. The parameter θ, representing infection via other non-specified infection pathways, is currently independent of genotype. It could be made dependent of genotypes, for instance by modelling θ as an increasing function of the number of ISA diagnoses of each genotype during the last 3 years (say) in the whole of Norway. Furthermore, in our model the infectiousness is now assumed to be constant from the infection event until the infected cohort is removed. It could instead be modelled as a function that varies over time since infection. Systematic screening for ISAV on at least a sub-sample of farms may give valuable information on how infectiousness may change over time.
Our model assumes a fixed time delay from infection to the recording of ISA on a farm, and this time delay is common for all infected farms. This assumption is un-realistic, but has been adopted to simplify the model fitting. In the future, we wish to reformulate the present model as Bayesian hierarchical model, with a latent, un-observed infection process and an additional disease outbreak process modelling the time from infection to a recorded diagnosis of ISA. Such a model would allow the time delay to be stochastic with a distribution that can be estimated from the data. It would then also be possible to take into account infection episodes that never lead to a diagnosis of ISA, but where the infected farms are still infectious until the infected cohort is removed. Finally, the model can be extended to allow for fish cohorts being infected when stocked into the marine environment with a probability that can be estimated from the data.
Acknowledgements
This work was funded by The Fishery and Aquaculture Industry Research Fund (project no. 199734) and by the Research Council of Norway projects ‘Tracing viral disease dissemination in aquaculture: an interdisciplinary approach between molecular virology and dispersal modelling’ (185262/S40), and ‘Modern Application driven Statistical Challenges’ (186951/I30). Monika Hjortaas, Elin Trettenes and Kaia Haugbro are acknowledged for the expert work in genotyping and sequencing ISA virus isolates at the National Veterinary Institute.
References
- 1.Archie E. A., Luikart G., Ezenwa V. O. 2009. Infecting epidemiology with genetics: a new frontier in disease ecology. Trends Ecol. Evol. 24, 21–30 10.1016/j.tree.2008.08.008 (doi:10.1016/j.tree.2008.08.008) [DOI] [PubMed] [Google Scholar]
- 2.Keeling M. J., et al. 2001. Dynamics of the 2001 UK foot and mouth epidemic: stochastic dispersal in a heterogeneous landscape. Science 294, 813–817 10.1126/science.1065973 (doi:10.1126/science.1065973) [DOI] [PubMed] [Google Scholar]
- 3.Diggle P. J. 2006. Spatio-temporal point processes, partial likelihood, foot and mouth disease. Stat. Methods Med. Res. 15, 325–336 10.1191/0962280206sm454oa (doi:10.1191/0962280206sm454oa) [DOI] [PubMed] [Google Scholar]
- 4.Höhle M. 2009. Additive-multiplicative regression models for spatio-temporal epidemics. Biomet. J. 51, 961–978 10.1002/bimj.200900050 (doi:10.1002/bimj.200900050) [DOI] [PubMed] [Google Scholar]
- 5.Scheel I., Aldrin M., Frigessi A., Jansen P. A. 2007. A stochastic model for infectious salmon anemia (ISA) in Atlantic salmon farming. J. R. Soc. Interface 4, 699–706 10.1098/rsif.2007.0217 (doi:10.1098/rsif.2007.0217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aldrin M., Storvik B., Frigessi A., Viljugrein H., Jansen P. A. 2010. A stochastic model for the assessment of the transmission pathways of heart and skeleton muscle inflammation, pancreas disease and infectious salmon anaemia in marine fish farms in Norway. Prev. Vet. Med. 93, 51–61 10.1016/j.prevetmed.2009.09.010 (doi:10.1016/j.prevetmed.2009.09.010) [DOI] [PubMed] [Google Scholar]
- 7.Wallace R. G., Hodac H., Lathrop R. H., Fitch W. M. 2007. A statistical phylogeography of influenza A H5N1. Proc. Natl Acad. Sci. USA 104, 4473–4478 10.1073/pnas.0700435104 (doi:10.1073/pnas.0700435104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cottam E. M., Thebaud G., Wadsworth J., Gloster J., Mansley L., Paton D. J., King D. P., Haydon D. T. 2008. Integrating genetic and epidemiological data to determine transmission pathways of foot-and-mouth disease virus. Proc. R. Soc. B 275, 887–895 10.1098/rspb.2007.1442 (doi:10.1098/rspb.2007.1442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.OIE 2010. Infectious salmon anaemia. In Manual of diagnostic tests for aquatic animals 2010, pp. 222–235 Paris, Italy: World Organization for Animal Health; See http://www.oie.int/eng/normes/fmanual/a_summry.htm. [Google Scholar]
- 10.Mardones F. O., Perez A. M., Carpenter T. E. 2009. Epidemiologic investigation of the re-emergence of infectious salmon anemia virus in Chile. Dis. Aquat. Org. 84, 105–114 10.3354/dao02040 (doi:10.3354/dao02040) [DOI] [PubMed] [Google Scholar]
- 11.FIS. 2010 (Fish Information & Services). Salmon industry lost USD 2,000 million due to ISA virus. See http://www.fis.com/fis/worldnews/worldnews.asp?l=e&id=39160&ndb=1 . [Google Scholar]
- 12.Kawaoka Y., et al. 2005. Infectious salmon anaemia virus. In Virus taxonomy—eight report of the international committee on taxonomy viruses (eds Fauquet C. M., Mayo M. A., Maniloff J., Desselberger U., Ball L. A.), pp. 681–693, New York, NY: Elsevier Academic Press [Google Scholar]
- 13.Mjaaland S., Rimstad E., Falk K., Dannevig B. H. 1997. Genomic characterization of the virus causing infectious salmon anemia in Atlantic salmon (Salmo salar L.): an orthomyxo-like virus in a teleost. J. Virol. 71, 7681–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krossøy B., et al. 2001. Cloning and identification of the infectious salmon anaemia virus haemagglutinin. J. Gen. Virol. 82, 1757–1765 [DOI] [PubMed] [Google Scholar]
- 15.Mjaaland S., Hungnes O., Teig A., Dannevig B. H., Thorud K., Rimstad E. 2002. Polymorphism in the infectious salmon anemia virus hemagglutinin gene: importance and possible implications for evolution and ecology of infectious salmon anemia disease. Virology 304, 379–391 10.1006/viro.2002.1658 (doi:10.1006/viro.2002.1658) [DOI] [PubMed] [Google Scholar]
- 16.Devold M., Falk K., Dale O. B., Krossøy B., Biering E., Aspehaug V., Nilsen F., Nylund A. 2001. Strain variation, based on the hemagglutinin gene, in Norwegian ISA virus isolates collected from 1987 to 2001: indications of recombination. Dis. Aquat. Org. 47, 119–128 10.3354/dao047119 (doi:10.3354/dao047119) [DOI] [PubMed] [Google Scholar]
- 17.Nylund A., Devold M., Plarre H., Isdal E., Aarseth M. 2003. Emergence and maintenance of infectious salmon anaemia virus (ISAV) in Europe: a new hypothesis. Dis. Aquat. Org. 56, 11–24 10.3354/dao056011 (doi:10.3354/dao056011) [DOI] [PubMed] [Google Scholar]
- 18.Kibenge F. S., et al. 2009. Infectious salmon anaemia virus (ISAV) isolated from the ISA disease outbreaks in Chile diverged from ISAV isolates from Norway around 1996 and was disseminated around 2005, based on surface glycoprotein gene sequences. Virol. J. 6, 88. 10.1186/1743-422X-6-88 (doi:10.1186/1743-422X-6-88) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyngstad T. M., Hjortaas M. J., Kristoffersen A. B., Markussen T., Karlsen E. T., Jonassen C. M., Jansen P. A. 2011. Use of molecular epidemiology to trace transmission pathways for infectious salmon anaemia virus (ISAV) in Norwegian salmon farming. Epidemics 3, 1–11 10.1016/j.epidem.2010.11.001 (doi:10.1016/j.epidem.2010.11.001) [DOI] [PubMed] [Google Scholar]
- 20.Kristoffersen A. B., Viljugrein H., Kongtorp R. T., Brun E., Jansen P. A. 2009. Risk factors for pancreas disease (PD) outbreaks in farmed Atlantic salmon and rainbow trout in Norway during 2003–2007 Prev. Vet. Med. 90, 127–136 10.1016/j.prevetmed.2009.04.003 (doi:10.1016/j.prevetmed.2009.04.003) [DOI] [PubMed] [Google Scholar]
- 21.Gustafson L. L., Ellis S. K., Beattie M. J., Chang B. D., Dickey D. A., Robinson T. L., Marenghi F. P., Moffett P. J., Page F. H. 2007. Hydrographics and the timing of infectious salmon anemia outbreaks among Atlantic salmon (Salmo salar L.) farms in the Quoddy region of Maine, USA and New Brunswick, Canada. Prev. Vet. Med. 78, 35–56 10.1016/j.prevetmed.2006.09.006 (doi:10.1016/j.prevetmed.2006.09.006) [DOI] [PubMed] [Google Scholar]
- 22.Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 10.1007/BF01731581 (doi:10.1007/BF01731581) [DOI] [PubMed] [Google Scholar]
- 23.Markussen T., Jonassen C. M., Numanovic S., Braaen S., Hjortaas M., Nilsen H., Mjaaland S. 2008. Evolutionary mechanisms involved in the virulence of infectious salmon anaemia virus (ISAV), a piscine orthomyxovirus. Virology 374, 515–527 10.1016/j.virol.2008.01.019 (doi:10.1016/j.virol.2008.01.019) [DOI] [PubMed] [Google Scholar]
- 24.Cox D. R. 1975. Partial likelihood. Biometrika 62, 269–276 10.1093/biomet/62.2.269 (doi:10.1093/biomet/62.2.269) [DOI] [Google Scholar]
- 25.Aalen O. O., Borgan Ø., Gjessing H. K. 2008. Survival and event history analysis—a process point of view. New York, NY: Springer; 10.1007/978-0-387-68560-1 (doi:10.1007/978-0-387-68560-1) [DOI] [Google Scholar]
- 26.Jarp J., Karlsen E. 1997. Infectious salmon anaemia (ISA) risk factors in sea-cultured Atlantic salmon Salmo salar. Dis. Aquat. Org. 28, 79–86 10.3354/dao028079 (doi:10.3354/dao028079) [DOI] [Google Scholar]
- 27.Murray A. G., Amundrud T. L., Gillibrand P. A. 2005. Models of hydrodynamic pathogen dispersal affecting Scottish salmon production: Modelling shows how Scotland eradicated ISA, but not IPN. Bull. Aquat. Assoc. Can. 105, 80–87 [Google Scholar]
- 28.Murray A. G., Munro L. A., Wallace I. S., Berx B., Pendrey D., Fraser D., Raynard R. S. 2010. Epidemiological investigation into the re-emergence and control of an outbreak of infectious salmon anaemia in the Shetland Islands, Scotland. Dis. Aquat. Org. 91, 189–200 10.3354/dao02262 (doi:10.3354/dao02262) [DOI] [PubMed] [Google Scholar]
- 29.Norwegian Food Safety Authority 2007. Contingency plan for control of ISA in Norway. See http://www.mattilsynet.no/smittevern_og_bekjempelse/fisk/ila/regelverk_bekjempelsesdokumenter/bekjempelsesplan_for_infeksi_s_lakseanemi__ila___20149 accessed 25 Nov 2010. [Google Scholar]
- 30.Viljugrein H., Staalstrom A., Molvaer J., Urke H. A., Jansen P. A. 2009. Integration of hydrodynamics into a statistical model on the spread of pancreas disease (PD) in salmon farming. Dis. Aquat. Org. 88, 35–44 10.3354/dao02151 (doi:10.3354/dao02151) [DOI] [PubMed] [Google Scholar]