

Abstract
Transient global ischemia, as with cardiac arrest, causes loss of CA1 hippocampal neurons 2–4 d later, whereas nearby dentate gyrus (DG) neurons are relatively resistant. Whether differential astrocyte vulnerability in ischemic injury contributes to CA1 neuronal death is uncertain. Here, we find that CA1 astrocytes are more sensitive to ischemia than DG astrocytes. In rats subjected to transient forebrain ischemia, CA1 astrocytes lose glutamate transport activity and immunoreactivity for GFAP, S100β, and glutamate transporter GLT-1 within a few hours of reperfusion, but without astrocyte cell death. Oxidative stress may contribute to the observed selective CA1 changes, because CA1 astrocytes show early increases in mitochondrial free radicals and reduced mitochondrial membrane potential. Similar changes were not observed in DG astrocytes. Upregulation of GLT-1 expression in astrocytes with ceftriaxone protected CA1 neurons from forebrain ischemia. We suggest that greater oxidative stress and loss of GLT-1 function selectively in CA1 astrocytes is central to the well known delayed death of CA1 neurons.
Keywords: astrocyte, global ischemia, glutamate transporter, hippocampus, mitochondria, oxidative stress
Introduction
Global cerebral ischemia, as seen with cardiac arrest, causes delayed loss of CA1 pyramidal neurons, whereas the nearby dentate gyrus (DG) area is relatively resistant. Patients who survive cardiac arrest face significant neurological disability because of the loss of CA1 neurons. Thus far, only hypothermia has been shown to improve neurological outcome in these patients (Bernard et al., 2002; THACAS Group, 2002). In animals, transient forebrain ischemia has been characterized using a global ischemia model; it causes delayed neuronal death selectively in hippocampal CA1 pyramidal neurons after 2–4 d of reperfusion, whereas the DG, CA3, and most cortical neurons remain essentially intact (Kirino, 1982; Pulsinelli, 1985). For many years, selective neuronal vulnerability has been presumed to reflect the inherent vulnerability and connectivity of the neuronal population. However, progress delineating these mechanisms has not been able to fully explain the long recognized selectivity of ischemic neuronal loss. We postulate that differences in astrocyte vulnerability or sensitivity to functional change are central to this regional neuronal loss.
Astrocytes are the most numerous cell type in the brain, dynamically involved in synaptic transmission, metabolic and ionic homeostasis, inflammatory response, antioxidant defense, trophic support of neurons, as well as in the establishment and maintenance of the blood–brain barrier (Kettenmann and Ransom, 2005). Hippocampal astrocytes occupy exclusive, nonoverlapping territories (Bushong et al., 2002), a pattern that develops in the early postnatal period in parallel with neuronal and vascular territories (Bushong et al., 2004). Astrocytes respond to signals from and signal actively to neurons, endothelium, and microglia (for review, see Ransom et al., 2003; Volterra and Meldolesi, 2005). Recent work has demonstrated a protective role of reactive astrocytes after spinal cord injury (Faulkner et al., 2004). One protective function of astrocytes during ischemia is glutamate uptake to limit excitotoxic injury of neighboring neurons (Rosenberg and Aizenman, 1989; Dugan et al., 1995). Glutamate excitotoxicity is one major mechanism underlying ischemic damage in the CNS (Benveniste, 1991; Choi, 1994; Siesjo et al., 1995; Swanson et al., 2004). Failure of uptake as well as release of glutamate by impaired astrocytes is thought to contribute to ischemic neuronal injury (Swanson et al., 2004).
Here, we investigated the early astrocyte response to ischemic injury comparing the selectively vulnerable CA1 region with the more resistant DG. This study provides evidence for the novel hypothesis that selective hippocampal astrocytic impairment is responsible for the selective loss of CA1 hippocampal neurons after global or forebrain ischemia. We demonstrate, for the first time, differential responses of astrocytes from different hippocampal subregions to ischemia: generation of reactive oxygen species, changes in mitochondrial membrane potential, and uptake of glutamate. Furthermore, we show that dramatic loss of GFAP immunoreactivity is not associated with astrocyte death. We provide evidence that early loss of glutamate transport contributes to neuronal loss because induction of higher GLT-1 levels in astrocytes before ischemia reduces neuronal death in slice and in vivo.
Materials and Methods
Forebrain ischemia, drug treatment, and assessment.
All experiments using animals were performed in accordance with a protocol approved by the Stanford University animal care and use committee and in keeping with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Forebrain ischemia in rats was induced with the two-vessel occlusion plus hypotension model (Smith et al., 1984) with some modifications. In brief, male Sprague Dawley rats weighing 270–330 g were anesthetized by face mask with isoflurane in oxygen:N2O (3:7). Hypotension (to 50 mmHg) was induced by removing blood through the jugular vein into heparinized sterile tubing before carotid clamping. At the end of 10 min, bilateral carotid occlusion recirculation was induced by reinfusing the shed blood and by releasing the clamps placed around the carotid arteries. At various time points after recirculation, the rats were killed, and the brains were removed. The treatment group was injected daily with ceftriaxone [200 mg/kg, i.p., for 5 d (Rothstein et al., 2005)] before 10 min forebrain ischemia.
Before removal, brains were perfused with saline then ice-cold 4% phosphate-buffered paraformaldehyde. Coronal sections were made with a vibratome at a thickness of 35 μm for immunohistochemistry or staining with cresyl violet to assess injury. In other experiments, acute brain slices obtained after forebrain ischemia were stained with sulforhodamine 101 (SR101) (Nimmerjahn et al., 2004). SR101 (2 μm) was dissolved in cell culture medium and applied for 5 min followed by two washes before observation.
Primary astrocyte cultures and organotypic brain slice cultures.
Primary astrocyte cultures were prepared from postnatal days 1–3 Swiss Webster mice (Simonsen, Gilroy, CA) as described previously (Dugan et al., 1995; Xu et al., 2001; Ouyang et al., 2006). Subregions of the hippocampus were isolated as described by Chen et al. (2005). In brief, hippocampi were isolated, and the dorsal region of the hippocampus containing primarily CA1 (Chen et al., 2005) was dissected free of the rest of the hippocampus. The ventral hippocampus containing DG was further reduced by removal of the CA3 region. Cultures were maintained at 37°C in a 5% CO2 incubator. The organotypic slice cultures were prepared as described previously (Ouyang et al., 2005).
Injury paradigms and assessment.
Two insults were studied in astrocyte and slice cultures. Glucose deprivation (GD) was done by removing glucose from the medium (Ouyang et al., 2006). Combined oxygen GD (OGD) was done in slice culture by anoxic exposure in medium meant to replicate ischemic extracellular fluid as described previously (Ouyang et al., 2005). For primary cultures, we used OGD to mimic ischemia (Xu et al., 2001). Ceftriaxone pretreatment of slice cultures was 100 μm for 5 d before OGD.
Cell death in primary astrocyte cultures was detected with the fluorescent cell death markers propidium iodide (PI) or SYTOX green. In hippocampal slice cultures, cell death was assessed by measuring mean fluorescence intensity of PI in a standardized area in the CA1 region subtracting the fluorescence in a square placed in an undamaged area outside of the CA2/CA3 cell band (background) as described previously (Ouyang et al., 2005).
Immunohistochemistry and Western blots.
After experiments, cell and hippocampal slice cultures were fixed with 4% phosphate-buffered paraformaldehyde. Fluorescence immunohistochemistry was performed as described previously (Ouyang et al., 2005) using antibodies against GFAP, GLT-1, S100β, and GLAST (glutamate–aspartate transporters) (Millipore, Bedford, MA). Western blots were performed using anti-GFAP and anti-GLT-1 antibodies.
Glutamate uptake.
Glutamate uptake was measured according to published methods (Vesce et al., 1997; Brooke and Sapolsky, 2003) using aspartate. Aspartate is taken up by the same transporters as glutamate because of their similar molecular weight but is not further metabolized (Drejer et al., 1983). Briefly, after ischemia-like injury, the cells were incubated in glucose-free medium with a mixture of 3H d-aspartate (0.5 μC/ml) and cold aspartate at 1 μm. After 7 min, the cells were washed four times with ice-cold PBS containing the transport inhibitor threo-β-hydroxyaspartic acid (1 mm), scraped with lysate buffer (1% Triton X-100), and counted by a scintillation counter for incorporated radioactivity.
Mitochondrial membrane potential and oxygen radical production.
Live cell imaging studies were performed as described previously (Ouyang et al., 2006) with some modifications. We used tetramethylrhodamine ethylester (TMRE) to assess mitochondrial membrane potential, and MitoSOX Red or hydroethidine (HEt) to assess oxygen radical production. Fluorescence changes were quantified (Ouyang et al., 2006) by selecting a cytoplasmic region of each cell that was strongly fluorescent at baseline and normalizing subsequent fluorescence measurements to the basal fluorescence for each cell at the start of the experiment.
Statistics.
All data represent three or more independent experiments. Data are reported as mean ± SD. Statistical differences were determined using ANOVA followed by Scheffes or Tukey test with p < 0.05, the level considered significant, using StatView by SAS Institute (Carey, NC) or Instat from GraphPad Software (San Diego, CA).
Results
Differences in immunoreactivity between CA1 and DG astrocytes after transient in vivo and in vitro ischemia
To define early changes in CA1 astrocytes, we performed immunohistochemical studies at early reperfusion times in rats subjected to 10 min forebrain ischemia. We evaluated immunoreactivity for GFAP (Fig. 1A), an astrocyte-specific intermediate filament protein, and for the astrocytic glutamate transporter GLT-1 (Fig. 1B), which plays a key role in limiting neuronal excitotoxicity. There were obvious differences observed between CA1 and DG. At 5 and 12 h of reperfusion, there was a marked reduction in GFAP staining of astrocytes in the CA1 region, whereas the neuronal layer still looked normal (Fig. 1A). By 2 d of reperfusion, there was clear evidence of astrocyte activation and hypertrophy with increased GFAP staining and the relative disappearance of the CA1 neuronal cell body layer evident in the PI-stained panel. The GFAP immunostaining in DG was not lost at early reperfusion intervals (Fig. 1A), although Western blot did show some reduction at 5 h of reperfusion (Fig. 1C), which was less than the reduction observed in CA1. At 5 h reperfusion after 10 min forebrain ischemia, we saw a marked reduction of GLT-1 staining in CA1 but not in DG (Fig. 1B). This was confirmed by Western blot of GLT-1 protein that demonstrated the characteristic wide, poorly defined bands (Fig. 1C) at ∼65 kDa (Danbolt, 2001). In our studies, GLT-1 staining in the CA1 region remained reduced for up to 24 h of reperfusion, with slow recovery to control values after 2–3 d (data not shown), by which time the CA1 pyramidal neuronal layer had already disappeared. Hippocampal slices subjected to transient in vitro ischemia (OGD) (Fig. 1D) exhibited reduced GLT-1 immunoreactivity at early recovery (2 h) after 15 min OGD in the CA1 region, whereas the DG region still showed strong staining, similar to the in vivo observations.
Figure 1.

Forebrain ischemia-induced changes in GFAP and GLT-1 immunoreactivity in CA1 and DG hippocampal regions. A, GFAP immunostaining in the hippocampal CA1 and DG regions of rats subjected to 10 min forebrain ischemia followed by the indicated durations of reperfusion (R). Green is GFAP immunoreactivity, whereas red is PI staining for nuclei. GFAP staining was strikingly decreased during early reperfusion in CA1 compared with DG. Scale bars: top, 80 μm; bottom, 60 μm. B, Changes in GLT-1 staining in hippocampal CA1 and DG regions of a rat subjected to 10 min forebrain ischemia followed by 5 h recovery (R). Scale bar, 40 μm. C, Quantification of the changes by Western blot in GFAP and GLT-1 staining in hippocampal CA1 and DG regions of rats subjected to 10 min forebrain ischemia followed by 5 h recovery (R). n = 3; *p < 0.05 indicates statistically different from control for the same region. Each lane shows protein from a different animal. D, GLT-1 immunoreactivity in hippocampal CA1 and DG regions of organotypic hippocampal brain slices subjected to 15 min OGD followed by 2 h recovery. Scale bars: (in bottom left panel) left panels, 2 mm; (in bottom right panel) middle and right panels, 25 μm. Ctrl, Control group.
We took advantage of subregional primary cultures of astrocytes to examine, in greater detail, the effects of varying ischemic severity (Fig. 2A). Astrocytes isolated from CA1 lost GLT-1 staining by the end of 1 h ischemia (OGD) with modest reduction of GFAP labeling. At 5 h recovery, GFAP staining was almost gone from CA1 astrocytes, whereas DG astrocytes still had no significant changes. Although there were no changes of GLT-1 or GFAP staining in DG astrocytes after 1 h OGD, after 2 h OGD and 2 h recovery, GLT-1 was decreased in DG astrocytes. GFAP staining, which was markedly decreased in CA1, was still normal in DG astrocytes despite the more severe insult. In summary, we observed that decreased immunoreactivity for GLT-1 occurred first, followed by loss of GFAP immunoreactivity in both CA1 and DG astrocytes, but less severe insults induced these changes in CA1 astrocytes. Greater sensitivity of CA1 astrocytes to functional change was observed in all three models of ischemic hippocampal injury. This suggests that the greater sensitivity is in part intrinsic to CA1 astrocytes, because it was still present in culture in the absence of neurons. Because immunoreactivity does not report directly about function, we also investigated glutamate transport activity. As shown in Figure 2B, a significant decrease of >40% in the rate of glutamate uptake by CA1 astrocyte cultures was observed after 2 h of OGD followed by 2 h of recovery, assessed by 3H aspartate uptake. The same treatment induced no significant change in uptake by DG cultures.
Figure 2.
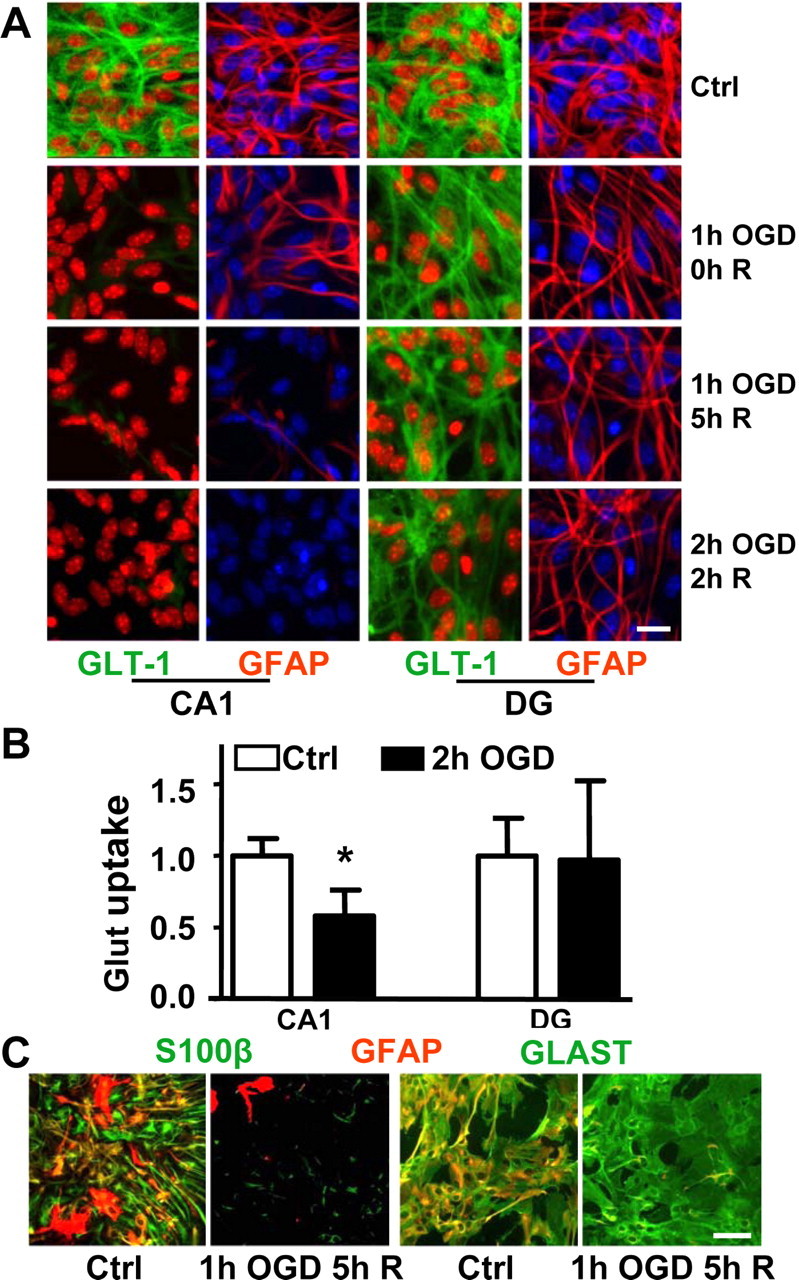
GFAP, GLT-1, S100β, and GLAST immunoreactivity and glutamate uptake in astrocytes from CA1 and DG hippocampal regions after OGD. A, Comparison of GFAP and GLT-1 immunostaining with different durations of OGD and recovery (R) time. Green, GLT-1; red, GFAP; blue, Hoechst for nuclei (for better contrast, the nuclei were pseudocolored red in the GLT-1 stained columns). Scale bar, 20 μm. B, Changes in glutamate (Glut) uptake activity in cultured astrocytes from hippocampal CA1 and DG regions after 2 h of OGD followed by 1 h of recovery. Values of glutamate uptake are a ratio to control and expressed as mean ± SD of three separate experiments, each performed in triplicate (*p < 0.05 compared with CA1 at 0 h time point). C, Comparison of GFAP, S100β (left two panels), and GLAST (right two panels) immunoreactivity at 5 h recovery (R) after 1 h OGD in astrocytes from the hippocampal CA1 region. Green, S100β and GLAST; red, GFAP; yellow, overlap. Scale bar, 60 μm. Ctrl, Control group.
To determine whether the rapid decrease of GLT-1 and GFAP staining is accompanied by the disappearance of other immunocytochemical epitopes in CA1 astrocytes, we tested additional astrocytic markers at 5 h after 1 h OGD (Fig. 2C). Both S100β, a calcium binding protein, and GFAP immunolabeling were markedly decreased at 5 h recovery (Fig. 2C). However, immunostaining for GLAST, another astrocytic glutamate transporter, did not change much at this time point (Fig. 2C), which is consistent with other reports (Yeh et al., 2005). This suggested that astrocytes were not dead because some astrocytic markers remained unchanged.
CA1 astrocyte death is minimal when GFAP and GLT-1 immunoreactivity is reduced after ischemia
Whether significant astrocyte death occurs after forebrain ischemia in vivo as well as in slice and culture models is an important question. We demonstrated, based on previous reports (Nimmerjahn et al., 2004), that SR101, an astrocyte-specific dye, is taken up by live but not dead astrocytes (Fig. 3A). We found that there was no significant increase in either apoptotic or necrotic death in CA1 astrocytic cultures after insults that reduced GFAP and GLT-1 immunoreactivity (Fig. 3B). Similar observations were made in acute slices of CA1 region after 10 min forebrain ischemia in vivo (Fig. 3C–E) by SR101 staining.
Figure 3.

Astrocytic viability at times corresponding to the loss of GFAP and GLT-1 staining. A, Left, SYTOX green specifically labels nuclei of dead cells in mildly injured primary cortical astrocyte cultures, whereas SR101 red staining is only observed in viable cells that exclude SYTOX green. Right, SR101 red staining colocalizes with the viability dye calcein green (resulting in yellow) in live cells; cell nuclei are stained with Hoechst (blue). Scale bars, 100 μm. B, Primary astrocyte cultures from the CA1 region subjected to ischemic injury sufficient to induce loss of GLT-1 and GFAP immunostaining do not demonstrate signs of apoptotic or necrotic cell death. Quantitation of cell death in astrocytic primary cultures after 2 and 3 h OGD (3 independent experiments; n = 600–700 cells per condition) is shown. The cells were stained with PI and polycaspase green fluorescent substrate to count necrotic and apoptotic cells, respectively, after 2 h of reperfusion. C, SR101 staining and nuclear DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) staining of brain sections from controls and rats subjected to 10 min of forebrain ischemia and 5 h of reperfusion (R). Acute slices subjected to a lethal insult (1 h OGD) demonstrate no SR101 astrocytic staining. The average SR101 intensity was quantified in 300 × 600 μm areas (white boxes). D, Higher-power views of 300 × 600 μm boxes indicated in CA1 regions show similar SR101 staining of viable astrocytes in forebrain ischemia and uninjured control. E, Quantification of astrocyte viability by SR101 staining in CA1 regions from rats subjected to dense forebrain ischemia (FI), sham control, and lethally injured acute hippocampal sections. Average SR101 intensity was measured in similarly positioned 300 × 600 μm areas of CA1 of 400 μm acute hippocampal slices −2.8 to −4.4 mm bregma, seven sections per condition from three or four animals per condition. *p < 0.05. Ctrl, Control group.
Possible mechanisms of selective CA1 astrocyte dysfunction: oxidative stress and mitochondrial dysfunction
GLT-1 is susceptible to oxidative damage (Lauderback et al., 2001). To test the hypothesis that differential mitochondrial response to stress and generation of reactive oxygen species (ROS) could contribute to the differential response of astrocytes from the two regions, we used cultured astrocytes isolated from the CA1 and DG regions and monitored mitochondrial membrane potential using TMRE (Fig. 4A) and generation of ROS using MitoSOX red (Fig. 4B) and HEt (Fig. 4C). CA1 astrocytes were more susceptible to reduction of mitochondrial membrane potential and increased generation of ROS compared with DG astrocytes challenged with the same insult. As shown in Figure 4A, after 1 h GD, the TMRE signal from DG astrocytes was constant, whereas the signal from CA1 astrocytes was reduced ∼15%, indicating partial mitochondrial depolarization. The use of the mitochondrially localized ROS sensitive dye MitoSOX red (Fig. 4B) showed increased ROS in CA1, but not in DG astrocytes after 15 min GD. After 30 min, the oxidized dye appeared to move into nuclei, and this occurred earlier in CA1 compared with DG astrocytes. We also used HEt to detect ROS (Fig. 4C). After 1 h GD, the HEt signal approximately doubled in DG astrocytes, whereas in CA1 astrocytes it was significantly higher, reaching ∼3.5 times the fluorescence intensity at baseline (Fig. 4C).
Figure 4.

Mitochondrial changes after GD. A, Changes in TMRE fluorescence with 1 h GD in cultured astrocytes. Left panels, micrographs of representative fields. Scale bar, 25 μm. *p < 0.05. The graph depicts the means of 10–15 cells per condition. B, Changes in mitochondrial ROS indicated by live imaging of MitoSOX Red fluorescence during GD in cultured astrocytes. Left panels, CA1 astrocytes; Right panels, DG. C, Changes in ROS indicated by HEt fluorescence during GD in cultured astrocytes. Left panels show representative micrographs. The graph depicts the means of 10–15 cells per condition. *p < 0.05 statistically different from CA1 0 h time point and from DG at the same time point using ANOVA followed by Scheffes test. D, Rotenone (50 nm) induced mitochondrial production of ROS and decreased GLT-1 immunostaining in CA1 subregion astrocytes. Red, HEt; green, GLT-1. Rot, Rotenone; Ctrl, control group. Scale bar, 25 μm.
One of the major sites of ROS production in cells is the mitochondrial respiratory chain (Turrens and Boveris, 1980). To further investigate the relationship between the activity of GLT-1 and mitochondrially generated ROS, we exposed cultured CA1 and DG astrocytes to rotenone, a classical inhibitor of mitochondrial complex I. After loading the astrocytes with HEt, control images were taken and then rotenone (50 nm) was added. After 30 min of rotenone exposure, there was a significant increase of HEt fluorescence and obvious decrease in GLT-1 immunostaining at the same time (Fig. 4D) in both CA1 and DG astrocytes. These results indicate that the greater generation of ROS from mitochondria observed in CA1 astrocytes after ischemia may contribute to the loss of GLT-1.
Upregulation of astrocytic GLT-1 decreases CA1 neuronal death
A study by Rothstein et al. (2005) demonstrated that the β-lactam antibiotic ceftriaxone could upregulate GLT-1 in astrocytes and resulted in some protection from excitotoxicity in vitro and from a model of amyotrophic lateral sclerosis in vivo. We therefore tested ceftriaxone and found that it did induce higher expression of GLT-1 in CA1 in hippocampal slice culture treated with 100 μm of ceftriaxone for 5 d (Fig. 5A). Note that in the slice control, GLT-1 staining shows a typical astrocytic pattern leaving the neuronal cell body layer relatively unstained. When overexpressed after drug treatment, astrocyte staining was so bright in CA1 that the neuronal cell body layer was relatively obscured. We found pretreatment with ceftriaxone reduced CA1 neuronal loss in slice cultures subjected to 15 min OGD followed by 24 h recovery (Fig. 5B,C). To determine whether this method of protection would also work in vivo, we pretreated animals with ceftriaxone before subjecting them to 10 min forebrain ischemia. We found that treatment with ceftriaxone increased expression of GLT-1 in the hippocampus (Fig. 5D) and reduced delayed CA1 neuronal death as shown by cresyl violet staining after 7 d of reperfusion (Fig. 5E,F). To further confirm that ceftriaxone specifically induces GLT-1, we also stained the slice cultures and brain sections from animals pretreated with ceftriaxone with GFAP, S100β, and GLAST antibodies. There were no significant differences in staining between untreated and ceftriaxone pretreated slice cultures or brain sections (data not shown). Thus, increased GLT-1 expression in astrocytes results in decreased susceptibility of neighboring neurons to ischemic injury after brief forebrain ischemia.
Figure 5.

Ceftriaxone induces GLT-1 expression and reduces CA1 delayed neuronal injury in hippocampal slice culture and in vivo. A, Ceftriaxone pretreatment (+Cef; 100 μm for 5 d) strongly induces expression of GLT-1 immunoreactivity (green) in hippocampal slice cultures, especially in the CA1 subregion, compared with the vehicle-treated control (−Cef). B, Bar graph shows the significant reduction of CA1 neuronal damage in ceftriaxone-treated (+Cef) slices compared with vehicle-treated (−Cef) ones by relative PI fluorescence in hippocampal slice culture 24 h after 15 min OGD. n = 9 slices per condition; *p < 0.05 indicates significant difference from control. C, Fluorescence images of representative PI-stained (red) hippocampal slices 24 h after 15 min OGD. Decreased PI fluorescence indicates decreased CA1 neuronal death in ceftriaxone-treated (+Cef) slices compared with vehicle-treated (−Cef) ones. Scale bar, 400 μm. D, Pretreatment of rats with ceftriaxone (200 mg/kg, i.p., daily for 5 d) strongly induces expression of GLT-1 throughout the hippocampus. E, Bar graph shows the reduction of CA1 neuronal damage in ceftriaxone-treated (+Cef) animals compared with vehicle treated (−Cef) rats by cresyl violet staining intensity. n = 4 in each group; *p < 0.05 significantly different from control. F, Cresyl violet staining of representative brain slices from control and ischemic rats pretreated with ceftriaxone (+Cef) or vehicle (−Cef). More pyramidal neurons were present in ceftriaxone-treated brains after cresyl violet staining compared with those in the vehicle-treated group after 7 d reperfusion. Ctrl, Control group; R, reperfusion.
Discussion
It is often said that neurons are the cells most vulnerable to ischemia. This view is based primarily on observations that astrocytes in culture show greater resistance than neurons to some ischemia-like insults (Goldberg and Choi, 1993; Xu et al., 2001) and that, as noted above, brief forebrain ischemia apparently results in selective loss of neurons. However, vulnerability of cultured astrocytes is markedly increased by acidic conditions relevant to ischemic injury (Giffard et al., 1990; Bondarenko and Chesler, 2001). Several laboratories have now pointed out that glial cells may be injured more readily than previously thought, sometimes before neuronal damage is obvious (Petito et al., 1998; Liu et al., 1999; Zhao et al., 2003). Astrocytic processes were fragmented and mitochondria inhibited after 15 min exposure to acidic conditions in hippocampal slice cultures (Hulse et al., 2001). Astrocytic demise was found to precede delayed neuronal death after focal ischemia (Liu et al., 1999) and was observed early after traumatic brain injury (Zhao et al., 2003). Selective glial vulnerability was seen after transient global ischemia primarily involving oligodendrocytes (Petito et al., 1998). In contrast to these findings, the present study shows that at early reperfusion times after transient forebrain ischemia, astrocytes displayed functional changes without any loss of viability. We previously observed that astrocytes, as well as neurons, isolated from different brain regions showed different responses to stress (Xu et al., 2001).
Although these studies suggest that astrocytic change can precede neuronal death in some situations, there is relatively little information currently available about differences in astrocyte response to ischemia in the selectively vulnerable CA1 region of the hippocampus, compared with the less readily injured DG region. In the present study, we investigated the early temporal and spatial profile of astrocyte changes in hippocampal CA1 and DG regions in response to ischemia. We demonstrated that (1) there is a more rapid decrease in GLT-1 immunoreactivity and glutamate uptake activity in CA1 astrocytes compared with DG astrocytes, (2) these early changes are functional and are not accompanied by CA1 astrocyte death, (3) early increases in mitochondrial ROS in CA1 astrocytes may in part explain the loss of GLT-1 transport activity, and (4) the loss of GLT-1 is a relevant mechanism of CA1 neuronal injury because upregulation of astrocytic GLT-1 decreased CA1-delayed neuronal death in vitro and in vivo. A diagram showing this model of injury is shown in Figure 6. We used rats for the forebrain ischemic model and mice for the primary astrocyte cultures and slice cultures. Although there are some species differences between rats and mice, the similarity of results obtained in the current study suggests that the mechanism of injury identified here is present in both species.
Figure 6.

Proposed mechanism of astrocyte contribution to delayed neuronal death. Transient forebrain ischemia selectively decreases mitochondrial membrane potential (Δψm) and increases ROS in CA1 astrocytes. The greater production of ROS leads to astrocyte impairment including oxidative damage of GLT-1 on the astrocyte membrane. The loss of GLT-1 contributes to the increase of extracellular glutamate and excitotoxicity of the pyramidal neurons.
To date, five brain glutamate transporters have been cloned, excitatory amino acid transporters 1–5 (EAAT1–5). Of these, GLT-1/EAAT2 and GLAST/EAAT1 are expressed in astrocytes and play a crucial role in preventing glutamate excitotoxicity (Gegelashvili and Schousboe, 1997). Reduction or loss of astroglial transporters, GLT-1 or GLAST, led to a tonic increase in extracellular glutamate concentration and subsequent neurodegeneration (Rothstein et al., 1996; Mitani and Tanaka, 2003). GLT-1 appears to be especially important in the hippocampus, because mice lacking GLT-1 showed spontaneous seizures, selective hippocampal neurodegeneration, and exacerbation of acute cortical injury (Tanaka et al., 1997), whereas mice deficient in GLAST were more susceptible to cerebellar injury (Watase et al., 1998). In addition, downregulation of glial EAAT1/GLAST or EAAT2/GLT-1 glutamate transporters, but not the neuronal subtype EAAT3/EAAC1 (excitatory amino acid carrier 1), has been shown to increase ischemic injury after focal cerebral ischemia in rats (Watase et al., 1998; Fukamachi et al., 2001; Rao et al., 2001).
Direct evidence has recently been provided that glial glutamate transporters are lost with hypoxia in the neonatal pig brain (Pow et al., 2004) and that hippocampal astrocytic glutamate uptake is impaired after global ischemia (Chen et al., 2005; Yeh et al., 2005). Our studies demonstrate, for the first time, that differential astrocytic glutamate transporter protein levels and activity contribute to the differential sensitivity of CA1 and DG regions to ischemia. Decrease in astrocyte glutamate uptake would likely be associated with increased susceptibility of neighboring neurons to excitotoxic injury. This provides a new variation on the old idea of excitotoxicity contributing to selective CA1 neuronal injury: we suggest that early loss of astrocyte glutamate uptake is likely attributable in part to oxidative damage in CA1 astrocytes, which contributes to the selective vulnerability of CA1 neurons (Fig. 6).
This mechanism is reminiscent of that suggested to occur in amyotrophic lateral sclerosis. Work by Weiss and colleagues (Rao et al., 2003) showed that motor neurons only died after glutamate transport activity was lost in astrocytes surrounding the neurons. This work suggested that oxidative stresses, possibly emanating from neurons, eventually compromised astrocyte function, which then precipitated neuronal death. Oxidative stress has also been shown to downregulate astrocytic glutamate uptake in ammonia-induced toxicity (Jayakumar et al., 2006). In CA1 after transient forebrain ischemia, increased generation of ROS, possibly in part resulting from CA1 neurons, but also, as suggested here, originating in astrocytes, compromises astrocyte function, which in turn compromises the ability of the CA1 pyramidal neurons to survive.
Mitochondrial dysfunction has been implicated in the selective vulnerability of CA1 cells after ischemia, because cell death in this region after global ischemia is inhibited by Cyclosporin A, a known inhibitor of the calcium-induced mitochondrial permeability transition in brain mitochondria (Uchino et al., 1995). The observation that Cyclosporin A can protect mitochondria from calcium insult in astrocytes but not in neurons (Bambrick et al., 2006) suggests astrocyte mitochondria are central to protection by this drug in vivo. The mechanism of selective CA1 astrocyte dysfunction we propose here is as a result of differences in mitochondrial response, leading to greater production of ROS in CA1 astrocytes. This leads to greater energetic compromise, oxidative damage, and loss of GLT-1 function. Oxidative stress is known to contribute to damage and misfolding of proteins, including glutamate transporters (Lauderback et al., 2001). The impaired CA1 astrocytes may be unable to carry out many of their normal functions, such as glutamate uptake, antioxidant defense, and regulation of extracellular ions. Consistent with our results is a report from Mattiason et al. (2003) who found that isolated mitochondria from the hippocampal CA1 subregion produced more ROS and were more sensitive to calcium-induced swelling than mitochondria isolated from the adjacent subregion, CA3 (Mattiasson et al., 2003). This work, however, did not separate neurons and astrocytes.
In conclusion, our study indicates that early changes in CA1 astrocyte immunoreactivity and function precede the degeneration of CA1 neurons by more than 1 d, so selective vulnerability of CA1 neurons may be secondary to selective ischemia-induced astrocyte dysfunction. It suggests that greater attention should be paid to developing strategies to protect normal astrocyte physiology and function. Although not a model of cardiac arrest, forebrain ischemia induces CA1 loss similar to that seen in global cerebral ischemia and cardiac arrest and may therefore be relevant. Furthermore, similar mechanisms of astrocyte impairment could contribute to neuronal demise in other types of brain injury or neurological disease.
Footnotes
This work was supported in part by National Institutes of Health Grants NS053898, GM49831, NS014543, and NS037520 to R.G.G. We thank Heng Zhao for help with the transient forebrain ischemia model, Bruce Winegar and Bruce MacIver for help with acute hippocampal slices, and Robert Sapolsky and Shiela Brooks for help with the glutamate uptake assays.
References
- Bambrick LL, Chandrasekaran K, Mehrabian Z, Wright C, Krueger BK, Fiskum G. Cyclosporin a increases mitochondrial calcium uptake capacity in cortical astrocytes but not cerebellar granule neurons. J Bioenerg Biomembr. 2006;38:43–47. doi: 10.1007/s10863-006-9004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste H. The excitotoxin hypothesis in relation to cerebral ischemia. Cerebrovasc Brain Metab Rev. 1991;3:213–245. [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Chesler M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001;34:134–142. doi: 10.1002/glia.1048. [DOI] [PubMed] [Google Scholar]
- Brooke SM, Sapolsky RM. Effects of glucocorticoids in the gp120-induced inhibition of glutamate uptake in hippocampal cultures. Brain Res. 2003;972:137–141. doi: 10.1016/s0006-8993(03)02517-4. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Ellisman MH. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci. 2004;22:73–86. doi: 10.1016/j.ijdevneu.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Chen JC, Hsu-Chou H, Lu JL, Chiang YC, Huang HM, Wang HL, Wu T, Liao JJ, Yeh TS. Downregulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology. 2005;49:703–714. doi: 10.1016/j.neuropharm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Drejer J, Larsson OM, Schousboe A. Characterization of uptake and release processes for D- and L-aspartate in primary cultures of astrocytes and cerebellar granule cells. Neurochem Res. 1983;8:231–243. doi: 10.1007/BF00963923. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Bruno VM, Amagasu SM, Giffard RG. Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–4555. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukamachi S, Furuta A, Ikeda T, Ikenoue T, Kaneoka T, Rothstein JD, Iwaki T. Altered expressions of glutamate transporter subtypes in rat model of neonatal cerebral hypoxia-ischemia. Brain Res Dev Brain Res. 2001;132:131–139. doi: 10.1016/s0165-3806(01)00303-0. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Schousboe A. High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol. 1997;52:6–15. doi: 10.1124/mol.52.1.6. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Monyer H, Choi DW. Selective vulnerability of cultured cortical glia to injury by extracellular acidosis. Brain Res. 1990;530:138–141. doi: 10.1016/0006-8993(90)90670-7. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse RE, Winterfield J, Kunkler PE, Kraig RP. Astrocytic clasmatodendrosis in hippocampal organ culture. Glia. 2001;33:169–179. doi: 10.1002/1098-1136(200102)33:2<169::aid-glia1016>3.0.co;2-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakumar AR, Panickar KS, Murthy Ch R, Norenberg MD. Oxidative stress and mitogen-activated protein kinase phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes. J Neurosci. 2006;26:4774–4784. doi: 10.1523/JNEUROSCI.0120-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Ransom B. Neuroglia. Ed 2. New York: Oxford UP; 2005. [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1–42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Liu D, Smith CL, Barone FC, Ellison JA, Lysko PG, Li K, Simpson IA. Astrocytic demise precedes delayed neuronal death in focal ischemic rat brain. Brain Res Mol Brain Res. 1999;68:29–41. doi: 10.1016/s0169-328x(99)00063-7. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Friberg H, Hansson M, Elmer E, Wieloch T. Flow cytometric analysis of mitochondria from CA1 and CA3 regions of rat hippocampus reveals differences in permeability transition pore activation. J Neurochem. 2003;87:532–544. doi: 10.1046/j.1471-4159.2003.02026.x. [DOI] [PubMed] [Google Scholar]
- Mitani A, Tanaka K. Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J Neurosci. 2003;23:7176–7182. doi: 10.1523/JNEUROSCI.23-18-07176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Xu L, Giffard RG. Geldanamycin treatment reduces delayed CA1 damage in mouse hippocampal organotypic cultures subjected to oxygen glucose deprivation. Neurosci Lett. 2005;380:229–233. doi: 10.1016/j.neulet.2005.01.055. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Xu LJ, Sun YJ, Giffard RG. Overexpression of inducible heat shock protein 70 and its mutants in astrocytes is associated with maintenance of mitochondrial physiology during glucose deprivation stress. Cell Stress Chaperones. 2006;11:180–186. doi: 10.1379/CSC-182R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petito CK, Olarte JP, Roberts B, Nowak TS, Jr, Pulsinelli WA. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol. 1998;57:231–238. doi: 10.1097/00005072-199803000-00004. [DOI] [PubMed] [Google Scholar]
- Pow DV, Naidoo T, Lingwood BE, Healy GN, Williams SM, Sullivan RK, O'Driscoll S, Colditz PB. Loss of glial glutamate transporters and induction of neuronal expression of GLT-1B in the hypoxic neonatal pig brain. Brain Res Dev Brain Res. 2004;153:1–11. doi: 10.1016/j.devbrainres.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M. New roles for astrocytes (stars at last) Trends Neurosci. 2003;26:520–522. doi: 10.1016/j.tins.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Rao SD, Yin HZ, Weiss JH. Disruption of glial glutamate transport by reactive oxygen species produced in motor neurons. J Neurosci. 2003;23:2627–2633. doi: 10.1523/JNEUROSCI.23-07-02627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VL, Dogan A, Todd KG, Bowen KK, Kim BT, Rothstein JD, Dempsey RJ. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Neurosci. 2001;21:1876–1883. doi: 10.1523/JNEUROSCI.21-06-01876.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex [Erratum (1990) 116:399] Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Katsura K, Zhao Q, Folbergrova J, Pahlmark K, Siesjo P, Smith ML. Mechanisms of secondary brain damage in global and focal ischemia: a speculative synthesis. J Neurotrauma. 1995;12:943–956. doi: 10.1089/neu.1995.12.943. [DOI] [PubMed] [Google Scholar]
- Smith ML, Bendek G, Dahlgren N, Rosen I, Wieloch T, Siesjo BK. Models for studying long-term recovery following forebrain ischemia in the rat. 2. A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385–401. doi: 10.1111/j.1600-0404.1984.tb07822.x. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- THACAS Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino H, Elmer E, Uchino K, Lindvall O, Siesjo BK. Cyclosporin A dramatically ameliorates CA1 hippocampal damage following transient forebrain ischaemia in the rat. Acta Physiol Scand. 1995;155:469–471. doi: 10.1111/j.1748-1716.1995.tb09999.x. [DOI] [PubMed] [Google Scholar]
- Vesce S, Bezzi P, Rossi D, Meldolesi J, Volterra A. HIV-1 gp120 glycoprotein affects the astrocyte control of extracellular glutamate by both inhibiting the uptake and stimulating the release of the amino acid. FEBS Lett. 1997;411:107–109. doi: 10.1016/s0014-5793(97)00674-1. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Xu L, Sapolsky RM, Giffard RG. Differential sensitivity of murine astrocytes and neurons from different brain regions to injury. Exp Neurol. 2001;169:416–424. doi: 10.1006/exnr.2001.7678. [DOI] [PubMed] [Google Scholar]
- Yeh TH, Hwang HM, Chen JJ, Wu T, Li AH, Wang HL. Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol Dis. 2005;18:476–483. doi: 10.1016/j.nbd.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG. Early loss of astrocytes after experimental traumatic brain injury. Glia. 2003;44:140–152. doi: 10.1002/glia.10283. [DOI] [PubMed] [Google Scholar]