

Abstract
Purpose
The inhibitory role of secreted Chondroitin-sulfate-proteoglycans (CSPGs) on Oncolytic viral (OV) therapy was examined. Chondroitinase ABC (Chase-ABC) is a bacterial enzyme that can remove chondroitin sulfate glycoso-amino glycans from proteoglycans without any deleterious effects in vivo. We examined the effect of Chase-ABC on OV spread and efficacy.
Experimental Design
Three dimensional glioma spheroids placed on cultured brain slices were utilized to evaluate OV spread. Replication-conditional OV expressing Chase-ABC (OV-Chase) was engineered using HSQuik technology, and tested for spread and efficacy in glioma spheroids. Subcutaneous and intracranial glioma xenograft, were utilized to compare anti-tumor efficacy of OV-Chase, rHsvQ (control) and PBS. Titration of viral particles was performed from OV treated subcutaneous tumors. Glioma invasion was assessed in collagen embedded glioma spheroids in vitro, and in intracranial tumors. All statistical tests were two sided.
Results
Treatment by Chase-ABC in cultured glioma cells significantly enhanced OV spread in glioma spheroids grown on brain slices (P< 0.0001). Inoculation of subcutaneous glioma xenografts with Chase-expressing OV significantly increased viral titer (> 10 times, P=0.0008), inhibited tumor growth and significantly increased overall animal survival (P<0.006) compared to treatment with parental rHsvQ virus. Single OV-Chase administration in intracranial xenografts also resulted in longer median survival of animals compared to rHsvQ (32 versus 21 days, P<0.018). Glioma cell migration and invasion were not increased by OV-Chase treatment.
Conclusions
We conclude that degradation of glioma ECM by OV expressing bacterial Chase-ABC enhanced OV spread and anti-tumor efficacy.
Introduction
Oncolytic viruses (OVs) are viruses that are genetically created or have a natural propensity to infect/replicate and destroy cancer cells with minimal damage to non-neoplastic tissues (1–2). While the recent approval by the Chinese State Food and Drug Administration for H101 (Oncorine, an OV functionally identical to ONYX-015) has resulted in the marketing of the world’s first OV, the regulatory approval of OVs in US and Europe is pending on the results of large randomized Phase-III studies (1, 3–4).
Inefficient OV dispersal through the tumor extracellular matrix (ECM) can be a significant barrier in its anti-tumor efficacy (4–5). Structural components of tumor ECM such as collagens and proteoglycans have been shown to hinder distribution of large therapeutic molecules (6–7). Protease or hyaluronidase mediated digestion of the ECM can improve intratumoral spread and efficacy of conditionally replication competent adeno and herpes viruses (8–12). Based on these observations, oncolytic adenoviruses expressing relaxin, a peptide hormone able to decrease the synthesis and secretion of interstitial collagens and increase the expression of matrix metalloproteinase (procollagenase) were tested and found to have enhanced spread and anti-tumor efficacy compared to control adenovirus (13). While the use of degrading tumor ECM enzymes can be an innovative strategy for enhancing spread of macromolecular therapeutic, such enzymes have not yet been tested in intracranial brain tumor models. Emerging evidence suggests that intracranial use of such strategies can be associated with serious complications. For example brain proteases are involved in neurodegenerative diseases (14), collagenase mediated ECM disruption can cause hemorrhagic necrosis of brain (15), and hyaluronidase elicits astrocytic reactivity which can promote optic glioma growth (16).
Secreted and membrane-bound chondroitin sulfate proteoglycans (CSPGs) linked to extra-cellular hyaluronan form a major component of the extracellular matrix in the brain (17). In CNS tumors expression of several CSPGs such as versican, brevican, phosphacan, and NG2 is increased and associated with increased tumor growth, angiogenesis and invasion (18). Apart from molecular signaling, the sugar side chains of chondroitin sulfate glycosaminoglycans (CS-GAGs) on CSPGs are responsible for biophysical properties that limit interstitial diffusion. Choindroitinase ABC is a bacterial enzyme that can cleave and remove the CS-GAG from CSPG leaving the core protein intact (19). While Chase-ABC has been studied for its effect on neuronal regeneration after injury, its impact on tumor ECM has not been previously examined. Here, we hypothesized that Chase-ABC-mediated digestion of glioma CS-GAGs would open glioma ECM, enhancing OV dissemination and efficacy without detrimental effects to surrounding brain. Treatment of glioma spheroids grown in organotypic cultures with purified Chase-ABC enhanced spread of oncolytic virus into the sphere. To investigate the inhibitory role of tumor CSPGs on OV therapy we have created a novel armed Chase-ABC- I expressing herpes simplex-1 OV, and tested its spread and anti tumor effects.
This is the first study investigating the use of bacterial chondroitinase to enhance anti-cancer therapy.
Materials and Methods
Cells, Viruses and antibodies
U343, U87, U87ΔEGFR, LN229, Gli36 EGFR-H2B-RFP, X12 human glioma cells and Vero cells were maintained as described (20–22). Mouse monoclonal anti-chondroitin-4-sulfate antibody (clone BE-123, MP Biomedicals Inc, Aurora, OH) was used to probe for Chase functionality. Tumor bearing sections were labeled with Wisteria floribunda lectin (WFL, Vector Labs Inc., Burlingame, CA), anti vimentin (clone SP20, Lab Vision, Fremont, CA). Oncolytic viruses rHsvQ, rHsvQLuc and rQNestin34.5 have been previously described (23–24). Genomic DNA from Proteus vulgaris (ATCC number 9920D) was used as template to clone Chase-ABC-I cDNA as described (25). The PCR product was sub-cloned into pSecTag/FRT/V5 His-Topo vector (Invitrogen, Carlsbad, CA) and used to generate OV-Chase as previously described (24). Cytotoxicity of viruses was assessed by a standard crystal violet assay (26).
Viral spread/replication assays
Spread of viral particles was visualized by fluorescence microscopy of OV-encoded GFP in infected cells using a Zeiss LSM 510 Meta confocal microscope and quantified using the software ImageJ (NIH, Bethesda, MD). For replication assays, infected cells and supernatant or tissue were collected and viral stocks were titrated by a standard plaque assay (21).
Invasion assays
To perform collagen invasion assays, glioma cell spheroids were treated with OV-Chase, rHsvQ or vehicle, and embedded in collagen (22). Migration of cells away from the core aggregate was monitored using a Nikon Eclipse TE2000-U fluorescent microscope.
Animal studies
All experiments with animals were performed according to the guidelines of the Subcommittee on Animal Research and Care of The Ohio State University. Female athymic mice (nu/nu) were used for all xenografts studies. For intracranial xenografts, tumor cells were implanted stereotactically at a position 2 mm lateral to bregma at a depth of 3 mm. Ten days post tumor cell implantation animals were stereotactically inoculated with PBS or virus. Mice bearing subcutaneous tumors received intra-tumoral injections of HBSS, or the indicated OV. Tumor volumes were measured regularly as described (26). Animals were observed daily and were euthanized at the indicated time points, or when they showed signs of morbidity (intracranial) or when tumor volumes exceeded 1,600 mm3 (subcutaneous).
Statistical Analysis
Two tailed Student’s t-test were used to assess effects of Chase-ABC on viral infection/replication, cytotoxicity and titration from subcutaneous tumors (Origin 7 statistical software, OriginLab Corporation, Northampton, MA). Two-way ANOVAs for repeated measures were used to evaluate viral spread on glioma spheroids. Kaplan-Meier curves were compared using the log-rank test, generalized Wilcoxon and Tarone-Ware tests (SPSS statistical software, version 17.0; SPSS, Inc., Chicago, IL). T-test from fitting linear mixed model was used to test slope difference in the tumor volume growth over the time. A p value lower than 0.05 was considered to be statistically significant for all tests.
Results
CSPGs detected in glioma cell lines and tumor samples interfere with OV spread in glioma spheroids
The presence of CSPG in glioma cell lines and tumor tissue was confirmed by western blotting and immuno-fluorescence staining of tumor tissue (Supplementary Fig. 1). To test whether purified Chase-ABC could affect the ability of oncolytic HSV-1 to infect and replicate in glioma cells, we first analyzed the ability of OVs: rQnestin34.5 (MOI=1, Fig. 1A) and rHsvQLuc (MOI=0.5, Fig. 2B) to infect glioma cells treated with purified Chase-ABC (Black bars) or vehicle(white bars). OV infection was evaluated by comparing the number of GFP positive cells/view field 12 hours post infection (Fig. 1, A) or by measuring the amount of virus encoded Luciferase/mg of protein in cell lysates harvested 6 hours after infection in the wells with treated- or untreated cells (Fig. 1B). The results indicated that treatment of cells with Chase-ABC did not affect ability of virus to infect glioma cells grown in monolayer.
Figure 1. Treatment with Chase-ABC does not affect OV infection/replication, and improves viral spread.
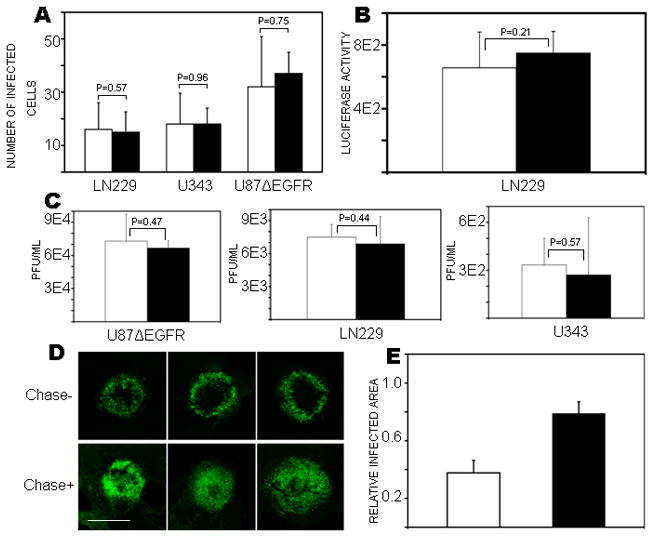
A) The indicated glioma cells were treated with purified Chase-ABC (Black Column) or vehicle (white column) for 24h prior to infection with rQnestin34.5 at an MOI=1. Viral infection was assessed by quantification of GFP-positive infected cells/field of view at 12h post-infection. Columns represent mean number of GFP positive cells/field and (± 95% CI). Results were obtained from at least 3 fields per well, and 3 wells per treatment. B) LN229 cells were treated with or without Chase-ABC (Black or white column respectively) prior to infection with luciferase-carrying OV rHsvQ-Luc. Cells were harvested 6h post-infection and the amount of viral-encoded luciferase/mg total protein was quantified. Columns represent mean value of luciferase activity/mg total protein (± 95% CI) (n=3/group). C) Oncolytic rHsvQ was treated with purified Chase (Black Column) or vehicle (White column) for 1h at 37 oC prior to infecting the indicated glioma cells. Seventy-two hours after viral infection, cells and supernatant were harvested and the amount of infectious viral particles was quantified by a standard titration assay. Columns represent mean pfu/ml (± 95% CI) (n=3/group). D) Spheroids of glioma U87ΔEGFR were cultured on organotypic brain slices for 72h, treated with purified Chase-ABC or vehicle for 24h before and after infection with rQnestin34.5. Viral spread was evaluated four days post OV treatment by detection of the GFP-positive infected cells. Images show three representative spheroids (n=6/group) treated with Chase-ABC (Chase +) or vehicle (Chase −), scale bar=1400μm. E) Quantification of results from (D). Columns indicate the mean GFP-positive area expressed relative to total spheroid area in spheroids treated with Chase (black column) or vehicle (white column) (± 95% CI) (p<0.0001 by Student’s t-test, n=6/group).
Figure 2. OV-Chase is a dual-armed OV encoding functional recombinant Chase-ABC-I.
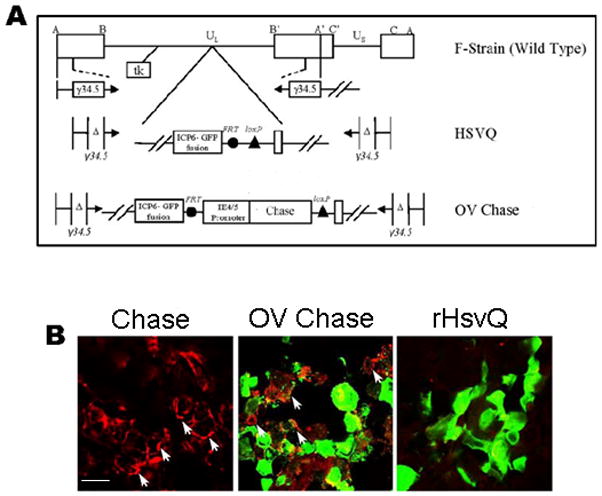
A) Schematic map of viruses used in this study. Top: genetic map of wild type HSV-1, indicating the location of both the copies of γ34.5 and the ICP6 locus. Middle: map of control rHsvQ with deletion of both copies of γ34.5 and in-frame gene disrupting insertion of GFP within the ICP6 gene. Bottom: map of OV-Chase showing the insertion of Chase-ABC cDNA under the viral IE4/5 promoter within the ICP 6 locus. B) Glioma spheroids derived from U87ΔEGFR were treated with purified Chase-ABC (Chase) or infected with GFP-encoding OV-Chase or rHsvQ. Spheroids were fixed 72h post infection and processed for immunohistochemistry with antibody against CS-stubs. Results show the appearance of red immunorreactive CS-stubs (arrows) in spheroids treated with purified Chase or OV-Chase, indicating that the virus produced a functional enzyme (scale bar = 5μm).
Next we tested if Chase-ABC treatment of OV could affect the ability of OVs to infect and replicate in glioma cells. The indicated glioma cells were infected with buffer (white bars) or Chase-ABC treated (black bars) OV and the amount of infectious viral particles obtained was quantified by a standard titration assay (Fig. 1C). The results indicated that Chase-ABC treatment of rHsvQ did not affect its ability to infect and/or replicate in glioma cells in vitro. Collectively these results indicated that Chase-ABC treatment of glioma or OV did not affect viral infection or replication in vitro.
To evaluate the effect of Chase-ABC treatment of glioma spheroids on OV spread in three-dimensional matrix; glioma spheroids were cultured on organotypic brain slices. This reproduced most of the ECM organization of the tumor. U87ΔEGFR spheroids treated with purified Chase-ABC or vehicle were infected with rQnestin34.5 (n=6/group), and OV spread was visualized by fluorescence microscopy of OV-encoded GFP. Visually, all Chase-treated spheroids showed more efficient spread of the OV throughout the spheroid compared to control-treated spheroids (Fig. 1D). Quantification revealed a statistically significant increase in OV spread after Chase-ABC treatment of glioma spheroids (p-value < 0.0001) (Fig. 1E).
Creation of OV-Chase encoding functional recombinant Chase-ABC
Based on the observed lack of detrimental effects of purified Chase-ABC treatment on OV infection of monolayer cells, combined with the enhanced spread of OV observed in treated glioma spheroids, we created OV-Chase: an oncolytic HSV-1 expressing bacterial Chase-ABC driven by the HSV-1 IE4/5 promoter within the backbone of the double attenuated rHsvQ (24) Figure 2A shows the genetic maps of w.t HSV-1, attenuated rHsvQ and the recombinant OV-Chase. The functionality of recombinant Chase-ABC produced by OV-Chase was tested in U87ΔEGFR spheroids grown on organotypic brain slices infected with control rHsvQ or OV-Chase (Fig. 2B). Seventy-two hours post-infection the spheroids were fixed and analyzed by immunofluorescent microscopy. Spheres treated with purified Chase ABC for twenty-four hours were utilized as a positive control. The appearance of red immuno-reactive sugar stubs (arrows) in OV-Chase infected spheres indicated the presence of functional Chase-ABC produced by infected cells (Figure 2B).
To evaluate if expression of Chase-ABC affected the ability of the OV to infect, replicate, and lyse glioma cells in vitro, we compared the cytotoxicity of OV-Chase and rHsvQ in three different glioma cell lines grown as mono-layers. To discriminate if the observed properties were a true reflection of OV-Chase or occurred due to a random non specific mutation that may have occurred during virus isolation we compared two different isolates of OV-Chase. Glioma cells were inoculated with rHsvQ or two different isolates of OV-Chase at the indicated MOI. Five days post infection cell viability was assessed by a standard crystal violet assay (Figure 3). No significant difference was observed between rHsvQ and either of the two independent viral isolates of OV-Chase (p-values > 0.14 for all rHsvQ-treated cells versus OV-Chase1- or OV-Chase2-treated cells).
Figure 3. Chase expression from OV-Chase does not interfere with viral cytotoxicity.
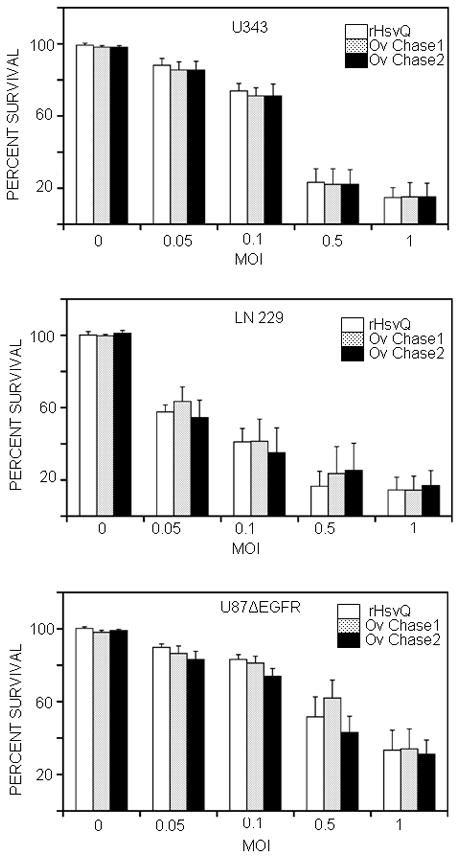
Glioma cells (U343, LN229 and U87ΔEGFR) were inoculated with rHsvQ or two different isolates of OV-Chase (OV-Chase 1 and OV-Chase 2) at MOI = 0, 0.05, 0.1, 0.5 or 1. Two independent isolates of OV-Chase were purified and used in this assay to test if changes in virus infection or replication were specific to the inserted Chase ABC expression cassette. Cell viability was assessed during five days post-infection, using a standard crystal violet assay. Columns (% of surviving cells at day 5) indicate mean values for each treatment (± 95% CIs) (n=4/treatment for each cell line). Results indicate that in vitro cytotoxicity of both the independent isolates of OV-Chase is comparable to control rHsvQ.
We next compared the spread of rHsvQ and OV-Chase in U87 EGFR glioma cells grown as three dimensional spheroids on organotypic brain slices. Briefly, glioma spheroids were infected with 104 pfu of rHsvQ or OV-Chase (n=6/group) and infected cells were visualized by appearance of OV-encoded GFP over time. Quantitative analysis of GFP fluorescence from spheres 24 and 60h post-infection (Fig. 4A) showed that rHsvQ infection did not spread within the spheroids. In contrast, OV-Chase spread to the core of the tumor spheroids in all cases, resulting in wide distribution of GFP fluorescence. These results were statistically significant at all time points (p<0.0001). Taken together our results indicated that OV-Chase infected and replicated in glioma cells as efficiently as the control rHsvQ virus, and exhibited a highly improved spread in three-dimensional glioma cultures.
Figure 4. OV-Chase has improved spread in glioma spheroids.
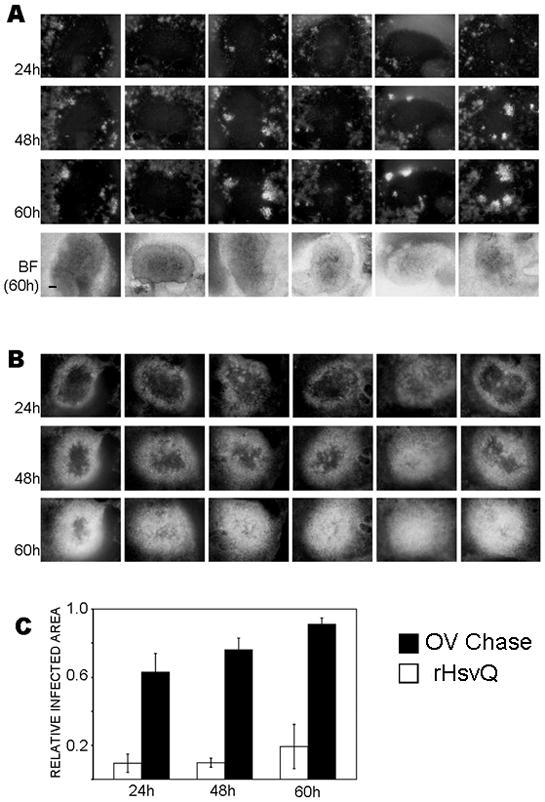
A and B) U87 EGFR glioma spheroids were cultured on organotypic brain slices for 72h and infected with 104 pfu of rHsvQ (A) or OV-Chase (B). Spheroids were imaged for GFP-fluorescence at 24, 48 and 60h post-infection. The figures show representative images of GFP-expressing, OV-infected spheroids at each time point (n=6/group), scale bar = 200μm. BF=brightfield images at t=60h. C) Viral spread was calculated as the GFP-positive area relative to the total area of the spheroids. Columns indicate mean relative infected area and (± 95% CI) (p<0.0001 at each time point).
OV-Chase has improved anti-tumor efficacy in vivo compared to control OV
To test the anti-tumor efficacy of OV-Chase, we assayed its effect on subcutaneous tumors formed by injection of Gli36ΔEGFR–H2B-RFP glioma cells in athymic mice. Animals were treated with PBS or 106 pfu of rHsvQ or OV-Chase when tumors reached a volume of 80–150 mm3. The mice were closely monitored for tumor growth and sacrificed when tumors reached a volume of 1,600 mm3 (endpoint condition). Survival analysis (Kaplan-Meier curves, Fig. 5A) showed a significant increase in survival of animals treated with OV-Chase compared to rHsvQ (median survival=28 and 16 days respectively, p< 0.0001). Two of the 8 animals treated with OV-Chase were long-term survivors (>80 days) and found to be tumor-free upon sacrifice and histological evaluation. The longer survival times correlated with smaller flank tumor burden, with PBS-treated tumors increasing over 14.7-fold in volume by 12-days post-treatment, versus 9.3-fold for rHsvQ -treated tumors and 2.7-fold for OV-Chase-treated tumors (Fig. 5B).
Figure 5. OV-Chase has improved efficacy against glioma in vivo.
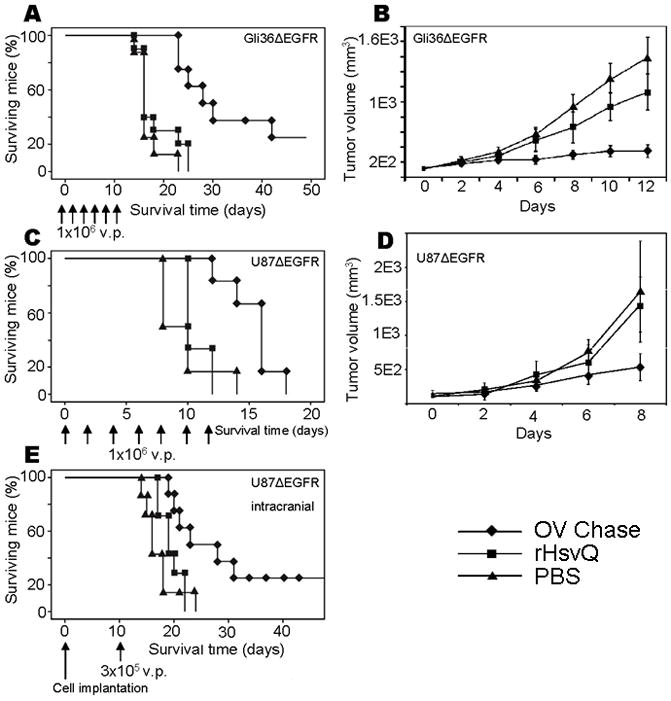
A and B) Mice injected with Gli36ΔEGFR–H2B-RFP cells subcutaneously were left untreated until the tumors reached an average volume of 80–150 mm3. Animals were then injected with PBS (triangles), 106 pfu rHsvQ (squares) or 106 pfu OV-Chase (diamonds) six times (day 0, 2, 4, 6, 8 and 10, arrows). Tumors were monitored daily and animals sacrificed when the tumors reached a volume of 1,600–2,000 mm3. Results show the Kaplan-Meier survival curves (A) and tumor growth (B) (± 95% CI). Animals treated with OV-Chase survived significantly longer in average than those treated with vehicle or the control OV (p<0.0001). Tumor volume values were log-transformed to make the trend over time linear. Slope difference between OV-Chase and PBS was 0.119 (p<0.0001 while slope difference between OV-Chase and rHsvQ was 0.094 (p = 0.0008). Trends were not different between PBS and rHsvQ (p = 0.162). C and D) Mice injected with U87ΔEGFR cells subcutaneously were left untreated until the tumors reached an average volume of 80–150 mm3 and then treated with PBS (triangles), rHsvQ (squares) or OV-Chase (diamonds) as above. Animals were monitored and tumors harvested using the same criteria as before for Gli36ΔEGFR tumors. Results show Kaplan-Meier survival curves (C) and tumor growth (D) (± 95% CI). Animals treated with OV-Chase survived again significantly longer in average than those treated with vehicle or the control OV (p<0.006 by log-rank test). OV-Chase treated tumors had statistically significant lower slope values than both rHsvQ (P=0.0008) and PBS treated tumors (P=0.0006). The slope difference between rHsvQ and PBS treated tumors was not significant (P=0.297). E) U87ΔEGFR cells were injected intracranially in mice and animals treated with a single dose of PBS, rHsvQ or OV-Chase 10 days after tumor implantation. Kaplan-Meier survival curves, indicate improved anti tumor efficacy of OV-Chase over the control treatments (p<0.05).
In a separate experiment, mice were inoculated with the highly proliferative U87ΔEGFR glioma cells and treated similarly with PBS (n=6) or rHsvQ or OV-Chase. Comparison of slopes revealed that OV-Chase has statistically significant lower slope values than both rHsvQ and PBS (Slope difference = −0.1976, p value = 0.0008). The median survival of OV-Chase treated mice was significantly more than mice treated with rHsvQ (median survival=16 days for OV-Chase-treated tumors and 10 days for rHsvQ -treated tumors, p < 0.006).
To confirm that OV-Chase had efficient anti-tumoral activity in an orthotopic xenograft model, we tested the virus against intracranial gliomas derived from U87ΔEGFR cells. Tumor-bearing animals were treated with PBS or 3×105 pfu of rHsvQ or OV-Chase 10 days after tumor cell implantation. Survival analysis (Fig. 5E) again showed a significant improvement in survival of animals treated with OV-Chase (median survival= 32 days versus 21 days for rHsvQ -treated animals, p< 0.05). Two of the eight mice treated with OV-Chase lived more than 80 days, at which point they were sacrificed and no evidence of intracranial tumor was found by histological examination. Collectively these results indicated that OV-Chase treatment had a significant anti-tumor effect in vivo compared to control rHsvQ.
OV-Chase has enhanced spread in vivo compared to control OV
We compared the spread and replication of OV-Chase to control rHsvQ in tumors. Mice with subcutaneous tumors (Gli36ΔEGFR–H2B-RFP cells) that reached 200–270 mm3 were treated with 107 pfu of OV-Chase or rHsvQ on day 0 and day 2 as described in the methodology section. Five days later the animals were sacrificed and the amount of infectious virus particles in each tumor was evaluated. Results (Supplementary figure S2A) showed a statistically significant increase in the titer of infectious particles from OV-Chase-treated tumors compared to rHsvQ -treated tumors (p=0.0008). Representative microphotographs of 50 μm-thick sections from these tumors (Supplementary figure S2B) depict the wider distribution of viral-encoded GFP in OV-Chase-treated tumors compared to rHsvQ treated tumors. In a parallel experiment we tested the spread of OV-Chase in intracranial (U87ΔEGFR) tumors in mice. HSV-1 staining of tumor bearing brain sections from mice three days after treatment with 1.5 × 105 pfu of rHsvQ or OV-Chase was performed to visualize viral spread in intracranial tumors. Supplementary Figure 2C shows representative images of tumor sections from rHsvQ (top panel), or OV-Chase (bottom panel) treated tumors. Marked increase in the spread of OV-Chase was observed in all the sections compared to rHsvQ treated tumors (Supplementary figure S2C).
Chase-ABC and OV-Chase do not increase glioma cell dispersal
CSPGs form large macromolecular complexes that collectively inhibit cell and axon motility in normal brain. In contrast, CSPGs are more soluble in gliomas and have been clearly shown to increase glioma cell dispersion (17). Because degradation of CS chains by Chase could solubilize CSPGs from the tumor extracellular matrix and facilitate cell dispersion, we investigated whether treatment with OV-Chase would affect the invasiveness of glioma cells into their surrounding matrix in vitro and in vivo. First we evaluated the effect of OV-Chase infection on invasion of glioma cells grown in a three-dimensional matrix. Spheroids of LN229, U87ΔEGFR, and X12 glioma cells (n=4/group) were treated with PBS, OV-Chase, or rHsvQ, implanted in collagen, and imaged at different time points (Fig. 6A, and supplementary Figure S3). Glioma cells from PBS-treated spheroids derived from all three cell lines dissociated from the spheroid and migrated into the collagen matrix. However this migration was largely reduced in both rHsvQ and OV-Chase treated spheroids compared to PBS treated spheres. In general OV-Chase treated spheroids showed a trend towards reduced migration compared to rHsvQ, and this may be a reflection of better tumor cell killing by OV-Chase. Importantly the results indicated that OV-Chase did not increase glioma cell invasion in vitro. To test if in vivo treatment of glioma with OV-Chase affected glioma invasion in vivo we treated mice bearing U87ΔEGFR (day 7), or Gli36-H2B-RFP-ΔEGFR (day 10) with PBS, or 3 × 105 pfu of rHsvQ or OV-Chase (Figure 6B and S3C respectively). Mice were sacrificed when they showed signs of tumor burden, and their brains were sectioned and stained for human glioma cells using anti-human vimentin. There was no obvious sign of increased glioma invasion in mice treated with OV-Chase compared to PBS or rHsvQ treated mice.
Figure 6. OV-Chase infection reduces glioma cell dispersion in vitro and does not affect tumor spread in vivo.
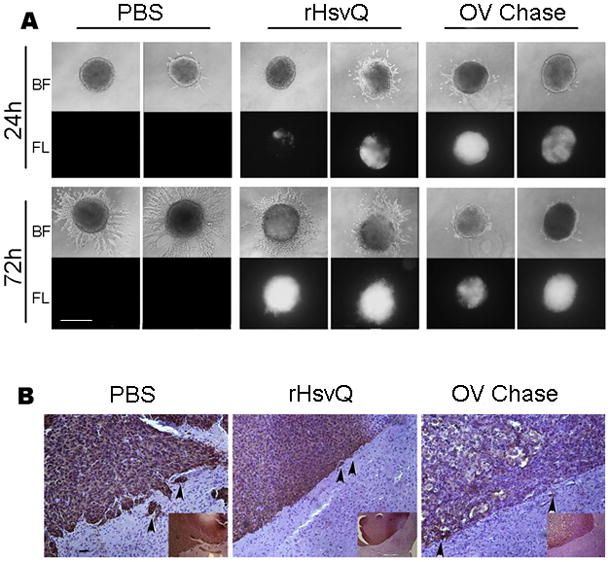
A) Spheroids of LN229 glioma cells were treated with PBS, rHsvQ or OV-Chase and embedded in collagen as indicated in the methodology. The images show representative bright-field and fluorescent images revealing total spheroid dispersal (bright-field) and total viral spread in the spheroid (scale bar = 500μm). B) Representative images of mouse brain sections bearing U87ΔEGFR glioma treated with PBS, rHsvQ or OV-Chase. The sections were stained with anti-human vimentin to highlight human glioma cells in mouse brain. Infiltrating human glioma cells are visualized as brown staining cells. There was no difference in invasion of glioma cells after treatment with PBS, rHsvQ or OV (Scale bar = 100μm).
Discussion
Malignant gliomas are resistant to traditional therapeutic approaches and, despite significant advances in research, their prognosis remains poor with a median survival of less than 15 months. Conditionally replicating OVs with the ability to replicate and destroy tumor cells are a promising therapeutic approach for this disease (3). Enzymatic degradation of some tumor ECM constituents has been shown to enhance viral spread and efficacy in vivo, suggesting that tumor ECM is one of the major limitating factors in viral spread and antitumor efficacy (4). However concerns about damaging normal brain tissue has limited the usage of matrix modulating enzymes in intracranial models (8, 10, 12). Here we show for the first time the ability of bacterial chondroitinase ABC-mediated digestion of glioma ECM to enhance OV spread and anti tumor efficacy in vitro and in vivo.
The composition and organization of the ECM directly affects tissue structural integrity, cell-cell communication, availability of trophic factors and diffusion of macromolecules in the interstitial space. CSPGs are major structural components of the neural ECM, produced mostly by astrocytes and are known to accumulate in aged brain as well as neural tissue after acute or chronic injury (27). These molecules are well-known inhibitors of cellular motility and axonal extension in the normal CNS (28) and can also regulate the bioavailability of growth factors and the biophysical properties of the extracellular space that facilitates particle diffusion (29). Removal of these CS-GAGS by Chase-ABC has been shown to result in improved re-growth of acutely injured axons in vivo and has been associated with some functional recovery with no evidence of toxicity (30–32). However, the impact of modifying tumoral ECM with Chase-ABC on tumor biology or therapeutic efficacy has not been previously studied.
Malignant gliomas exhibit a dense ECM that is rich in the components found in neural ECM, including HA and CSPGs (7). Several CSPGs such as versican, brevican, phosphacan, and NG2 are highly up-regulated in gliomas compared to normal adult neural tissue, and are associated with increased tumor growth, angiogenesis and invasion (18). Thus, while CSPG in normal brain are inhibitory towards axonal regeneration, they have been shown to promote glioma cell invasion and angiogenesis (33–34).
In addition to their novel functions in glioma, accumulation of CSPGs in the glioma matrix leads to increased tortuosity of the extracellular space and increased interstitial pressure in the tumor. These changes cause resistance for diffusion and limit the spread of large-sized therapeutic agents (35). For these reasons, ECM macromolecules have been repeatedly identified as potential therapeutic targets for adjuvant therapy (36–38). Targeting of CSPGs, using blocking antibodies against versican (36) and RNA-interference against phosphacan (39) has shown that reduction or inhibition of these molecules may impair tumor proliferation and dispersion. In addition, degradation of CSPGs using MMP-1 and MMP-8 has been shown to increase hydraulic conductivity and particle diffusion in solid tumors (40). Together these results suggest that reduction of CSPGs levels in gliomas by enzymatic manipulation could inhibit their pro-tumoral effects and at the same time improve diffusion of therapeutic agents within the tumor.
The bacterial enzyme Chase-ABC I (chondroitin lyase), derived from Proteus vulgaris, has been shown to selectively depolymerize CS-GAG chains present on CSPGs into soluble disaccharides. Digestion of these GAGs in normal brain reduces the level of glycanated CSPGs (41), overcomes many inhibitory effects of CSPGs and enables in part the ability of neuronal processes to extend past site of injury to form new synaptic connections (19). This enzyme has been widely used to promote regeneration of injured axonal tracts in rodent models, and has been shown to provide a long-lasting ‘loosening’ effect of the ECM scaffold, promoting synaptic plasticity without noticeable deleterious effects (42). Based on extensive evidence from preclinical models, highly purified, protease-free preparations of recombinant Chase-ABC I have recently been used in phase 1–2 trials to treat herniated lumbar disks in Japan (43). Collectively, these studies highlight the safety of Chase-ABC I in preclinical models and human trials.
In the present work, we hypothesized that Chase-ABC I- mediated digestion of CSPGs could enhance viral dissemination in gliomas without the deleterious effects associated with protease treatment of neural tissue. To test our hypothesis, we first studied the effect of Chase-ABC treatment on viral infection/replication and spread on cultured glioma cells and glioma spheroids. Our results demonstrated that enzymatic digestion of CS-GAGs by Chase-ABC did not affect the ability of OVs to infect/replicate and lyse glioma cells, and significantly enhanced OV diffusion through matrix-containing glioma spheroids. This result is particularly relevant because other studies have suggested that CSPGs can modulate virus-cell interactions and affect viral infection and gene transfer efficiency (44–47). Our observation that CS removal did not reflect in changes in viral efficiency in glioma cells suggests that the predominant effect of the enzyme was an improvement of viral spread through the tumor matrix.
Based on our initial results, we generated an armed OV expressing recombinant Chase-ABC under the control of an immediate-early viral promoter. This strategy allowed us to directly remove CS from the surrounding matrix of infected cells independently of co-injection with purified enzyme. Expression of recombinant Chase-ABC using this approach did not interfere with viral oncolysis and enhanced the spread of OV-Chase in glioma spheroids. More importantly, the enhanced spread of OV-Chase observed in organotypic cultures translated into increased viral efficacy against subcutaneous and intracranial glioma xenografts compared to a control OV.
Because CS degradation in the brain may remove major components which inhibit cellular movement in adult neural tissue, we further investigated if Chase-ABC treatment could affect glioma cell motility and invasion. Cell migration assays performed in modified Boyden chambers (Transwell assay) revealed that treatment of CSPGs with Chase-OV did not increase glioma cell motility (not shown). This was an interesting result and suggested that exogenous CSPGs do not impair glioma cell migration compared to migration of neural growth cones (17). Therefore removal of CSPGs should likely not affect tumor cell dispersal. Our results suggest that treatment of glioma spheroids with OV-Chase did not increase glioma cell dispersion through a three-dimensional collagen matrix. Further tests in vivo indicated that direct treatment of intracranial xenografts with OV-Chase did not increase cell dispersion, and tumor borders were comparable between tumors treated with OV-Chase and a control OV.
In summary, our results show for the first time that degradation of glioma CS-GAGs by OV-expressed recombinant Chase-ABC is sufficient to increase viral spread through the tumor ECM, and underscore the potential of utilizing Chase-ABC-armed viruses for the enhancement of tumor oncolysis.
Supplementary Material
Translational Significance.
Despite the current standard of care malignant astrocytomas remain a devastating disease with a median survival of less than fifteen months. Thus there is an urgent need to improve current therapeutic modalities. Oncolytic viruses represent a promising biological therapy currently being evaluated in patients for safety and efficacy. Here we show that tumoral chondroitin sulafte proteoglycans (CSPG) present a formidable barrier for the spread of OV through the tumor. Based on these results, we have created a novel oncolytic virus armed with bacterial Choindroitinase ABC (Chase-ABC). Chase-ABC mediated removal of chondroitin-sulfated glycoso-amino glycan sugar chains from glioma CSPG enhances virus spread, without increasing glioma invasion. This is the first report investigating the effects of Chase-ABC on tumor biology. These preclinical results will facilitate future clinical testing of this oncolytic virus.
Acknowledgments
Funding Sources:
This work was supported by grants of the National Institutes of Health, R21NS066299 and R01NS064607 (to BK).
References
- 1.Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–76. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- 2.Msaouel P, Dispenzieri A, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: an overview. Curr Opin Mol Ther. 2009;11:43–53. [PMC free article] [PubMed] [Google Scholar]
- 3.Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaur B, Cripe TP, Chiocca EA. “Buy one get one free”: armed viruses for the treatment of cancer cells and their microenvironment. Curr Gene Ther. 2009;9:341–55. doi: 10.2174/156652309789753329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker JN, Bauer DF, Cody JJ, Markert JM. Oncolytic viral therapy of malignant glioma. Neurotherapeutics. 2009;6:558–69. doi: 10.1016/j.nurt.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–503. [PubMed] [Google Scholar]
- 7.Zamecnik J. The extracellular space and matrix of gliomas. Acta Neuropathol (Berl) 2005;110:435–42. doi: 10.1007/s00401-005-1078-5. [DOI] [PubMed] [Google Scholar]
- 8.Ganesh S, Gonzalez Edick M, Idamakanti N, et al. Relaxin-expressing, fiber chimeric oncolytic adenovirus prolongs survival of tumor-bearing mice. Cancer Res. 2007;67:4399–407. doi: 10.1158/0008-5472.CAN-06-4260. [DOI] [PubMed] [Google Scholar]
- 9.Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. 2010;18:1275–83. doi: 10.1038/mt.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuriyama N, Kuriyama H, Julin CM, Lamborn K, Israel MA. Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum Gene Ther. 2000;11:2219–30. doi: 10.1089/104303400750035744. [DOI] [PubMed] [Google Scholar]
- 11.Hong CS, Fellows W, Niranjan A, et al. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010 doi: 10.1038/gt.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKee TD, Grandi P, Mok W, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66:2509–13. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- 13.Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst. 2006;98:1482–93. doi: 10.1093/jnci/djj397. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Luo W, Reiser G. Trypsin and trypsin-like proteases in the brain: proteolysis and cellular functions. Cell Mol Life Sci. 2008;65:237–52. doi: 10.1007/s00018-007-7288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenberg GA, Estrada E, Kelley RO, Kornfeld M. Bacterial collagenase disrupts extracellular matrix and opens blood-brain barrier in rat. Neurosci Lett. 1993;160:117–9. doi: 10.1016/0304-3940(93)90927-d. [DOI] [PubMed] [Google Scholar]
- 16.Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16:1098–112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- 17.Viapiano MS, Matthews RT. From barriers to bridges: chondroitin sulfate proteoglycans in neuropathology. Trends Mol Med. 2006;12:488–96. doi: 10.1016/j.molmed.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Viapiano MS, Lawler SE. Glioma Invasion: Mechanisims and Therapeutic Challenges. In: Vm EG, editor. CNS Cancer Models, Prognostic factors and targets. New Jersey: Humana Press; 2009. [Google Scholar]
- 19.Rhodes KE, Fawcett JW. Chondroitin sulphate proteoglycans: preventing plasticity or protecting the CNS? J Anat. 2004;204:33–48. doi: 10.1111/j.1469-7580.2004.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hardcastle J, Kurozumi K, Dmitrieva N, et al. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol Ther. 2010;18:285–94. doi: 10.1038/mt.2009.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ichikawa T, Chiocca EA. Comparative analyses of transgene delivery and expression in tumors inoculated with a replication-conditional or -defective viral vector. Cancer Res. 2001;61:5336–9. [PubMed] [Google Scholar]
- 22.Nowicki MO, Dmitrieva N, Stein AM, et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro Oncol. 2008;10:690–9. doi: 10.1215/15228517-2008-041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34. 5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832–9. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- 24.Terada K, Wakimoto H, Tyminski E, Chiocca EA, Saeki Y. Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 2006;13:705–14. doi: 10.1038/sj.gt.3302717. [DOI] [PubMed] [Google Scholar]
- 25.Prabhakar V, Capila I, Bosques CJ, Pojasek K, Sasisekharan R. Chondroitinase ABC I from Proteus vulgaris: cloning, recombinant expression and active site identification. Biochem J. 2005;386:103–12. doi: 10.1042/BJ20041222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaur B, Brat DJ, Devi NS, Van Meir EG. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene. 2005;24:3632–42. doi: 10.1038/sj.onc.1208317. [DOI] [PubMed] [Google Scholar]
- 27.Jones LL, Margolis RU, Tuszynski MH. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol. 2003;182:399–411. doi: 10.1016/s0014-4886(03)00087-6. [DOI] [PubMed] [Google Scholar]
- 28.Bandtlow CE, Zimmermann DR. Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol Rev. 2000;80:1267–90. doi: 10.1152/physrev.2000.80.4.1267. [DOI] [PubMed] [Google Scholar]
- 29.Vargova L, Homola A, Zamecnik J, Tichy M, Benes V, Sykova E. Diffusion parameters of the extracellular space in human gliomas. Glia. 2003;42:77–88. doi: 10.1002/glia.10204. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Alias G, Barkhuysen S, Buckle M, Fawcett JW. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat Neurosci. 2009;12:1145–51. doi: 10.1038/nn.2377. [DOI] [PubMed] [Google Scholar]
- 31.Rolls A, Cahalon L, Bakalash S, Avidan H, Lider O, Schwartz M. A sulfated disaccharide derived from chondroitin sulfate proteoglycan protects against inflammation-associated neurodegeneration. Faseb J. 2006;20:547–9. doi: 10.1096/fj.05-4540fje. [DOI] [PubMed] [Google Scholar]
- 32.Crespo D, Asher RA, Lin R, Rhodes KE, Fawcett JW. How does chondroitinase promote functional recovery in the damaged CNS? Exp Neurol. 2007 doi: 10.1016/j.expneurol.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Zheng PS, Wen J, Ang LC, et al. Versican/PG-M G3 domain promotes tumor growth and angiogenesis. FASEB J. 2004;18:754–6. doi: 10.1096/fj.03-0545fje. [DOI] [PubMed] [Google Scholar]
- 34.Hu B, Kong LL, Matthews RT, Viapiano MS. The proteoglycan brevican binds to fibronectin after proteolytic cleavage and promotes glioma cell motility. J Biol Chem. 2008;283:24848–59. doi: 10.1074/jbc.M801433200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moon LD, Asher RA, Fawcett JW. Limited growth of severed CNS axons after treatment of adult rat brain with hyaluronidase. J Neurosci Res. 2003;71:23–37. doi: 10.1002/jnr.10449. [DOI] [PubMed] [Google Scholar]
- 36.Arslan F, Bosserhoff AK, Nickl-Jockschat T, Doerfelt A, Bogdahn U, Hau P. The role of versican isoforms V0/V1 in glioma migration mediated by transforming growth factor-beta2. Br J Cancer. 2007;96:1560–8. doi: 10.1038/sj.bjc.6603766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viapiano MS, Hockfield S, Matthews RT. BEHAB/brevican requires ADAMTS-mediated proteolytic cleavage to promote glioma invasion. J Neurooncol. 2008;88:261–72. doi: 10.1007/s11060-008-9575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gladson CL. The extracellular matrix of gliomas: modulation of cell function. J Neuropathol Exp Neurol. 1999;58:1029–40. doi: 10.1097/00005072-199910000-00001. [DOI] [PubMed] [Google Scholar]
- 39.Grumet M, Friedlander DR, Sakurai T. Functions of brain chondroitin sulfate proteoglycans during developments: interactions with adhesion molecules. Perspect Dev Neurobiol. 1996;3:319–30. [PubMed] [Google Scholar]
- 40.Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007;67:10664–8. doi: 10.1158/0008-5472.CAN-07-3107. [DOI] [PubMed] [Google Scholar]
- 41.Lin R, Kwok JC, Crespo D, Fawcett JW. Chondroitinase ABC has a long-lasting effect on chondroitin sulphate glycosaminoglycan content in the injured rat brain. J Neurochem. 2008;104:400–8. doi: 10.1111/j.1471-4159.2007.05066.x. [DOI] [PubMed] [Google Scholar]
- 42.Bruckner G, Bringmann A, Hartig W, Koppe G, Delpech B, Brauer K. Acute and long-lasting changes in extracellular-matrix chondroitin-sulphate proteoglycans induced by injection of chondroitinase ABC in the adult rat brain. Exp Brain Res. 1998;121:300–10. doi: 10.1007/s002210050463. [DOI] [PubMed] [Google Scholar]
- 43.Thuret S, Moon LD, Gage FH. Therapeutic interventions after spinal cord injury. Nat Rev Neurosci. 2006;7:628–43. doi: 10.1038/nrn1955. [DOI] [PubMed] [Google Scholar]
- 44.Banfield BW, Leduc Y, Esford L, Visalli RJ, Brandt CR, Tufaro F. Evidence for an interaction of herpes simplex virus with chondroitin sulfate proteoglycans during infection. Virology. 1995;208:531–9. doi: 10.1006/viro.1995.1184. [DOI] [PubMed] [Google Scholar]
- 45.Le Doux JM, Morgan JR, Snow RG, Yarmush ML. Proteoglycans secreted by packaging cell lines inhibit retrovirus infection. J Virol. 1996;70:6468–73. doi: 10.1128/jvi.70.9.6468-6473.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Doux JM, Morgan JR, Yarmush ML. Removal of proteoglycans increases efficiency of retroviral gene transfer. Biotechnol Bioeng. 1998;58:23–34. doi: 10.1002/(sici)1097-0290(19980405)58:1<23::aid-bit3>3.3.co;2-s. [DOI] [PubMed] [Google Scholar]
- 47.Le Doux JM, Morgan JR, Yarmush ML. Differential inhibition of retrovirus transduction by proteoglycans and free glycosaminoglycans. Biotechnol Prog. 1999;15:397–406. doi: 10.1021/bp990049c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.