

Oxocarbenium ions are intermediates in a number of synthetic processes including Prins cyclizations,[1] acid-mediated additions to acetals,[2] allyl group transfers,[3] and additions of carbonyls to electrophiles.[4] Stereocontrol in these transformations can be quite high as a result of the strong preference, calculated at approximately 2 kcalmol−1,[5] for monosubstituted oxocarbenium ions to exist in E configurations. However, reports of geometric control for 1,1-disubstituted oxocarbenium ions are rare[6] because the steric difference between the alkyl groups is generally smaller than the steric difference between an alkyl group and a hydrogen atom. General models that predict the geometry of disubstituted oxocarbenium ions would be valuable for designing syntheses of natural products or natural-product-like libraries[7] that contain tertiary ether groups. Recently, our research group reported[8] that intramolecular nucleophilic additions to alkynyl-substituted oxocarbenium ions proceed with minimal stereocontrol to provide cis- and trans-2,6-disubstituted tetrahydropyrans. This unusual lack of stereo-control results from the approximate energetic equivalence of the E and Z oxocarbenium ions, which is a result of the small steric difference between an alkynyl group and a hydrogen atom (Scheme 1). Herein, we describe a rare application of carbon–hydrogen bond functionalization for stereoselective syntheses of molecules that contain fully substituted carbon atoms. The approach is based on the development of a model that is able to predict the geometries of 1,1-disubstituted oxocarbenium ions involved in nucleophilic additions that form tertiary ethers with high stereocontrol. We also report a model that illustrates stereocontrol in intramolecular additions to monosubstituted oxocarbenium ions relative to a tertiary ether.
Scheme 1.

Alkynyl-substituted oxocarbenium ions.
We postulated that 1,1-disubstituted oxocarbenium ions containing an alkyl group and an alkynyl group should exist in a conformation in which the two alkyl groups have a trans relationship in consideration of the minimal steric demands of alkynyl groups. We chose to employ a DDQ-mediated ether oxidation[9] protocol for carbocation formation to test this hypothesis because these conditions eliminate the potential for acid-induced solvolytic product decomposition[10] The synthesis of the ether linkage between the two branched carbon atoms in 2 (Scheme 2) was readily constructed by applying Yamamoto’s Me3Al-mediated acetal opening protocol[11] to alkynyl acetal 1. Functional group manipulations provided the cyclization substrate 3, which was exposed to DDQ at room temperature and provided tetrahydropyran 4 as a single stereoisomer in 72% yield. This stereochemical outcome is consistent with our hypothesis that the reaction proceeds through the oxocarbenium ion 5. Although tertiary and spirocyclic ethers have been prepared through intramolecular Prins-type additions to disubstituted oxocarbenium ions,[12] stereoselectivity has not been addressed aside from reactions that proceed through a specific isatin-derived ion.[6d–f]
Scheme 2.

Stereocontrolled tertiary ether synthesis. Reagents and conditions: a) Me3Al, toluene, 75%. b) Py·SO3, Et3N, DMSO, 92%. c) Bestmann–Ohira reagent,[13] K2CO3, MeOH. d) HOAc, [{(p-cyme-ne)RuCl2}2], Fur3P, Na2CO3, toluene,[14] 65%, two steps. e) DDQ, M.S. (4 Å), 2,6-Cl2Py, 1,2-dichloroethane, 72%. DDQ = 2,3-dichloro-5,6-6, dicyano -1,4-benzoquinone, DMSO = dimethyl sulfoxide, Fur3P = tri(2-furyl)phosphine, M.S.=molecular sieves, Py =pyridine.
This result led us to devise more generalized models of oxocarbenium ion geometry to guide substrate design for the stereocontrolled formation of cyclic tertiary ethers. One approach was based on expanding the model for the geometric control over 1,1-disubstituted oxocarbenium ions, and the other was based on control over the trajectory of nucleophilic approach by preexisting tertiary stereocenters (Figure 1). The generalized model for 1,1-disubstituted oxocarbenium ion geometric control (model A) places the smaller group (RS) in a cis relationship with the opposite alkyl group and the larger group (RL) in a trans relationship. Model B illustrates the manner by which preexisting tertiary stereocenters can influence the sense of nucleophilic addition into oxocarbenium ions. Thus RS occupies an eclipsed orientation with the hydrogen atom of a monoalkyl oxocarbenium ion. These models are similar to the reactive conformation for the well-studied Corey–Bakshi–Shibata (CBS) ketone reduction (model C),[15] and successful structural classes for CBS reactions were used to guide substrate design in this study.
Figure 1.

Models for geometrical control in highly substituted oxocarbenium ions. The large, small, and nucleophilic groups on the tertiary ether are labeled as RL, RS, and RN, respectively.
Alkynes serve as the RS substituent when compared to alkyl groups in model A, but alkenes and arenes could serve as the RL group. Thus, secondary allylic and benzylic ethers should yield products that place the unsaturated groups in equatorial orientations. These substrates reacted with moderate stereocontrol in dichloroethane (not shown), but changing the reaction solvent to nitromethane led to a substantial improvement in selectivity. We postulate that the more polar solvent stabilizes the intermediate oxocarbenium ion, thus slowing the cyclization and lowering the barrier of oxocarbenium-ion rotation,[16] and thereby allowing the ions to equilibrate to their more stable isomers prior to ring closure.
Representative examples of these cyclization reactions are shown in Table 1. In each of these examples the alkenyl or aryl group is the RL group. Allylic ethers 6 and 8 reacted quite efficiently and provided tetrahydropyrans 7 and 9 with excellent stereocontrol (Table 1, entries 1 and 2). Aryl analogues were effective substrates, with diastereomeric ethers 10 and 12 yielding 11 as a single stereoisomer (Table 1, entries 3 and 4). The formation of the same isomer starting from diasteromeric reagents provides support for a planar oxocarbenium ion intermediate. Spirocyclic ethers, which are subunits in many natural products,[17] are also accessible through this protocol as shown in entries 5, 6, and 7 of Table 1. These reactions were quite fast and extremely stereoselective. Entries 8 and 9 of Table 1 show the effect of using trans-1,2-disubstituted alkene substrates. Allylic ether 19 reacted with excellent efficiency, though the diastereocontrol was modest (3:1). This outcome is consistent with the lower steric demand of a disubstituted alkene relative to a trisubstituted alkene. The diastereomeric products were separable, however, and the major product could be isolated as a pure compound in good yield. Cinnamyl ethers are significantly more reactive than allylic ethers, and allowed the reaction of 21 to be conducted at −60°C in nitroethane to provide 22 in 82% yield as a 10:1 mixture of diastereomers within 4 hours.
Table 1.
Synthesis of tertiary ethers from oxidation of allylic and benzylic ethers.[a]
| Entry | Substrate[b] | Product | t [h] | d.r.[c] | Yield [%][d] |
|---|---|---|---|---|---|
| 1 |
 6 |
 7 |
12 | 26:1 | 84 |
| 2 |
 8 |
 9 |
2.5 | 100:0 | 86 |
| 3 |
 10 |
 11 |
2 | 100:0 | 78 |
| 4 |
 12 |
 11 |
2 | 100:0 | 74 |
| 5 |
 13 |
 14 |
0.67 | 100:0 | 82 |
| 6 |
 15 |
 16 |
0.5 | 100:0 | 84 |
| 7 |
 17 |
 18 |
7 | 100:0 | 67 |
| 8 |
 19 |
 20 |
5.5 | 3:1 | 85[e] |
| 9[f] |
 21 |
 22 |
4 | 10:1 | 82 |
Typical procedure: a 0.1 M solution of the substrate and 2,6-dichloropyridine in MeNO2 was treated with DDQ and stirred at 0 °C or at RT for the indicated time period. See the Supporting Information for details on specific reactions.
See the Supporting Information for details on substrate synthesis.
d.r. =diastereomer ratio normalized to 100.
Yields refer to isolated, purified material unless otherwise noted.
Yield corresponding to the mixture of stereoisomers.
Reaction was conducted at −60°C in EtNO2.
Our success in this phase of the project led us to explore applications of model B, in which tertiary ethers serve to direct the generation of new stereocenters. Prenyl ethers were used in this study because of their exceptional efficiency in these reactions. The results of these cyclization reactions are shown in Table 2. Propargylic ether 23 smoothly reacted to form 24 as a single stereoisomer (Table 2, entry 1). The stereochemical outcome of this reaction is consistent with the alkynyl group serving as the RS substituent. In contrast to model A, alkenyl groups act as the RS substituent in this model, as shown in the cyclizations of 25 and 27 to form tetrahydropyrans 26 and 28, respectively (Table 2, entries 2 and 3). The potential for alkyl groups to occupy the RS site was demonstrated through the cyclization of 29 to form spirocycle 30 (Table 2, entry 4). While diastereocontrol was not an issue for this reaction, the transformation was important for expanding the scope of the process to include saturated tertiary ether substrates. Diastereocontrol was demonstrated in the cyclization of 31 to 32 (Table 2, entry 5), in which the unbranched group was the RS substituent and the branched group was the RL substituent. These spirocyclization reactions were quite rapid, and allowed the transformations to be conducted at −30°C to maximize stereocontrol. Unfortunately, attempts to prepare tetrahydropyrans with two quaternary stereocenters failed, as demonstrated by the DDQ oxidation of 33 (Table 2, entry 6). No reaction was observed at room temperature and nonspecific decomposition of the starting material was observed at elevated temperatures. Presumably the steric interactions in this system are simply too significant to overcome through ether oxidation.
Table 2.
Tertiary ether substrates in stereoselective cyclization reactions.[a]
| Entry | Substrate[b] | Product | t [h] | d.r.[c] | Yield [%][d] |
|---|---|---|---|---|---|
| 1 |
 23 |
 24 |
2.5 | 100:0 | 79 |
| 2 |
 25 |
 26 |
2 | 15.7:1 | 95 |
| 3 |
 27 |
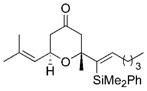 28 |
0.5 | 100:0 | 76 |
| 4 |
 29 |
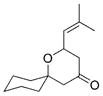 30 |
0.3 | – | 88 |
| 5[e] |
 31 |
 32 |
5 | 100:0 | 81 |
| 6[f] |
 33 |
– | – | – | – |
Typical procedure: a solution of the substrate and 2,6-dichloropyridine in 1,2-dichloroethane was treated with DDQ and stirred for the indicated time period.
See the Supporting Information for details on substrate synthesis.
d.r. = diastereomer ratio normalized to 100.
Yields refer to isolated, purified material.
Reaction was conducted at −30°C.
no reaction occurred.
A postulate for the steric differences of alkenes in models A and B is shown in Figure 2. The conjugation between the alkene and the oxocarbenium ion in model A forces the substituent on the alkene to project across the ether linkage in the disfavored configuration. The alkenyl groups in model B can orient their sterically undemanding flat faces toward the substituents across the ether linkage to minimize steric interactions. An electrostatic attraction between the π electrons and the electron deficient formyl hydrogen atom[18] could further stabilize this conformation. From a synthesis perspective the fact that alkenyl groups serve as the smaller substituent in this model illustrates that stereochemically complementary unsaturated products can be prepared by judicious substrate design.
Figure 2.

Steric interactions for alkenes in models A and B.
The applicability of models A and B to related reactions is shown in Scheme 3. The condensation of acetophenone (34) with homoallylic alcohol 35 in the presence of TMSCl and NaI[12c] provided, after radical-mediated removal of iodide, tetrahydropyran 36 as a single isomer. Aldehyde 38 condensed with tertiary alcohol 37 and AlCl3[19] and yielded chlorotetrahydropyran 39, also as a single stereoisomer. The yields of these reactions were rather low because of the competitive ionization and of the oxonia-Cope-type reactions,[20] but the procedures were not optimized because our main interest was focused on determining the stereochemical outcomes of these transformations.
Scheme 3.

Applications to acid-mediated Prins reactions. AIBN=azo-bisisobutyronitrile, TMS =trimethylsilyl.
In conclusion, we have shown that two models can be applied to design stereocontrolled reactions that yield tetra-hydropyrans with tertiary ethers. In one model the geometry of unsaturated 1,1-disubstituted oxocarbenium ions can be predicted based on the steric differences between the two substituents. The other model uses preexisting quaternary centers to control the geometry of oxocarbenium ions. While oxidative carbocation formation was employed to initiate the majority of the reactions in this study, these models are applicable to oxocarbenium ions derived from other processes and are consistent with the results of previously reported reactions that proceed through highly substituted ions.[3a,b,6] The generality of the models described here will serve as a guide for future efforts in the synthesis of molecules that contain fully substituted stereocenters.
Supplementary Material
Footnotes
We thank the National Institutes of Health and the Institute of General Medicine (GM062924) for their generous support of this work. We thank Prof. Paul Wender for valuable discussions.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201002281.
References
- 1.Recent reviews: Olier C, Kaafarani M, Gastaldi S, Bertrand MP. Tetrahedron. 2010;66:413.Pastor IM, Yus M. Curr Org Chem. 2007;11:925.
- 2.a) Denmark SE, Willson TM, Almstead NG. J Am Chem Soc. 1989;111:9258. [Google Scholar]; b) Sammakia T, Smith RS. J Am Chem Soc. 1992;114:10998. [Google Scholar]; c) Overman LE, Pennington LD. J Org Chem. 2003;68:7143. doi: 10.1021/jo034982c. [DOI] [PubMed] [Google Scholar]; d) Smith AB, III, Fox RJ, Razler TM. Acc Chem Res. 2008;41:675. doi: 10.1021/ar700234r. [DOI] [PubMed] [Google Scholar]
- 3.a) Nokami J, Ohga M, Nakamoto H, Matsubara T, Hussain I, Kataoka K. J Am Chem Soc. 2001;123:9168. doi: 10.1021/ja011257f. [DOI] [PubMed] [Google Scholar]; b) Lee CLK, Lee CHA, Tan KT, Loh TP. Org Lett. 2004;6:1281. doi: 10.1021/ol049633z. [DOI] [PubMed] [Google Scholar]; c) Chen YH, McDonald FE. J Am Chem Soc. 2006;128:4568. doi: 10.1021/ja061082f. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yuan Y, Lai AJ, Kraml CM, Lee C. Tetrahedron. 2006;62:11391. doi: 10.1016/j.tet.2006.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Barluenga J, Vázquez-Villa H, Ballesteros A, González JM. J Am Chem Soc. 2003;125:9028. doi: 10.1021/ja0355372. [DOI] [PubMed] [Google Scholar]; b) Pohlhaus PD, Sanders SD, Parsons AT, Li W, Johnson JS. J Am Chem Soc. 2008;130:8642. doi: 10.1021/ja8015928. [DOI] [PubMed] [Google Scholar]
- 5.Broeker JL, Hoffmann RW, Houk KN. J Am Chem Soc. 1991;113:5006. [Google Scholar]
- 6.a) Sassaman MB, Kotian KD, Prakash GKS, Olah GA. J Org Chem. 1987;52:4314. [Google Scholar]; b) Tietze LF, Kinzel T, Schmatz S. J Am Chem Soc. 2008;130:4386. doi: 10.1021/ja078032a. [DOI] [PubMed] [Google Scholar]; c) Checa B, Gálvez E, Parelló R, Sau M, Romea P, Urpí F, Font-Bardia M, Solans X. Org Lett. 2009;11:2193. doi: 10.1021/ol9005135. [DOI] [PubMed] [Google Scholar]; d) Castaldi MP, Troast DM, Porco JA., Jr Org Lett. 2009;11:3362. doi: 10.1021/ol901201k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang Y, Panek JS. Org Lett. 2009;11:3366. doi: 10.1021/ol901202t. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) McQuaid KM, Sames D. J Am Chem Soc. 2009;131:402. doi: 10.1021/ja806068h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar K, Waldmann H. Angew Chem. 2009;121:3272. doi: 10.1002/anie.200803437. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:3224. [Google Scholar]
- 8.Liu L, Floreancig PE. Angew Chem. 2010;122:3133. [Google Scholar]; Angew Chem Int Ed. 2010;49:3069. [Google Scholar]
- 9.a) Tu W, Liu L, Floreancig PE. Angew Chem. 2008;120:4252. doi: 10.1002/anie.200706002. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:4184. [Google Scholar]; b) Liu L, Floreancig PE. Org Lett. 2009;11:3152. doi: 10.1021/ol901188q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jung HH, Floreancig PE. Tetrahedron. 2009;65:10830. doi: 10.1016/j.tet.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Wan S, Gunaydin H, Houk KN, Floreancig PE. J Am Chem Soc. 2007;129:7915. doi: 10.1021/ja0709674. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Green ME, Rech JC, Floreancig PE. Angew Chem. 2008;120:7427. doi: 10.1002/anie.200802548. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:7317. [Google Scholar]
- 11.Ishihara K, Hanaki N, Yamamoto H. J Am Chem Soc. 1993;115:10695. [Google Scholar]
- 12.a) Li JK, Li CJ. Heterocycles. 2000;53:1691. [Google Scholar]; b) Ghosh AK, Shin D, Schiltz G. Heterocycles. 2002;58:659. doi: 10.3987/COM-02-S(M)63. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sabitha G, Reddy KB, Bhikshapathi M, Yadav JS. Tetrahedron Lett. 2006;47:2807. [Google Scholar]; d) Reddy UC, Bondalapati S, Saikia AK. J Org Chem. 2009;74:2605. doi: 10.1021/jo802531h. [DOI] [PubMed] [Google Scholar]
- 13.a) Ohira S. Synth Commun. 1989;19:561. [Google Scholar]; b) Müller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521. [Google Scholar]
- 14.Goossen LJ, Paetzold J, Koley D. Chem Commun. 2003:706. [PubMed] [Google Scholar]
- 15.Corey EJ, Helal CJ. Angew Chem. 1998;110:2092. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1998;37:1986. [Google Scholar]
- 16.Cremer D, Gauss J, Childs RF, Blackburn C. J Am Chem Soc. 1985;107:2435. [Google Scholar]
- 17.Rosenberg S, Leino R. Synthesis. 2009:2651. [Google Scholar]
- 18.Corey EJ, Lee TW. Chem Commun. 2001:1321. [Google Scholar]
- 19.Coppi L, Ricci A, Taddei M. J Org Chem. 1988;53:911. [Google Scholar]
- 20.a) Crosby SR, Harding JR, King CD, Parker GD, Willis CL. Org Lett. 2002;4:577. doi: 10.1021/ol0102850. [DOI] [PubMed] [Google Scholar]; b) Jasti R, Rychnovsky SD. J Am Chem Soc. 2006;128:13640. doi: 10.1021/ja064783l. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.