

Abstract
Novel heteroatom-incorporated antofine and cryptopleurine analogs were designed, synthesized, and tested against a panel of five cancer cell lines. Two new S-13-oxo analogs (11 and 16) exhibited potent cell growth inhibition in vitro (GI50: 9 nM and 20 nM). Interestingly, both compounds displayed improved selectivity among different cancer cell lines, in contrast to the natural products antofine and cryptopleurine. MOAa studies suggested that R-antofine promotes dysregulation of DNA replication during early S phase, while no similar effects were observed for 11 and 15 on corresponding replication initiation complexes. Compound 11 also showed greatly reduced cytotoxicity against normal cells and moderate antitumor activity against HT-29 human colorectal adenocarcinoma xenograft in mice without overt toxicity.
Introduction
Phenanthroindolizidines and phenanthroquinolizidines, which are natural alkaloids isolated from several plant species such as the Asclepiadacae and Moracae family,1 have been investigated intensively in recent years, due to their interesting biological characteristics, including antimicrobial, anti-angiogenic, and anti-inflammatory effects,2–8 as well as significant cytotoxicity as demonstrated in the NCI 60 cell-line assay (Figure 1).9 Further studies have shown that these natural products not only exhibit strong inhibitory activity against cancer cell growth, but also significant effects on cancer cells resistant or cross-resistant to many anticancer drugs in the market.10 Thus, this important class of chemical entities may potentially augment our present arsenal of anticancer drugs. 11, 12
Figure 1.

Representative structures of phenanthroindolizidines and phenanthroquinolizidines
Although the biological target(s) and MOA of these natural products are currently still unclear, some interesting findings have been reported. A possible mechanism of action might be inhibition of NF-κB signaling, a well-known pathway in the anti-apoptosis and survival of cancer cells, as well as regulation of P-glycoprotein.13 Other hypotheses, such as inhibition of protein synthesis during chain elongation stage,14 targeting ribosomal subunits (low-affinity binding pockets have been identified in the 40S, 60S, 70S, and 80S subunits),15–17 inhibition of hypoxia-inducible factor 1 (HIF-1),18 inhibition of thymidylate synthase (TS) and dihydrofolate reductase (DHFR),19–21 suppression of activator protein-1 (AP-1) and cAMP response element (CRE) signaling pathway, and reduction of cell cycle regulatory proteins such as cyclin D1, cyclin B1, CDK1, CDK2, and CDK4 etc.10 have also been reported. In addition, evidence has suggested that certain structural analogs might not be functional analogs,22 and thus, multiple biological targets might exist.
Despite their promise, the potential development of antofine, cryptopleurine, or related natural or synthetic alkaloids as promising drug candidates has been limited, to a large degree, by severe CNS toxicity, such as disorientation and ataxia,23 and low natural availability. All of these factors combine to justify an urgent need for improving the pharmacokinetic and pharmacodynamic properties, such as polarity, of this compound class through rational structural modifications. However, only limited studies have been reported in this regard, e.g., introduction of a hydroxy group at C14 of phenanthroindolizidines,24 construction of phenanthrene-based tylophorine analogs,25–27 and N atom incorporation at C13a of tylophorine,28 probably due to the lack of a convergent and efficient synthetic methodology. Recently, our group reported a new strategy suitable for producing numerous new phenanthroindolizidine and phenanthroquinolizidine analogs with E-ring modifications.29 The versatility and significance of this approach lie in the fact that not only does it facilitate the SAR study of E-ring variations, but also, even more importantly, a group of analogs carrying a heterocyclic E-ring or other polar moieties can be generated to potentially overcome CNS toxicity. CNS toxicity is closely associated with the blood-brain barrier (BBB) penetration of a molecule, which is in large part related to its physicochemical properties, such as lipophilicity (cLogP) or polarity, molecular size and charge, polar surface area (PSA), or hydrogen bonding potential, etc., although other factors may also play a role.30–32 Herein, we report the design, synthesis, in vitro anticancer activity, SAR, and mechanistic studies of new antofine and cryptopleurine derivatives with a N or O atom incorporated in the E-ring. The in vivo antitumor activity for the most active compound (11) is also reported.
Results and discussion
Initially, antofine analogs bearing a N atom at position C12 were designed and synthesized. The key intermediate 1 was prepared via a procedure recently reported by our group. 27 The amino group of 1 (S or R isomer) was protected initially with a Boc group to give 2. Then the hydroxy group was oxidized with Py•SO3 to an aldehyde, which was then converted to various secondary amines through reductive amination using NaBH3CN. After removal of the Boc group, 3a-3l were obtained through cyclization with formaldehyde (Scheme 1).
Scheme 1.

Reagents and conditions: (a) (Boc)2O, Et3N, CH2Cl2 (b) i) Py•SO3, DMSO, Et3N, CH2Cl2; ii) RNH2, HOAc, NaBH3CN, MeOH; iii) TFA, CH2Cl2; iv) K2CO3, MgSO4, HCHO, CH2Cl2
Compounds 3a-3l were then screened against four cancer cell lines, A549 (lung), DU-145 (prostate), KB (nasopharyngeal), and HCT-8 (colon). The screening results are shown in Table 1. In comparison with R-antofine, all 12 compounds exhibited substantially decreased activity with an average GI50 over 1 μM. In addition, no cell-line selectivity was observed. Compound 3f (R = Ph) was the least potent (inactive) with an average GI50 value greater than 20 μM. Insertion of a single methylene group between the N and phenyl ring (3a and 3j, R = CH2Ph) resulted in greater activity (with the R isomer approximately two-fold better than the S isomer), while addition of a second methylene group (3d, R = CH2CH2Ph) did not improve activity any further. Generally, the compounds with an aliphatic amino moiety [cyclic (3g), straight chain (3c, 3h, and 3l) or branched chain (3b)] showed slightly better activity than those bearing an aromatic moiety. Compounds 3h and 3l (R = Me) showed the greatest potency among all compounds, and the R isomer (3l) was slightly more potent than its S enantiomer (3h). Conversely, 3i (S isomer) was two-fold more active than its R isomer (3k). These data implied that introduction of a N atom at position C12 of antofine might not improve the cytotoxicity against cancer cell lines, even though it did increase the polarity as predicted by PreADMET.33
Table 1.
GI50 values of 12-aza-antofines 3a-l against four cancer cell lines
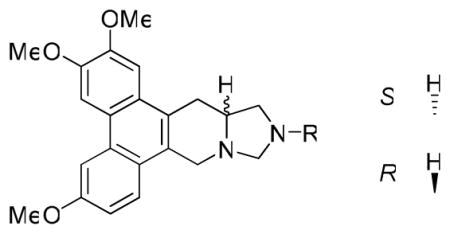 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R | Config. | A549 (μM) | DU145 (μM) | KB (μM) | HCT-8 (μM) |
| 3a | -Bn | S | 5.30 ± 1.02 | 4.95 ± 1.24 | 5.70 ± 1.52 | 3.63 ± 0.81 |
| 3b | -iBu | S | 2.00 ± 0.36 | 6.60 ± 1.45 | 4.40 ± 1.23 | 1.82 ± 0.45 |
| 3c | -nPr | S | 3.94 ± 0.78 | 7.38 ± 1.55 | 6.64 ± 1.73 | 2.71 ± 0.49 |
| 3d | -(CH2)2Ph | S | 6.62 ± 2.04 | 10.20 ± 3.13 | 10.03 ± 2.11 | 7.47 ± 1.68 |
| 3e | -2′-OH-Et | S | 4.41 ± 0.97 | 6.36 ± 1.29 | 7.83 ± 1.59 | 10.77 ± 2.44 |
| 3f | -Ph | S | > 20 | > 20 | > 20 | > 20 |
| 3g | -cPr | S | 1.80 ± 0.36 | 3.70 ± 0.81 | 2.71 ± 0.89 | 1.85 ± 0.48 |
| 3h | -Me | S | 0.66 ± 0.11 | 2.03 ± 0.38 | 1.72 ± 0.55 | 1.00 ± 0.19 |
| 3i | -NMe2 | S | 1.67 ± 0.37 | 2.26 ± 0.47 | 2.31 ± 0.65 | 2.01 ± 0.54 |
| 3j | -Bn | R | 2.68 ± 0.51 | 4.62 ± 1.16 | 3.41 ± 0.78 | 1.87 ± 0.43 (KBvin) |
| 3k | -NMe2 | R | 4.15 ± 0.87 | 6.01 ± 1.50 | 2.38 ± 0.60 | 10.98 ± 2.92 (KBvin) |
| 3l | -Me | R | 0.66 ± 0.10 | 1.00 ± 0.23 | 0.79 ± 0.21 | 0.66 ± 0.16 |
| R-antofine | - | R | 22 ± 7 nM | 25 ± 5 nM | 36 ± 8 nM | - |
Next, we studied the effect of N-incorporation in cryptopleurine, both at positions C12 and C13. For the latter compound series, 2 (S or R) was converted to 4 through oxidation with Py•SO3 and subsequent reductive amination using glycine methyl ester hydrochloride. The E ring was formed by sequential deprotection with HCl/MeOH and cyclization with Et3N/MeOH. The resulting intermediate was further protected with a Boc group to give 5 for easy purification. The lactam carbonyl was then reduced to a methylene using BMS/THF to afford 6. After removal of the Boc group, 13-aza-cryptopleurines (7a-q) were obtained through either acylation or reductive amination (Scheme 2).
Scheme 2.

Reagents and conditions: (a) i) Py•SO3, DMSO, Et3N, CH2Cl2; ii) glycine methyl ester hydrochloride, Et3N, HOAc, NaBH3CN, MeOH (b) i) HCl, MeOH; ii) MeOH, Et3N; iii) (Boc)2O, Et3N, CH2Cl2 (c) BMS, THF (d) i) TFA, CH2Cl2; ii) RCOCl, Et3N, CH2Cl2 or RCHO, Et3N, HOAc, NaBH3CN, MeOH
The GI50 values of 7a-q are listed in Table 2. Although the 13-aza analogs showed significantly reduced activity as compared with R-cryptopleurine, some interesting results were observed. Compound 7g, with a N-cyclopropylmethyl substituent, was the most potent analog against the HCT-8 cell line with a GI50 value of 0.25 μM, in addition to better selectivity against A549 and HCT-8 versus DU145 and KB. However, 7h, with a N-cyclopropyl substituent, one -CH2- shorter than 7g, showed dramatically reduced activity. Similar results were observed from the comparison of 7c and 7f, as well as antofine analogs 3f, 3a, and 3d, suggesting that the length of the side chain on nitrogen affects the anticancer activity. It is also interesting to note that 7g was much more potent than its R isomer 7q, whereas 7m and 7p exhibited the reversed order of activity, although to a less significant degree. Of all the tested analogs in this series, compound 7a, the hydrochloride salt of 13-aza-cryptopleurine, showed considerable anticancer activity against all four tested cell lines, indicating that 7a might be a promising lead meriting further investigation.
Table 2.
GI50 values of 13-aza-cryptopleurines 7a-q against four cancer cell lines
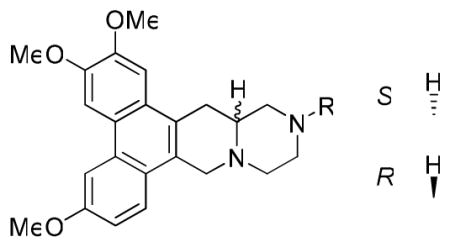 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R | Config. | A549 (μM) | DU145 (μM) | KB (μM) | HCT-8 (μM) |
| 7a | H, HCl salt | S | 1.73 ± 0.52 | 2.08 ± 0.27 | 1.79 ± 0.37 | 1.62 ± 0.56 |
| 7b | -Et | S | 7.13 ± 3.86 | 10.48 ± 2.51 | 8.24 ± 1.89 | 9.45 ± 3.21 |
| 7c | -CO2Me | S | 10.93 ± 5.10 | 12.19 ± 3.04 | 11.32 ± 2.25 | 12.05 ± 2.58 |
| 7d | -Ac | S | 11.65 ± 1.88 | 11.94 ± 2.22 | 15.46 ± 2.78 | 13.82 ± 2.80 |
| 7e | -Ms | S | 14.52 ± 3.02 | 16.97 ± 3.14 | 17.52 ± 3.10 | 17.83 ± 3.63 |
| 7f | -CH2CO2Me | S | 14.74 ± 2.49 | > 20 | 14.65 ± 2.88 | 14.03 ± 2.12 |
| 7g | -CH2cPr | S | 0.79 ± 0.11 | 2.94 ± 0.45 | 6.57 ± 1.56 | 0.25 ± 0.11 |
| 7h | -cPr | S | > 20 | 17.92 ± 5.28 | > 20 | 13.02 ± 3.03 |
| 7i | -Bz | S | 15.00 ± 3.77 | 15.71 ± 4.32 | > 20 | 18.11 ± 4.91 |
| 7j | -2′-OH-Et | S | 14.08 ± 5.49 | 12.62 ± 5.61 | 8.62 ± 2.44 | 7.41 ± 1.30 |
| 7k | -PO(OMe)2 | S | 12.18 ± 3.18 | 14.66 ± 4.59 | 8.09 ± 1.02 | 10.42 ± 2.21 (KBvin) |
| 7l | -Me | S | 9.73 ± 1.81 | 7.13 ± 1.05 | 5.30 ± 0.46 | 6.45 ± 1.03 |
| 7m | -Bn | S | > 20 | > 20 | 16.85 ± 5.64 | 15.24 ± 3.35 (KBvin) |
| 7n | -CONMe2 | R | 14.08 ± 1.86 | 13.12 ± 1.98 | 12.32 ± 2.15 | 11.41 ± 2.10 (KBvin) |
| 7o | -iBu | R | 12.86 ± 2.31 | 12.24 ± 4.04 | 11.25 ± 2.36 | 9.96 ± 1.89 (KBvin) |
| 7p | -Bz | R | 16.43 ± 6.08 | 11.87 ± 3.92 | 12.14 ± 2.91 | 7.64 ± 1.07 (KBvin) |
| 7q | -CH2cPr | R | > 20 | > 20 | > 20 | 13.44 ± 2.42 (KBvin) |
| crypto-pleurine | - | R | 1.38 ± 0.56 nM | 1.59 ± 0.53 nM | 1.51 ± 0.33 nM | 1.09 ± 0.20 nM |
In the following studies, a series of cryptopleurine analogs (10a-j) with N replacement at position C12 were synthesized to explore their anticancer activity. Compound 2 was converted to vinylmethyl ether 8 in two steps, oxidation followed by a Wittig reaction using Ph3P=CH2OMe. Compound 8 was then hydrolyzed with Hg(OAc)2 to give aldehyde 9. The targets, 12-aza-cryptopleurines 10a-j, were prepared using a similar strategy as described in the synthesis of compounds 3a-l (Scheme 3).
Scheme 3.

Reagents and conditions: (a) i) Py•SO3, DMSO, Et3N, CH2Cl2; ii) Ph3P+CH2OMeCl−, THF, KOtBu (b) Hg(OAc)2, THF, H2O (c) i) RNH2, HOAc, NaBH3CN, MeOH; ii) TFA, CH2Cl2; iii) K2CO3, MgSO4, HCHO, CH2Cl2
The GI50 values for 10a-j are listed in Table 3. Overall, the results suggested that N-incorporation at position C12 of cryptopleurine diminished the anticancer activity (lowest GI50 value was 1.12 μM for a 12-aza analog 10j versus nM values for cryptopleurine). Similar anticancer activity patterns were observed to those described above with the other compound series. Compounds with bulky or aromatic groups, such as a phenyl or benzyl moiety (10b or 10d), exhibited lower activity (> 20 μM), as also observed in the above two series of analogs. Introducing a polar group, such as hydroxyethyl in 10a/10i and N,N-dimethylamino in 10f/10h, did not lead to increased activity, although some moderate cell-line selectivity was found. The stereochemistry of the analogs also affected the anticancer activity (compare 10a/10i, 10c/10g, and 10f/10h), as was also observed in the above two analog series, although the trends were neither significant nor consistent. Interestingly, in the comparison of 10j and 7q (N12 vs. N13 substitution), the former compound was significantly more potent than the latter. Likewise, 10g was more potent than 7o, likely indicating that the relocation of the N atom could cause a possible conformational change, which led to a differentiation in biological activity.
Table 3.
GI50 values of analogs 10a-j against four cancer cell lines
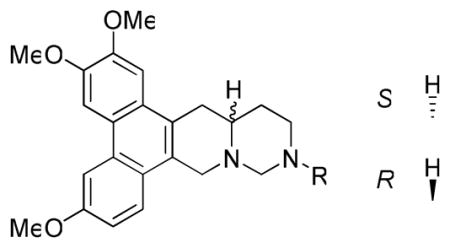 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R | Config. | A549 (μM) | DU145 (μM) | KB (μM) | HCT-8 (μM) |
| 10a | -2′-OH-Et | S | 5.68 ± 1.36 | 1.99 ± 0.52 | 1.59 ± 0.36 | 10.65 ± 3.73 |
| 10b | -Ph | S | > 20 | > 20 | > 20 | > 20 |
| 10c | -iBu | S | 1.52 ± 0.38 | 2.30 ± 0.55 | 5.06 ± 1.82 | 1.36 ± 0.29 |
| 10d | -Bn | S | > 20 | > 20 | > 20 | > 20 |
| 10e | -cPr | S | 4.30 ± 0.77 | 14.33 ± 3.80 | 11.47 ± 3.72 | 5.73 ± 1.14 |
| 10f | -NMe2 | S | 1.78 ± 0.43 | 2.97 ± 0.51 | 3.08 ± 0.50 | 2.37 ± 0.78 |
| 10g | -iBu | R | 5.11 ± 1.62 | 1.66 ± 0.43 | 1.45 ± 0.22 | 7.32 ± 1.90 (KBvin) |
| 10h | -NMe2 | R | 5.20 ± 1.02 | 3.00 ± 0.65 | 2.84 ± 0.56 | 5.30 ± 1.18 (KBvin) |
| 10i | -2′-OH-Et | R | 2.48 ± 0.36 | 1.46 ± 0.33 | 2.22 ± 0.41 | > 20 (KBvin) |
| 10j | -CH2cPr | R | 1.12 ± 0.29 | 1.20 ± 0.28 | 2.10 ± 0.39 | 5.68 ± 1.53 (KBvin) |
| crypto-pleurine | - | R | 1.38 ± 0.56 nM | 1.59 ± 0.53 nM | 1.51 ± 0.33 nM | 1.09 ± 0.20 nM |
Even though the above modifications of antofine and cryptopleurine by N incorporation in the E-ring did not provide analogs with enhanced or at least comparable anticancer activity to the natural alkaloids, we extended our studies by investigating the effect of O atom incorporation at corresponding positions. Unfortunately, introduction of the oxygen atom at position C12 of antofine was not successful due to the instability of the resulting analog. However, incorporation of O atom into the E-ring of cryptopleurine at position C12 or C13 was achieved, as described in Scheme 4.
Scheme 4.

Reagents and conditions: (a) i) ClCH2COCl, Et3N, CH2Cl2; ii) NaH, THF, reflux; iii) BMS, THF (b) i) NaBH4, MeOH; ii) TFA, CH2Cl2; iii) K2CO3, MgSO4, HCHO, CH2Cl2
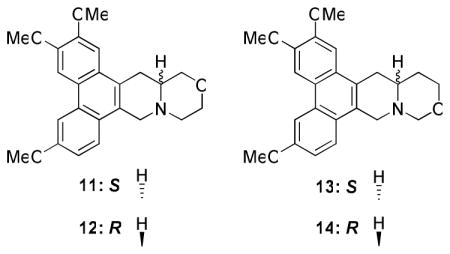
Reaction of 1 with chloroacetyl chloride furnished the intermediate amide, which underwent intramolecular nucleophilic attack using NaH/THF and subsequent reduction using BMS/THF to afford S-13-oxo-cryptopleurine 11 and its R isomer 12. For the O replacement at position C12 of cryptopleurine, the aldehyde of 9 was first converted to an hydroxymethyl group by NaBH4 reduction, followed by removal of Boc and subsequent cyclization using HCHO to give S-12-oxo-cryptopleurine 13 and its R isomer 14 (Scheme 4).
A cytotoxicity evaluation of 11–14 against four cancer cell lines was conducted, and the results are summarized in Table 4. It is exciting to note that 11, with O at position C13, exhibited potent nanomolar anticancer activity with greater selectivity towards the HCT-8 tumor cell line (GI50 = 9 nM). In contrast, compound 12, the R-enantiomer of 11, showed significantly decreased activity. As for the incorporation of O at position C12, both analogs 13 and 14 exhibited much lower potency than R-crytopleurine with GI50 >1 μM. The slight structural variation among 11–14 resulted in significantly different biological activity. Compound 11 might achieve a specific structural conformation that is required for interacting with the target protein leading to the observed anticancer activity. However, further studies are needed to verify such an hypothesis, as the decreased activity of 13 and 14 could also possibly be due to molecular instability (the hemiaminal ether E-ring).
Table 4.
GI50 values of analogs 11–14 against four cancer cell lines
 | |||||
|---|---|---|---|---|---|
| Compd. | Config. | A549 | DU145 | KB | HCT-8 |
| 11 | S | 23 ± 5 nM | 67 ± 10 nM | 37 ± 9 nM | 9 ± 5 nM |
| 12 | R | 13.95 ± 4.74 μM | 7.05 ± 1.25 μM | 6.68 ± 1.52 μM | 3.45 ± 0.56 μM |
| 13 | S | 1.79 ± 0.27 μM | 1.82 ± 0.31 μM | 1.66 ± 0.33 μM | 1.45 ± 0.24 μM |
| 14 | R | 1.37 ± 0.29 μM | 1.29 ± 0.36 μM | 1.10 ± 0.28 μM | 1.26 ± 0.20 μM |
| crypto-pleurine | R | 1.38 ± 0.56 nM | 1.59 ± 0.53 nM | 1.51 ± 0.33 nM | 1.09 ± 0.20 nM |
Our study of O incorporation in antofine/cryptopleurine-type compounds was also extended to the previously reported 7-membered E-ring analog 15.29 Its O-containing analog 16 was synthesized using similar procedures to those for 11 (Scheme 5). Firstly, the aldehyde of 9 was reduced to a hydroxymethyl group followed by removal of Boc. The resulting amino intermediate was then reacted with chloroacetyl chloride at 0 °C to provide the chloroacetamide, which was subjected to ring closure and subsequent reduction of the amide carbonyl to a methylene with BMS to give compound 16 in 24% yield over five steps.
Scheme 5.

Reagents and conditions: (a) i) NaBH4, MeOH; ii) TFA, CH2Cl2; iii) ClCH2COCl, Et3N, CH2Cl2, 0 °C; iv) NaH, THF, reflux; v) BMS, THF, 24% for 5 steps
When tested in parallel for cytotoxicity, both 15 and 16 were more active against A549 and HCT-8 cancer cell lines than against DU145 and KB. Compound 16 was slightly more active than 15 against DU145 and KB cell proliferation, while it was slightly less active against A549 and HCT-8. These results suggested that the remarkable cell-line selectivity of 15 was somewhat decreased by introducing an oxygen atom at C13. Nevertheless, both analogs exhibited strong potency against the HCT-8 cell line, which indicated that phenanthroindolizidine and phenanthroquinolizidine derivatives with a 7-membered E-ring have a new and interesting structural scaffold and could have a great potential for further development as selective anti-colorectal cancer agents.
In consideration of the impressive cytotoxicity, we next aimed our study at the potential mechanism(s) of action of the representative analogs 11 and 15, as well as R-antofine. Our preliminary cDNA microarray data indicated that R-antofine had a profound influence on the major replication initiation complexes ORC (origin recognition complex) and MCM (DNA replication licensing factor) in early S phase. The DNA replication licensing factors Cdt1 (DNA replication factor) and CDC6 (cell division control protein 6) were also up-regulated in a dose- or time-dependent manner when CL1-5 cells were exposed to R-antofine for 24 and 48 h, as confirmed by RT-PCR (0.01 μg/mL, Figure 2) and Western blot analysis (Figure 3). These data indicated that R-antofine promotes dysregulation of DNA replication in lung cancer cells. However, it is not clear whether the cytotoxic effect of R-antofine is directly correlated with promoting dysregulation of DNA replication. In general, the effects of 11 and 15 were not significant, falling in between those produced by R-antofine and control. An in-depth investigation on diverse cell events should be helpful for further clarification of their mechanism of action. Obviously, the difference in the E-ring structure is involved in these observed effects, and the E-ring is not totally indispensable.
Figure 2.

RT-PCR results of R-antofine, 11, and 15 at 0.01 μg/mL on major DNA replication complexes in CL1-5 cells at 24 and 48 h
Figure 3.

Western blot analysis of R-antofine, 11, and 15 on major DNA replication complexes in CL1-5 cells at 24 and 48 h
Compound 11 was then selected for an in vivo study in mice against HT-29 human colorectal adenocarcinoma xenograft. Mice in group 1 received vehicle and served as control for all treatment groups. The median time to endpoint (TTE) for mice in the control group was 16.3 days with a range of 13.8–21.9 days. Compound 11, i.v. at 20 mg/kg (qd to end), produced a median TTE of 21.3 days and caused 1.1% maximum group body weight loss on day 12, indicating statistically significant antitumor activity (P < 0.05) and no obvious toxicity. When dosed i.v./i.p. at 20 mg/kg (bid to end), 11 produced a median TTE of 20.9 days and caused 7.2% maximum group body weight loss occurring on day 19. One tumor remained in the study on day 29 and one treatment-related (TR) death occurred on day 20 (Figures 4 and 5). When dosing frequency was increased, no improved efficacy was observed. Interestingly, a similar phenomenon was also reported in previous studies. For instance, the effectiveness of phenanthroindolizidine analogs with C14-OH was found to be highly schedule dependent, e.g., the therapeutic index of DCB-3503 may be lost if given daily instead of every three days.33 Therefore, a more rationally designed dosing regimen is being investigated in order for the compounds to exert their best tumor inhibitory effects, and meanwhile, reduce toxicity that could result from unnecessary repetitive dosing. Moreover, we tested compound 11 against human umbilical vein endothelial cells (HUVECs, normal cells), and the results indicated that the cytotoxicity of R-cryptopleurine was reduced by 30-fold through our modification, as shown in Figure 6 (GI50 320 nM vs. 11 nM). Such improved selectivity against cancer cells over normal cells demonstrates that our newly synthesized analogs can potentially reduce unwanted side effects in vivo. In summary, compound 11 exhibited potent in vitro anticancer activity with improved cell line selectivity and moderate antitumor activity against HT-29 human colorectal adenocarcinoma in male nude-athymic mice at a dose of 20 mg/kg without causing obvious toxicity.
Figure 4.

Individual animal body weight change for control and treatment groups
Figure 5.

In vivo antitumor activity of compound 11
Figure 6.

Comparison of compound 11 and R-cryptopleurine against Human Umbilical Vein Endothelial Cells (HUVECs)
Conclusions
New synthetic antofine and cryptopleurine analogs with varied E-ring size or heteroatom incorporation were designed and prepared. Two analogs, 11 and 16, were the most active compounds against tested cancer cell lines, with interestingly improved selectivity against HCT-8 cell growth. Mechanistic studies indicated that R-antofine is capable of promoting dysregulation of DNA replication in early S phase, whereas no similar results were found for compounds 11 and 15. It is still unclear how this activity correlates with the potent cytotoxicity of antofine, and further investigation is ongoing. In our in vivo antitumor study, we found that 11 had improved activity on HUVECs and moderate antitumor effect on human HT-29 adenocarcinoma xenograft in mice. However, further studies in other tumor models are warranted to find out which type of tumor affords the most sensitivity and what treatment protocol provides the best therapeutic index. Moreover, compound 16 appears to be an interesting analog that necessitates in-depth investigation, and results will be reported in due course. Additionally, CNS toxicity will also be explored in the future.
Experimental section
All chemicals were used as purchased. Melting points were measured using a Fisher Johns melting apparatus without correction. Proton nuclear magnetic resonance (1H NMR) spectra were measured on a 300 MHz Gemini or a Varian Inova (400 MHz) NMR spectrometer with TMS as the internal standard. The solvent used was CDCl3 unless otherwise indicated. Mass spectra were recorded on a Shimazu-2010 LC/MS/MS instrument equipped with a TurboIonsSpray ion source. All final target compounds were characterized and determined as at least >95% pure by analytical HPLC.
(S)-N-Boc-(6,7,10-trimethoxy-1,2,3,4-tetrahydrodibenzo[f,h]isoquinolin-3-yl)m ethanol (2)
(Boc)2O (3.14 g, 14.40 mmol) was added to the amino-alcohol (4.24 g, 12 mmol) in 100 mL of CH2Cl2 and Et3N (6 mL), and the mixture was stirred for 2 h. HCl (50 mL, 1 N) was added and the organic layer was separated, washed with sat. NaHCO3 and brine, and dried over MgSO4. Column chromatography eluting with CH2Cl2/MeOH gave 5.20 g of compound 2 as a white solid. Yield: 95.6%; mp 105–107 °C; [α]23D= 85.5 ° (c 0.42, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.89 (s, 2H), 7.81 (d, J = 9.0 Hz, 1H), 7.28 (s, 1H), 7.22 (d, J = 8.7 Hz, 1H), 5.29 (br s, 1H), 4.84 (m, 1H), 4.61 (br s, 1H), 4.09 (s, 3H), 4.05 (s, 3H), 4.01 (s, 3H), 3.73-3.62 (m, 2H), 3.27 (dd, J = 16.5 Hz, J = 6.3 Hz, 1H), 3.18 (t, J = 16.5 Hz, 1H), 1.55 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 157.9, 156.2, 149.7, 148.8, 130.4, 126.7, 124.0, 123.9, 123.8, 123.4, 122.8, 115.2, 105.1, 104.2, 104.0, 80.6, 62.7, 56.2, 56.0, 55.7, 50.6, 41.0, 28.6 (3×C), 26.5; ESI-HRMS ([M + H]+) calcd for C26H32NO6 454.2230, found 454.2246.
General procedures for the synthesis of 12N-substituted-12-aza-antofine (3a-l)
The aldehyde was obtained using the same procedure as for compound 8. Various amines were reacted with the aldehyde in MeOH, followed by addition of HOAc and NaBH3CN, which was stirred at r.t. for 4 h. Sat. NaHCO3 was added to quench the reaction and CH2Cl2 was used for extraction. The residue was quickly purified through a column and TFA was used to remove the Boc group. Then TFA was removed by evaporation and the residue was dissolved in CH2Cl2 (10 mL), to which K2CO3 (100 mg) and MgSO4 (0.5 g) were added, followed by HCHO (37%, 0.1 mL). The mixture was stirred at r.t. overnight. Chromatography gave light yellow to white solids. Yield: 40%–80%.
(S)-tert-Butyl-6,7,10-trimethoxy-3-((2-methoxy-2-oxoethylamino)methyl)-3,4-dih ydrodibenzo[f,h]isoquinoline-2(1H)-carboxylate (4)
The Boc-aldehyde was obtained using the same procedure as compound 8. Glycine methyl ester hydrochloride (314 mg, 2.5 mmol) and Et3N (0.35 ml, 2.5 mmol) were added to the aldehyde in MeOH (30 mL), which was stirred for 0.5 h. Then HOAc (0.70 mL, 12 mmol) and NaBH3CN (165 mg, 2.5 mmol) were added. The mixture was stirred for 2 h until all the aldehyde disappeared. Sat. NaHCO3 was used to quench the reaction and CH2Cl2 was used for extraction, and washed with brine, dried over MgSO4. Chromatography gave 635 mg of compound 4 as a white solid. Yield: 61% over two steps. mp 78–80 °C; [α]23D= 91.6 ° (c 0.37, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.92 (s, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.87 (brs, 1H), 7.44-7.42 (m, 2H), 7.29 (s, 1H), 7.25-7.23 (m, 1H), 5.40-5.29 (m, 1H), 4.92-4.78 (m, 1H), 4.56 (m, 1H), 4.11 (s, 3H), 4.05 (s, 3H), 4.02 (s, 3H), 3.66 (s, 3H), 3.47-3.36 (m, 2H), 3.27 (dd, J = 16.4 Hz, J = 6.4 Hz, 1H), 3.14 (d, J = 16.4 Hz, 1H), 2.81 (dd, J = 12.0 Hz, J = 8.8 Hz, 1H), 2.64 (dd, J = 12.0 Hz, J = 6.4 Hz, 1H), 1.54 (s, 9H); ESI MS m/z 525.20 (M+H)+.
(S)-13N-Boc-11-oxo-13-aza-cryptopleurine (5)
The ester (635 mg, 1.21 mmol) was dissolved in HCl (1.25M in methanol, 15 mL), which was stirred at 50 °C for 1 h. The solvent was removed. 30 mL of MeOH and Et3N (0.3 mL) were added. The mixture was stirred at r.t. for 2 h. After normal workup, chromatography afforded 404 mg of ring-closed intermediate as a white solid. Using similar procedure as compound 2, the intermediate was protected by (Boc)2O to give 456 mg of 5 as a white solid. Yield: 76% over three steps. mp 125–127 °C; [α]23D= −202.8 ° (c 0.50, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.89 (s, 1H), 7.88 (d, J = 2.8 Hz, 1H), 7.87 (d, J = 9.2 Hz, 1H), 7.23 (dd, J = 9.2 Hz, J = 2.8 Hz, 1H), 7.19 (s, 1H), 5.88 (d, J = 17.2 Hz, 1H), 4.49 (d, J = 17.2 Hz, 1H), 4.40 (d, J = 18.0 Hz, 1H), 4.10 (s, 4H), 4.05 (s, 4H), 4.01 (s, 3H), 3.87-3.77 (m, 2H), 3.16 (m, 2H), 1.51 (s, 9H); ESI MS m/z 493.10 (M+H)+, 985.20 (2M+H)+.
(S)-13N-Boc-13-aza-cryptopleurine (6)
To a solution of the amide (456 mg, 0.93 mmol) in THF (30 mL) was added BMS (2M in THF, 2.79 mL), which was stirred at r.t. overnight. 5 mL of MeOH was added and the mixture was refluxed for 1 h. Chromatography afforded 400 mg of 6 as a white solid. Yield: 90%. mp 116–118 °C; [α]23D= −182.5 ° (c 0.28, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.88-7.87 (m, 2H), 7.76 (d, J = 8.8 Hz, 1H), 7.20 (s, 1H), 7.19 (dd, J = 8.8 Hz, J = 2.8 Hz, 1H), 4.44 (d, J = 15.2 Hz, 1H), 4.29 (m, 1H), 4.11 (m, 1H), 4.09 (s, 3H), 4.05 (s, 3H), 4.00 (s, 3H), 3.66 (d, J = 15.2 Hz, 1H), 3.15 (m, 2H), 3.04 (dd, J = 16.4 Hz, J = 3.2 Hz, 1H), 2.87 (m, 1H), 2.80 (dd, J = 16.4 Hz, J = 11.8 Hz, 1H), 2.57-2.52 (m, 1H), 2.45 (dt, J = 12.0 Hz, 3.2 Hz, 1H); ESI MS m/z 479.10 (M+H)+.
General procedures for the synthesis of 13N-substituted-13-aza-cryptopleurine (7a-q)
(a) 13N-Boc-13-aza-cryptopleurine 6 was stirred in HCl/MeOH for 2 h before the solvent was removed in vacuo. The solid was collected and washed sequentially with cold MeOH and ether. Then the solid was dissolved in 10 mL of anhydrous CH2Cl2 and Et3N (0.1 mL), to which RCOCl was added at 0 °C. The mixture was stirred at r.t. for 4 h before 1N HCl was added. After routine workup, chromatography afforded from light orange to white solids. Yield: 70% – 80%. (b) Using the same procedures (reductive amination) as compound 4. Yield: 50% – 80%.
(S,E/Z)-6,7,10-Trimethoxy-3-(2-methoxyvinyl)-1,2,3,4-tetrahydrodibenzo[f,h]isoq uinoline (8)
N-Boc-alcohol 2 (1.812 g, 4 mmol) was dissolved in CH2Cl2 (50 mL) and Et3N (1.95 mL, 14 mmol) at 0°C, to which Py•SO3 (2.23 g, 14 mmol) in 10 mL of DMSO was added dropwise. The ice bath was then removed and the mixture was stirred at r.t. until 2 disappeared by TLC. HCl (1N) was added, and the organic layer was then separated and washed with sat. NaHCO3, brine, and dried over MgSO4. In another flask, Ph3P+CH2OMeCl− (2.74 g, 8 mmol) was added to KOtBu (875 mg, 7.8 mmol) at 0 °C under N2, which was stirred for 0.5 h. The aldehyde in 20 mL of CH2Cl2 was added dropwise, followed by removal of ice bath. The reaction was kept for 2–3 h before sat. NH4Cl was poured into the mixture. CH2Cl2 was used for extraction and the organic layers were washed with sat. NaHCO3 and brine, dried over MgSO4. Chromatography afforded 1.5 g of compound 8 as a light yellow solid. Yield: 78% (Z/E = 1/2.5) for two steps. mp 150–152 °C; [α]23D= 108.1 ° (c 0.59, CHCl3); 1H NMR (400 MHz, CDCl3, E/Z = 2.5:1): δ 7.94-7.91 (m, 2H), 7.88 (d, J = 9.2 Hz, 1H), 7.29 (s, 1H), 7.26-7.22 (m, 1H), 6.30 (d, J = 12.8 Hz, 0.68H, E isomer), 5.86 (dd, J = 6.4 Hz, J = 1.6 Hz, 0.28 Hz, Z isomer), 5.72 (m, 0.31H, Z isomer), 5.30-5.25 (d, J = 16.8 Hz, 1H+0.67H, E isomer), 4.80 (dd, J = 12.8 Hz, J = 8.4 Hz, 0.71 H, E isomer), 4.59 (d, J = 17.2 Hz, 1H), 4.42 (dd, J = 8.0 Hz, J = 6.4 Hz, 0.29H, Z isomer), 4.12-4.11 (m, 3H), 4.05-4.04 (m, 3H), 4.03-4.02 (m, 3H), 3.62 (s, 0.71H, Z isomer), 3.44-3.32 (m, 1H), 3.36 (s, 2H, E isomer), 3.13 (d, J = 16.0 Hz, 1H), 1.55 (s, 9H); ESI MS m/z 480.05 (M+H)+.
(S)-2-(6,7,10-Trimethoxy-1,2,3,4-tetrahydrodibenzo[f,h]isoquinolin-3-yl)acetal dehyde (9)
Compound 8 (1.5 g, 3.12 mmol) was dissolved in THF (50 mL) and H2O (5mL), to which Hg(OAc)2 (3.0 g, 9.36 mmol) was added at 0 °C. Then the ice bath was removed and the mixture was stirred overnight. Freshly prepared sat. KI (50 ml) was added dropwise at 0 °C for 10 min, and CH2Cl2 was used for extraction. The organic layers were collected and washed with brine, dried over MgSO4. Chromatography furnished 1.09 g of compound 9 as light yellow foam. Yield: 75%. mp 98–100 °C; [α]23D= 94.4 ° (c 0.68, CHCl3); 1H NMR (400 MHz, CDCl3): δ 9.79 (s, 1H), 7.93 (s, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.85 (brs, 1H), 7.26-7.24 (m, 2H), 5.38-5.29 (m, 2H), 4.58 (d, J = 16.0 Hz, 1H), 4.11 (s, 3H), 4.05 (s, 3H), 4.02 (s, 3H), 3.38 (dd, J = 16.0 Hz, J = 6.4 Hz, 1H), 3.12 (d, J = 16.4 Hz, 1H), 2.70-2.59 (m, 2H), 1.53 (s, 9H); ESI MS m/z 466.10 (M+H)+, 488.15 (M+Na)+.
General procedures for the synthesis of 12N-substituted-12-aza-cryptopleurine (10a-j)
using similar procedures as compound 3a-l. Yield: 50% – 70%.
(S)-13-Oxa-cryptopleurine (11) and (R)-13-Oxa-cryptopleurine (12)
Compound 2 (177 mg, 0.5 mmol) was suspended in dry CH2Cl2 (15 mL) and Et3N (0.14 mL, 1.00 mmol) at 0 °C, to which chloroacetyl chloride (40 μL, 0.50 mmol) in 1 mL of CH2Cl2 was added dropwise slowly. The mixture was stirred at 0 °C for 5 h before 1N HCl was added. The organic layer was separated and washed with sat. NaHCO3 and brine, dried over MgSO4. CH2Cl2 was removed and the residue was dissolved in anhydrous THF (5 mL), to which NaH (2.1 equiv) was added at r.t. followed by reflux for 2 h. Sat. NH4Cl was added and CH2Cl2 was used for extraction. After workup, the organic layer was dried over MgSO4. Then the residue was dissolved in 10 mL anhydrous THF, BMS (1.50 mL, 6 mmol) was added, which was stirred at r.t. overnight. MeOH (5 mL) was added and the mixture was refluxed for 1 h. Chromatography gave 73 mg of 11 as a white solid. Yield: 38.5% for three steps; white solid; mp 199–201 °C; [α]23D= −134.6 ° (c 0.52, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.86-7.85 (m, 2H), 7.74 (d, J = 9.0 Hz, 1H), 7.18 (dd, J = 9.0 Hz, J = 2.4 Hz, 1H), 7.14 (s, 1H), 4.39 (d, J = 15.6 Hz, 1H), 4.12 (m, 2H), 4.08 (s, 3H), 4.03 (s, 3H), 4.00 (s, 3H), 3.91-3.83 (m, 1H), 3.66 (d, J = 15.6 Hz, 1H), 3.51 (dd, J = 11.1 Hz, J = 9.0 Hz, 1H), 3.06 (d, J = 11.7 Hz, 1H), 2.91-2.87 (m, 1H), 2.76-2.59 (m, 3H); ESI MS m/z 380.05 (M+H)+. For 12: mp 196–198 °C; [α]23D= 139.6 ° (c 0.26, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.90-7.89 (m, 2H), 7.78 (d, J = 9.0 Hz, 1H), 7.20 (dd, J = 9.0 Hz, J = 2.7 Hz, 1H), 7.20 (s, 1H), 4.44 (d, J = 15.6 Hz, 1H), 4.17-4.08 (m, 1H), 4.10 (s, 3H), 4.05 (s, 3H), 4.01 (s, 3H), 4.04-4.00 (m, 1H), 3.92-3.84 (m, 1H), 3.72 (d, J = 15.3 Hz, 1H), 3.54 (dd, J = 11.1 Hz, J = 9.0 Hz, 1H), 3.09 (d, J = 11.4 Hz, 1H), 2.98-2.94 (m, 1H), 2.77-2.61 (m, 3H); 13C NMR (75 MHz, CDCl3): δ 157.5, 149.4, 148.4, 130.2, 126.4, 125.1, 124.1, 123.6, 123.4, 123.3, 115.0, 104.7, 103.8, 103.6, 72.3, 67.4, 56.1, 56.0, 55.9, 55.5, 55.1, 54.4, 28.5; ESI MS m/z 380.05 (M+H)+.
(S)-12-Oxa-cryptopleurine (13) and (R)-12-Oxa-cryptopleurine (14)
The aldehyde 9 (100 mg, 0.21 mmol) was dissolved in MeOH, to which NaBH4 (19 mg, 0.50 mmol) was added in one portion. The mixture was stirred for 1 h and sat. NaHCO3 was added. After normal workup, 80 mg of white solid was obtained, which was used without further purification. The residue was dissolved in CH2Cl2 (10 mL), to which MgSO4, K2CO3, and HCHO were added sequentially. The mixture was stirred overnight. After workup, chromatography gave 32 mg of 13 as a white solid. Yield: 40% for two steps; light yellow solid; mp 195–197 °C; [α]23D= −56.8 ° (c 0.60, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.89-7.88 (m, 2H), 7.75 (d, J = 9.3 Hz, 1H), 7.22 (s, 1H), 7.19 (dd, J = 9.0 Hz, J = 2.7 Hz, 1H), 4.79 (d, J = 8.4 Hz, 1H), 4.41 (d, J = 15.3 Hz, 1H), 4.19 (dd, J = 11.1 Hz, J = 5.1 Hz, 1H), 4.10 (s, 3H), 4.05 (s, 3H), 4.03 (m, 1H), 4.01 (s, 3H), 3.75-3.65 (m, 2H), 3.17 (dd, J = 15.9 Hz, J = 3.6 Hz, 1H), 2.95-2.80 (m, 2H), 2.06-1.92 (m, 1H), 1.74 (m, 1H); ESI MS m/z 380.05 (M+H)+. For 14: mp 191–192°C; [α]23D= 51.2 ° (c 0.60, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.87-7.86 (m, 2H), 7.73 (d, J = 9.0 Hz, 1H), 7.20 (s, 1H), 7.18 (dd, J = 9.6 Hz, J = 3.0 Hz, 1H), 4.77 (d, J = 8.1 Hz, 1H), 4.38 (d, J = 15.3 Hz, 1H), 4.18 (dd, J = 11.1 Hz, J = 4.8 Hz, 1H), 4.09 (s, 3H), 4.04 (s, 3H), 3.99 (s, 3H), 3.98 (d, J = 8.1 Hz, 1H), 3.73-3.61 (m, 2H), 3.13 (dd, J = 15.9 Hz, J = 3.6 Hz, 1H), 2.91-2.72 (m, 2H), 2.02-1.90 (m, 1H), 1.72-1.67 (m, 1H); 13C NMR (75 MHz, CDCl3): δ 157.7, 149.6, 148.5, 130.3, 126.6, 124.7, 124.2, 123.8, 123.7, 123.6, 115.0, 104.9, 104.5, 103.9, 86.5, 68.1, 56.1, 56.0, 55.7, 55.4, 48.0, 33.5, 31.3; ESI MS m/z 380.05 (M+H)+.
Compound 16
The aldehyde 9 (140 mg, 0.30 mmol) was dissolved in MeOH, to which NaBH4 (76 mg, 2 mmol) was added. The resulting mixture was stirred at r.t. for 1 h before sat. NaHCO3 was added. The mixture was extracted with CH2Cl2 and the organic layers were combined, washed with brine, and dried over Na2SO4. Then the solvent was removed under reduced pressure and redissolved in TFA/CH2Cl2 (1:1, 10 mL), which was stirred for 0.5 h. The mixture was concentrated to remove TFA. The residue was subject to treatment with chloroacetyl chloride and the rest of the synthesis was similar as that of compound 11. After chromatography, 37 mg of 16 was obtained as a light yellow solid in 24% yield over five steps. mp 183–185 °C; [α]23D= −114.7 ° (c 0.19, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.90 (s, 1H), 7.88 (d, J = 2.4 Hz, 1H), 7.75 (d, J = 9.2 Hz, 1H), 7.24 (s, 1H), 7.19 (dd, J = 9.2 Hz, J = 2.4 Hz, 1H), 4.43 (d, J = 15.2 Hz, 1H), 4.10 (s, 3H), 4.05 (m, 4H), 4.01 (m, 4H), 3.98-3.90 (m, 3H), 3.20-3.13 (m, 1H), 3.08-3.01 (m, 3H), 2.96-2.91 (m, 1H), 2.25-2.19 (m, 1H), 2.17-2.11 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 157.7, 149.6, 148.6, 130.3, 126.9, 126.6, 125.4, 124.2, 123.9, 123.7, 115.1, 104.9, 104.1, 104.0, 69.9, 66.5, 59.0, 57.6, 57.0, 56.2, 56.1, 55.7, 36.7, 35.5; ESI MS m/z 394.10 (M+H)+.
Supplementary Material
Table 5.
GI50 values of compounds 15 and 16
| Compd. | Config. | A549 (nM) | DU145 (nM) | KB (nM) | HCT-8 (nM) |
|---|---|---|---|---|---|
| 15 | R | 25 ± 5 | 179 ± 36 | 102 ± 19 | 10 ± 3 |
| 16 | S | 41 ± 9 | 119 ± 33 | 68 ± 10 | 20 ± 6 |
Acknowledgments
This study was supported by grant CA17625-32 from the National Cancer Institute awarded to K. H. Lee and grant DOH98-TD-G-111-007 from the National Research Program for Genomic Medicine awarded to P.C. Yang. This study was also supported in part by the Department of Health Cancer Research Center of Excellence (DOH-100-TD-C-111-05).
Abbreviations used
- MOA
mechanism of action
- NCI
National Cancer Institute
- HIF-1
hypoxia-inducible factor 1
- TS
thymidylate synthase
- DHFR
dihydrofolate reductase
- AP-1
activator protein-1
- CRE
cAMP response element
- CDK
cyclin-dependent kinase
- CNS
central neural system
- SAR
structure-activity relationship
- BBB
blood-brain barrier
- PSA
polar surface area
- ORC
origin recognition complex
- CDC6
cell division control protein 6
- RT-PCR
reverse transcription polymerase chain reaction
- HUVECs
human umbilical vein endothelial cells
Footnotes
Supporting Information Available. Compound data for 3a-l, 7a-q, and 10a-j, biological studies, and HPLC analysis of final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gellert E. The indolizidine alkaloids. J Nat Prod. 1982;45(1):50–73. [Google Scholar]
- 2.Xi Z, Zhang R, Yu Z, Ouyang D. The interaction between tylophorine B and TMV RNA. Bioorg Med Chem Lett. 2006;16(16):4300–4304. doi: 10.1016/j.bmcl.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 3.Wang K, Su B, Wang Z, Wu M, Li Z, Hu Y, Fan Z, Mi N, Wang Q. Synthesis and antiviral activities of phenanthroindolizidine alkaloids and their derivatives. J Agric Food Chem. 58(5):2703–2709. doi: 10.1021/jf902543r. [DOI] [PubMed] [Google Scholar]
- 4.Baumgartner B, Erdelmeier CAJ, Wright AD, Rali T, Sticher O. An antimicrobial alkaloid from Ficus septica. Phytochemistry. 1990;29(10):3327–3330. [Google Scholar]
- 5.Bhutani KK, Sharma GL, Ali M. Plant based antiamoebic drugs; Part I. Antiamoebic activity of phenanthroindolizidine alkaloids; common structural determinants of activity with emetine. Planta Med. 1987;53(6):532–536. doi: 10.1055/s-2006-962803. [DOI] [PubMed] [Google Scholar]
- 6.Banwell MG, Bezos A, Burns C, Kruszelnicki I, Parish CR, Su S, Sydnes MO. C8c-C15 monoseco-analogues of the phenanthroquinolizidine alkaloids julandine and cryptopleurine exhibiting potent anti-angiogenic properties. Bioorg Med Chem Lett. 2006;16(1):181–185. doi: 10.1016/j.bmcl.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 7.Yang C-W, Chen W-L, Wu P-L, Tseng H-Y, Lee S-J. Anti-inflammatory mechanisms of phenanthroindolizidine alkaloids. Mol Pharmacol. 2006;69(3):749–758. doi: 10.1124/mol.105.017764. [DOI] [PubMed] [Google Scholar]
- 8.Yang C-W, Chuang T-H, Wu P-L, Huang W-H, Lee S-J. Anti-inflammatory effects of 7-methoxycryptopleurine and structure-activity relations of phenanthroindolizidines and phenanthroquinolizidines. Biochem Biophys Res Commun. 2007;354(4):942–948. doi: 10.1016/j.bbrc.2007.01.065. [DOI] [PubMed] [Google Scholar]
- 9.NCI 60-cell assay results can be found at http://dtp.nci.nih.gov/dtpstandard/dwindex/index.jsp
- 10.Gao W, Lam W, Zhong S, Kaczmarek C, Baker David C, Cheng Y-C. Novel mode of action of tylophorine analogs as antitumor compounds. Cancer Res. 2004;64(2):678–688. doi: 10.1158/0008-5472.can-03-1904. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Jin Z, Huang R. Isolation, total synthesis and biological activity of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Synthesis. 2001;(16):2365–2378. [Google Scholar]
- 12.Chemler SR. Phenanthroindolizidines and phenanthroquinolizidines: promising alkaloids for anti-cancer therapy. Curr Bioact Compd. 2009;5(1):2–19. doi: 10.2174/157340709787580928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cusack JC, Jr, Liu R, Baldwin AS. Jr., Inducible chemoresistance to 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothe cin (CPT-11) in colorectal cancer cells and a xenograft model is overcome by inhibition of nuclear factor-kappaB activation. Cancer Res. 2000;60(9):2323–2330. [PubMed] [Google Scholar]
- 14.Huang MT, Grollman AP. Mode of action of tylocrebrine - effects on protein and nucleic-acid synthesis. Mol Pharmacol. 1972;8(5):538–550. [PubMed] [Google Scholar]
- 15.Gupta RS, Siminovitch L. Mutants of CHO cells resistant to the protein synthesis inhibitors, cryptopleurine and tylocrebrine: genetic and biochemical evidence for common site of action of emetine, cryptopleurine, tylocrebrine, and tubulosine. Biochemistry. 1977;16(14):3209–3214. doi: 10.1021/bi00633a026. [DOI] [PubMed] [Google Scholar]
- 16.Gupta RS, Krepinsky JJ, Siminovitch L. Structural determinants responsible for the biological activity of (−)-emetine, (−)-cryptopleurine, and (−)-tylocrebrine: structure-activity relationship among related compounds. Mol Pharmacol. 1980;18(1):136–143. [PubMed] [Google Scholar]
- 17.Dolz H, Vazquez D, Jimenez A. Quantitation of the specific interaction of [14a-3H]cryptopleurine with 80S and 40S ribosomal species from the yeast Saccharomyces cerevisiae. Biochemistry. 1982;21(13):3181–3187. doi: 10.1021/bi00256a023. [DOI] [PubMed] [Google Scholar]
- 18.Cai XF, Jin X, Lee D, Yang YT, Lee K, Hong Y-S, Lee J-H, Lee JJ. Phenanthroquinolizidine alkaloids from the roots of Boehmeria pannosa potently inhibit hypoxia-inducible factor-1 in AGS human gastric cancer cells. J Nat Prod. 2006;69(7):1095–1097. doi: 10.1021/np060081y. [DOI] [PubMed] [Google Scholar]
- 19.Rao KN, Bhattacharya RK, Venkatachalam SR. Inhibition of thymidylate synthase and cell growth by the phenanthroindolizidine alkaloids pergularinine and tylophorinidine. Chem Biol Interact. 1997;106(3):201–212. doi: 10.1016/s0009-2797(97)00065-3. [DOI] [PubMed] [Google Scholar]
- 20.Rao KN, Bhattacharya RK, Veankatachalam SR. Inhibition of thymidylate synthase by pergularinine, tylophorinidine and deoxytubulosine. Indian J Biochem Biophys. 1999;36(6):442–448. [PubMed] [Google Scholar]
- 21.Rao KN, Venkatachalam SR. Inhibition of dihydrofolate reductase and cell growth activity by the phenanthroindolizidine alkaloids pergularinine and tylophorinidine: the in vitro cytotoxicity of these plant alkaloids and their potential as antimicrobial and anticancer agents. Toxicol In Vitro. 2000;14(1):53–59. doi: 10.1016/s0887-2333(99)00092-2. [DOI] [PubMed] [Google Scholar]
- 22.Gao W, Chen AP-C, Leung C-H, Gullen EA, Fuerstner A, Shi Q, Wei L, Lee K-H, Cheng Y-C. Structural analogs of tylophora alkaloids may not be functional analogs. Bioorg Med Chem Lett. 2008;18(2):704–709. doi: 10.1016/j.bmcl.2007.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suffness M, Douros J. Anticancer agents based on natural product models. Academic press; pp. 465–487. [Google Scholar]
- 24.Gao W, Bussom S, Grill SP, Gullen EA, Hu Y-C, Huang X, Zhong S, Kaczmarek C, Gutierrez J, Francis S, Baker DC, Yu S, Cheng Y-C. Structure-activity studies of phenanthroindolizidine alkaloids as potential antitumor agents. Bioorg Med Chem Lett. 2007;17(15):4338–4342. doi: 10.1016/j.bmcl.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Wei L, Shi Q, Bastow Kenneth F, Brossi A, Morris-Natschke Susan L, Nakagawa-Goto K, Wu T-S, Pan S-L, Teng C-M, Lee K-H. Antitumor agents 253. Design, synthesis, and antitumor evaluation of novel 9-substituted phenanthrene-based tylophorine derivatives as potential anticancer agents. J Med Chem. 2007;50(15):3674–3680. doi: 10.1021/jm061366a. [DOI] [PubMed] [Google Scholar]
- 26.Lin JC, Yang SC, Hong TM, Yu SL, Shi Q, Wei L, Chen HY, Yang PC, Lee KH. Phenanthrene-based tylophorine-1 (PBT-1) inhibits lung cancer cell growth through the Akt and NF-kappaB pathways. J Med Chem. 2009;52(7):1903–1911. doi: 10.1021/jm801344j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Shi Q, Liu YN, Zhao G, Bastow KF, Lin JC, Yang SC, Yang PC, Lee KH. Antitumor agents 268. Design, synthesis, and mechanistic studies of new 9-substituted phenanthrene-based tylophorine analogues as potent cytotoxic agents. J Med Chem. 2009;52(16):5262–5268. doi: 10.1021/jm9009263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang K, Wang W, Wang Q, Huang R. Efficient synthesis of aza-phenanthroindolizidine and aza-phenanthroquinolizidine and anticancer activities. Lett Org Chem. 2008;5(5):383–390. [Google Scholar]
- 29.Yang XM, Shi Q, Bastow KF, Lee KH. Antitumor Agents. 274. A New Synthetic Strategy for E-Ring SAR Study of Antofine and Cryptopleurine Analogues. Org Lett. 2010;12(7):1416–1419. doi: 10.1021/ol902819j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx. 2005;2(4):554–571. doi: 10.1602/neurorx.2.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark DE. In silico prediction of blood-brain barrier permeation. Drug Discov Today. 2003;8(20):927–933. doi: 10.1016/s1359-6446(03)02827-7. [DOI] [PubMed] [Google Scholar]
- 32.van de Waterbeemd H, Camenisch G, Folkers G, Chretien JR, Raevsky OA. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J Drug Target. 1998;6(2):151–165. doi: 10.3109/10611869808997889. [DOI] [PubMed] [Google Scholar]
- 33.http://preadmet.bmdrc.org
- 34.Wang Y, Gao W, Svitkin YV, Chen AP, Cheng YC. DCB-3503, a tylophorine analog, inhibits protein synthesis through a novel mechanism. PLoS One. 5(7):e11607. doi: 10.1371/journal.pone.0011607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.