

Abstract
Previous findings suggest a potential therapeutic action of relaxin, the putative vasodilatory signal of normal pregnancy, in some forms of cardiovascular disease. However, the mechanisms underlying the beneficial effects of relaxin have not been fully elucidated. The purpose of this study was to determine if the vasodilatory effects of relaxin are dependent upon activation of nitric oxide synthase (NOS). We examined the effect of relaxin in male Sprague-Dawley rats given Angiotensin II (ANGII, 200ng/kg/min SC by minipump), the NOS inhibitor L-NAME (1.5 mg/100g iv followed by 150 mg/L in drinking water), or vehicle for 3 weeks. After 7 days of ANGII or L-NAME, mean arterial pressure (MAP) was elevated compared to baseline. Relaxin was administered (4 μg/h, SC by minipump) for the next 2 weeks of ANGII, L-NAME, or vehicle treatment. Two-week relaxin treatment alone slightly reduced MAP in normotensive rats. Three weeks of either ANGII or L-NAME treatment alone produced hypertension, albuminuria, mild glomerular sclerosis, reduced NOx excretion, and increased oxidative stress (excretion of hydrogen peroxide and TBARS and renal cortex nitrotyrosine abundance). Relaxin reduced MAP, albumin excretion, and oxidative stress markers and preserved glomerular structure and NOx excretion in ANGII treated rats; however, relaxin did not attenuate these changes in the rats treated with L-NAME. None of the treatments affected protein abundance of neuronal or endothelial NOS in the kidney cortex. These data suggest that the vasodilatory effects of relaxin are dependent upon a functional NOS system and increased NO bioavailability possibly due to a reduction in oxidative stress.
Keywords: nitric oxide synthase, blood pressure, albuminuria, oxidative stress
Introduction
Relaxin may be useful in the treatment of cardiovascular disease in both males and females due to its vasodilatory, anti-fibrotic, and angiogenic properties (for review, see 1-3). Relaxin is a potent, endothelium-dependent vasodilator of human resistance arteries obtained from gluteal biopsies (4), and in rodents relaxin promotes dilation of microvessels in the kidney and heart as well as in reproductive organs. In addition, acute administration of relaxin opposes the actions of angiotensin II (ANGII) in vitro and in vivo (5-7). While some animal studies have shown that relaxin can reduce blood pressure when infused into spontaneously hypertensive rats (SHR) (8) and rats with 5/6 renal ablation/infarction (9), others have shown no effect of relaxin infusion on blood pressure in the SHR after 1-7 days or 2 weeks of relaxin treatment in aged (17 months) SHR despite reduced vascular resistance and improved arterial compliance (5, 10). In addition, Lekgabe, et al, demonstrated that relaxin reduces cardiac and renal fibrosis in the SHR and suggested that relaxin has therapeutic potential in hypertension (11). Therefore, the first goal of this study was to determine if chronic relaxin treatment can reduce blood pressure during chronic ANGII induced hypertension.
There is evidence that some of the cardiovascular benefits of relaxin treatment are mediated by increased production of nitric oxide (NO) (12) and that the vasodilatory effects of relaxin are dependent on NO (6,13). Baccari and Bani have recently reviewed the actions of relaxin to stimulate the NO pathway in both reproductive and non-reproductive organs (14). NO, produced by NO synthase (NOS), is an important regulator of blood pressure and regional blood flow, vascular smooth muscle proliferation, platelet aggregation, and leukocyte adhesion (15). NO deficiency is associated with endothelial dysfunction and the development of hypertension and associated target organ damage (16,17). Therefore, agents that increase NO bioavailability are of potential therapeutic use in the treatment of hypertension. Various studies have implicated all 3 of the NOS enzymes in the NO-dependent actions of relaxin according to the cell type studied (14). Within the kidney and heart, relaxin is a potent vasodilator and improves renal and coronary blood flow in a NOS dependent manner (6, 13, 18), and in vitro data suggest that relaxin stimulates NO production in endothelial and smooth muscle cells (19-22). We hypothesize that the antihypertensive effects of relaxin are dependent on activation of NOS and increased production of NO. To determine the importance of the endogenous NO system in the response to relaxin, we assessed the effects of relaxin treatment on blood pressure during chronic NOS inhibition.
Methods
Animal Models
All experiments were performed using male Sprague-Dawley rats (400 to 500 g; Harlan Laboratories, Indianapolis, IN) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved and monitored by the University of Florida Institutional Animal Care and Use Committee. Animals were housed under conditions of constant temperature and humidity and exposed to a 12:12-h light-dark cycle. All rats were given free access to regular rat chow and water. All surgeries were conducted with rats under general isoflurane anesthesia (Abbott, North Chicago, IL) using full sterile technique. A vascular catheter (tygon) was placed in the left femoral artery, threaded under the skin by trocar, exteriorized at the back of the neck, and primed and plugged. After recovery from anesthesia, rats were returned to individual cages in which they moved freely. All catheterized rats were trained to accept handling and the activity in the laboratory.
After 1 week, all rats were placed on a low-NOx diet (ICN AIN 76C, MP Biomedicals, Aurora, OH), and 24h later were placed in metabolic cages to facilitate 24-h urine collection, following which an arterial blood sample and mean arterial blood pressure (MAP) measurement was obtained from the indwelling arterial catheter, and then rats were randomly assigned to 1 of 3 groups: Sham, angiotensin II infused (ANGII), and NG-nitro-L-arginine methyl ester (L-NAME, a NOS inhibitor) treated as shown in Table 1. After the first week of treatment, recombinant human relaxin (relaxin) was then given to half of the rats in each of the 3 cohorts: ANGII and L-NAME were from Sigma (St. Louis, MO), and relaxin was a gift from Corthera, Inc. (San Mateo, CA). In both ANGII treated groups, ALZET Model-2001 minipumps (Durect Corporation, Cupertino, CA) were used to deliver ANGII at 200ng/kg/min for 1 week. For the L-NAME treated groups, a bolus dose was given intravenously (1.5 mg/100 g BW bolus via tail vein) followed by L-NAME ad libitum in the drinking water (150 mg/l) for the duration of the experiment. After 7 days, minipumps were replaced with ALZET 2ML2 minipumps containing ANGII II (200ng/kg/min) or relaxin (4 μg/h) alone or in combination for 2 weeks. Sham minipumps containing vehicle only were used in rats not receiving ANGII or relaxin. Rats were placed on a low NOx diet 24h prior to being placed in metabolic cages, and then MAP and blood samples were taken at the end of each week. At the end of the study, both mean and systolic blood pressures were measured. Rats were anesthetized with isoflurane, and an aortic blood sample was collected via the abdominal aorta. The kidneys were perfused blood free with cold PBS, and the right kidney was separated into cortex and medulla and snap frozen in liquid nitrogen. Then, the left kidney was then perfused with 2% paraformaldehyde, lysine, meta-periodate (PLP) for 8-10 min and removed.
Table 1.
Treatment protocol.
| Group | Week 1 | Weeks 2 - 3 | |
|---|---|---|---|
| 1 | CON (n=9) | Sham minipump | Sham minipump |
| 2 | relaxin (n=11) | Relaxin minipump | |
| 3 | ANGII (n=10) | ANGII minipump | ANGII minipump |
| 4 | ANGII+relaxin (n=10) | ANGII+Relaxin minipump | |
| 5 | L-NAME(n=9) | L-NAME (initial iv bolus, then via water) | L-NAME (water) + Sham minipump |
| 6 | L-NAME+relaxin (n=8) | L-NAME (water) + Relaxin minipump | |
Renal Histology
Paraffin embedded kidneys were sectioned at a thickness of 5 μm onto Superfrost plus slides, and kidney sections were stained using a periodic acid-Schiff stain kit (Sigma) with hematoxylin as the secondary stain. Up to one hundred glomeruli were scored, blinded, as follows: 0=healthy glomeruli, +1=<25% damage, +2=25-50% damage, +3=51-74% damage, +4=>75% damage. A glomerulosclerosis index score (GSI) was calculated using the following equation: (#of+1)+ 2(#of+2)+ 3(#of+3)+ 4(#of +4)/ total glomeruli observed as previously described (23).
Western Blotting
Protein abundances were detected using Western blotting, as previously described (24). Briefly, 200 μg of kidney cortex homogenate was loaded on 6% or 7.5% polyacrylamide gels and separated by electrophoresis. Membranes were incubated overnight with specific antibodies: mouse monoclonal NOS1 (1:50 dilution, Santa Cruz Biotechnology, SC-5302), rabbit polyclonal NOS1 (1:500 dilution, Thermo Scientific, PAI-033), mouse monoclonal NOS3 (1:250 dilution, BD Transduction, 610297), and mouse monoclonal nitrotyrosine (1:500, Upstate, 05-233). The membranes were then incubated with corresponding secondary antibodies: goat anti-rabbit antibody (1:3,000 dilution; Bio-Rad 170-6515) or goat anti-mouse antibody (1:2000 dilution; Bio-Rad 170-6516). Bands of interest were visualized using enhanced chemiluminescence reagent and quantified by densitometry (VersaDoc imaging system and Quantity One Analysis software, Bio-Rad) as integrated optical density (IOD) after subtraction of background. IOD was factored for Ponceau Red staining (Sigma) to correct for any variations in total protein loading and for an internal standard, and protein abundance is represented as IOD/Ponceau Red/standard.
Urine and Plasma Analysis
Urine albumin concentrations were measured using a commercially available kit (Cayman Chemical) according to the manufacturer's specifications. Total NO production (from NOx = NO3− + NO2−) was measured in urine Griess reaction (25). Plasma and urine creatinine concentrations were measured by HPLC as described by us previously (26). Urine concentrations of hydrogen peroxide and thiobarbituric acid reactive substances (TBARS) were measured using commercially available kits (Amplex Red, Molecular Probes and Oxitek, Zeptometrix, respectively) according to the manufacturer's specifications.
Statistical analysis
Results are presented as mean ± SE. For multiple comparisons, ANOVA with Newman-Keuls post hoc analysis was used. Mean arterial pressure and urinary NOx excretion were compared using repeated-measures ANOVA with Bonferroni posttest to compare the effects of both time (within group comparisons) and treatment (between group comparisons) using Prism 4 software (Graph Pad Software, San Diego, CA). Histologic (non-parametric) data were analyzed by Kruskal-Wallis test with Dunn's posttest. p<0.05 was considered statistically significant.
Results
As shown in Figure 1, the control rats maintained a constant MAP over the three week study period. After one week, ANGII and L-NAME treatments increased MAP to a similar extent (145±4 mm Hg in all rats receiving ANGII treatment and 156±3 mm Hg in all rats receiving L-NAME treatment; 127±3 mm Hg in vehicle treated rats, p<0.05 vs CON for both). After the first week of hypertension, relaxin was then given randomly to half of the rats in each treatment cohort (vehicle, ANGII and L-NAME). MAP remained elevated in the groups that received either ANGII or L-NAME alone throughout the study period. Seven days of relaxin treatment had no effect on MAP in any of these 3 groups. After 2 weeks, however, relaxin treatment slightly reduced MAP in normotensive rats (p<0.05 vs baseline and p<0.01 vs week 1); however, MAP in the relaxin treated group was not statistically lower than control treated rats. Relaxin reduced MAP in the ANGII+relaxin group so that MAP was similar to values observed in the control group (p<0.001 vs ANGII alone) but had no effect on MAP in the L-NAME+relaxin group. Similarly, systolic blood pressure (Table 2) was elevated after 3 week treatment with ANGII or L-NAME (p<0.01 vs CON). Relaxin treatment normalized systolic pressure in the ANGII hypertensive group (p<0.001 vs ANGII alone) but had no effect on systolic pressure in the control or L-NAME treated rats.
Figure 1.

Two-week relaxin treatment normalized blood pressure during ANG II but not L-NAME induced hypertension. (A) Effects of ANGII and relaxin and (B) effects of L-NAME and relaxin on mean arterial pressure (MAP) in conscious rats. Values are mean ± SEM, n=8-11. * p<0.05 vs respective baseline. # p<0.05 vs ANGII.
Table 2.
Systolic blood pressure and creatinine clearance in rats treated with ANGII, L-NAME and/or relaxin.
| Group | Systolic Blood Pressure (mm Hg) | Creatinine Clearance (ml/min/kg) | |
|---|---|---|---|
| 1 | CON | 128 ±6 | 6.8 ± 1.7 |
| 2 | relaxin | 118 ± 6 | 7.4 ± 0.9 |
| 3 | ANGII | 168 ± 6 * | 5.4 ± 0.6 |
| 4 | ANGII+relaxin | 123± 7 † | 6.7 ± 1.1 |
| 5 | L-NAME | 176 ± 9 * | 5.5 ± 1.1 |
| 6 | L-NAME+relaxin | 178 ± 10 * | 5.4 ± 0.7 |
Values are mean ± SEM, n=8-11,
denotes p< 0.01 vs CON,
denotes p<0.01 vs ANG II alone.
As shown in Table 2, there was no change in creatinine clearance with relaxin treatment with or without ANGII or LNAME. Urinary albumin excretion, a marker of renal injury, was elevated in both ANGII (p<0.05 vs CON) and LNAME (p<0.01 vs CON) treated groups (Figure 2). Relaxin treatment normalized albuminuria in the ANGII treated rats, but had no effect in control or L-NAME treated rats. Mild glomerular injury was also evident in both ANGII (p<0.05 vs CON) and LNAME (p<0.01 vs CON) treated groups. Relaxin treatment preserved glomerular structure during chronic ANGII treatment but had no effect during L-NAME treatment. Please see http://hyper.ahajournals.org for representative images of the renal histology (Figure S1).
Figure 2.

Two-week relaxin treatment prevents the development of renal injury in ANG II but not L-NAME induced hypertension. Effects of ANGII and relaxin on (A) the urinary excretion of albumin and (B) glomerular injury and effects of L-NAME and relaxin on (C) the urinary excretion of albumin and (D) glomerular injury. Values are mean ± SEM, n=8-11. * p<0.05 vs control.
Urinary excretion of NO metabolites, NOx, was not changed by one week of treatment with ANGII (p=0.14 vs baseline) but was significantly reduced by one week of L-NAME treatment (p=0.04 vs baseline, Figure 3). After two weeks of hypertension, NOx excretion was decreased in both ANGII and L-NAME treated rats (p<0.01 vs baseline), and remained low at week three in these groups. Relaxin treatment alone had no effect on NOx excretion (p=0.35 vs baseline); however, rats that received relaxin along with ANGII showed restoration of NOx excretion to normal values (p<0.05 vs ANG II alone at weeks 2 and 3). The fall in NOx excretion in rats given L-NAME persisted despite relaxin administration. Western blot analysis revealed that neither hypertensive treatment nor relaxin had an effect on the protein abundance of NOS1 alpha or beta or NOS3 in the kidney cortex (Figure 4).
Figure 3.

Relaxin treatment preserves urinary excretion of NO metabolites during ANG II but not L-NAME induced hypertension. (A) Effects of ANGII and relaxin and (B) effects of L-NAME and relaxin on excretion of nitric oxide metabolites (UNOxV). Values are mean ± SEM, n=8-11. * p<0.05 vs respective baseline. # p<0.05 vs ANGII.
Figure 4.
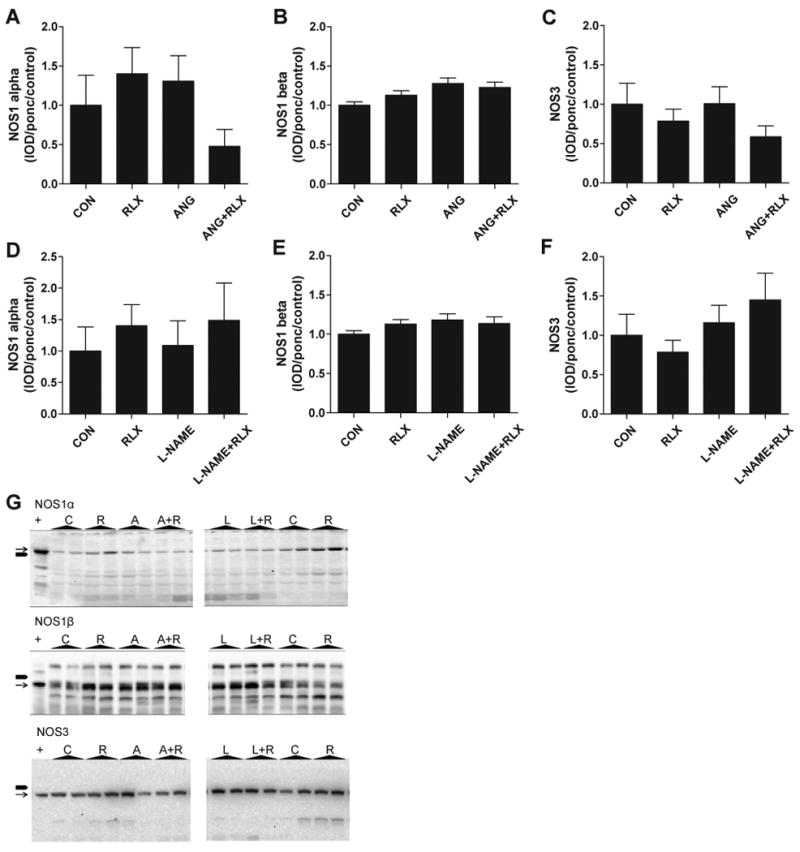
Relaxin treatment does not affect renal cortex abundance of NOS1 or NOS3. Effects of ANG II and relaxin (panels A-C) and effects of L-NAME and relaxin (panels D-F) on abundance of the constitutive NOS enzymes in the renal cortex. Values are mean ± SEM, n=7-10. (G) Representative images of Western blots for NOS1α, NOS1β, and NOS3 in kidney cortex homogenates from Control (C), Relaxin (R), ANGII (A), ANGII + Relaxin (A+R), L-NAME (L), and L-NAME + Relaxin (L+R) treated rats. + indicates positive control. Black block arrow (■) on left side of image indicates 150 kDa as measured using the BioRad Kaleidoscope Precision Plus Protein Standards. Thin black arrow (→) indicates specific protein band.
After three weeks of treatment with either ANGII or L-NAME, renal excretion of two markers of oxidative stress, hydrogen peroxide and TBARS (p<0.05 vs CON for both markers), was increased compared to control (Figure 5). In addition, Western blot analysis showed that there is increased nitrotyrosine content in the renal cortex of both hypertensive groups (p<0.05 for ANGII vs CON, p<0.01 for L-NAME vs CON,Figure 5 E-G). Two-week relaxin treatment in normotensive rats had no effect on these markers; however, relaxin treatment normalized the excretion of both hydrogen peroxide (p<0.05 vs CON) and TBARS (p<0.01 vs CON) and normalized nitrotyrosine immunoreactivity in rats treated with ANGII. No effect of relaxin treatment was observed in the L-NAME treated group.
Figure 5.
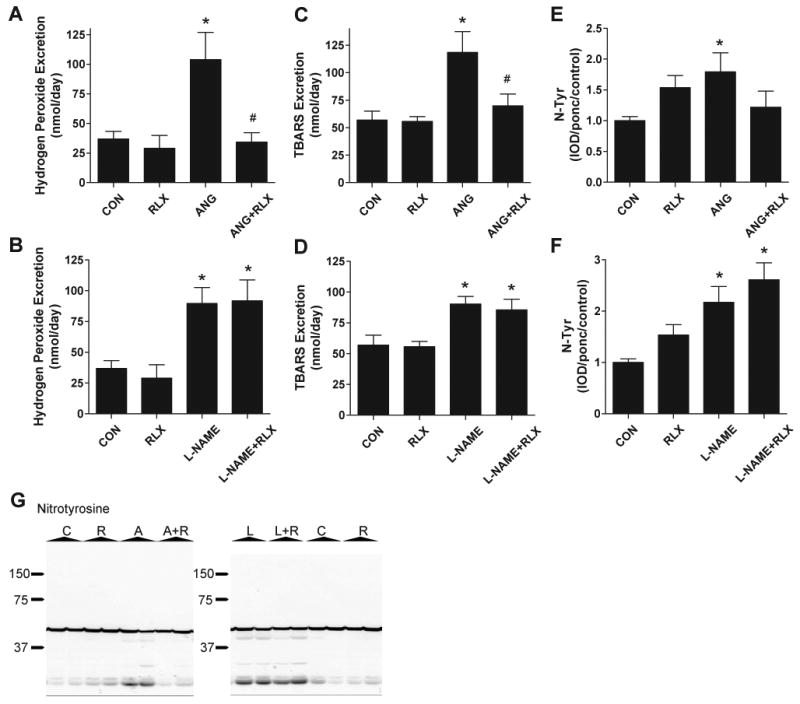
Relaxin reduces oxidative stress during ANG II but not L-NAME induced hypertension. Effects of ANG II and relaxin (panels A,C, and E) and effects of L-NAME and relaxin (panels B,D, and F) on excretion of hydrogen peroxide (panels A-B) and TBARS (panels C-D) and renal cortex nitro-tyrosine abundance. Values are mean ± SEM, n=8-9. * p<0.05 vs control. # p<0.05 vs ANGII. (G) Representative images of Western blots for nitrotyrosine in kidney cortex homogenates from Control (C), Relaxin (R), ANGII (A), ANGII + Relaxin (A+R), L-NAME (L), and L-NAME + Relaxin (L+R) treated rats. Black block arrows on right side of image illustrate molecular weights as measured using the BioRad Kaleidoscope Precision Plus Protein Standards.
Discussion
The main novel findings of this study are that relaxin lowers blood pressure, reduces albuminuria and glomerular sclerosis, and increases NOx excretion during chronic ANGII infusion. In contrast, relaxin was ineffective in preventing hypertension or renal injury during LNAME administration, demonstrating that the antihypertensive and renoprotective effects of relaxin are dependent upon a functional NOS system. Relaxin treatment had no effect on the protein abundance of the constitutive NOS enzymes, NOS1 and NOS3, in the kidney cortex indicating that the protective effect of relaxin in ANGII hypertension is not simply due to an increase in NOS enzyme abundance. Relaxin treatment reduced oxidative stress in rats treated with ANGII suggesting that the beneficial effects of relaxin are due to an antioxidant mechanism (see below).
Conrad et al demonstrated a reduction in systemic vascular resistance in normal non-pregnant female rats but without a change in blood pressure after 10 days of relaxin treatment (27). In the present study, relaxin treatment in normotensive rats had no effect on any of the measured parameters after 7 days, but 14 days of relaxin reduced MAP compared to baseline levels. The current study is the first to determine the effect of chronic relaxin treatment during ANGII induced hypertension. Here, we found that relaxin reduced blood pressure in rats with ANGII induced hypertension after 2 weeks of treatment. Our findings are consistent with in vitro and in vivo evidence that relaxin blunts the constrictor response to ANGII (5-7). Debrah et al found that acute relaxin treatment in ANGII hypertensive rats reduced arterial load and increased cardiac output within 2 hours of treatment but did not affect blood pressure (5). Likewise, we did not observe an anti-hypertensive effect of relaxin during the first week of treatment, but relaxin normalized blood pressure after two weeks.
There is some controversy in the literature regarding the effects of long-term relaxin treatment on blood pressure regulation in animal models of hypertension. Evidence for the vasodilatory and anti-hypertensive role of relaxin has been reported in both male and female SHR and in the 5/6 renal ablation/infarction model in male rats (8-9, 11) while others found no effect of relaxin on blood pressure in male SHR despite increased arterial compliance (5, 10). We cannot speculate on the reasons for the differences in the blood pressure lowering effects of relaxin in the SHR since we have not used this model; however, even when blood pressure did not fall with relaxin there was evidence of some cardiovascular benefit in every study.
The decrease in blood pressure in ANGII treated rats with relaxin was associated with a restoration of NOx excretion, a marker of NO bioavailability, to normal values. In contrast, relaxin had no effect on either blood pressure or NOx excretion during chronic NOS inhibition with LNAME. This is the first study that illustrates that the blood pressure lowering effects of relaxin require a functional NOS system. Our findings are consistent with previous work by Danielson et al that showed that acute NOS inhibition abolished the renal vasodilatory response to relaxin (6), and the effects of relaxin on coronary blood flow are also dependent on stimulation of NO (13). The molecular mechanisms by which relaxin activates the NO system are currently unknown. Based on the current studies, we are unable to determine which NOS isoform(s) are involved in the response to relaxin as L-NAME inhibits all NOS. In addition, the specific isoform of NOS activated by relaxin varies according to cell type (20, 21, 28, 29). For example, relaxin activates NOS II in cultured coronary endothelial cells (20) and aortic vascular smooth muscle cells (21); and upregulates NOS III in uterus (28). In the kidney, the hemodynamic effects of relaxin occur via NO (6) possibly via activation of endothelin B receptors (30), but the source of this NO has not been defined. In the current study, we saw no effect of relaxin on level of the constitutive NOS enzymes (NOS1 and NOS3) in the renal cortex. Of course, in addition to protein abundance, the activity of the NOS enzymes is regulated by many factors including substrate and cofactor availability, post-translational modifications such as phosphorylation, and the presence of oxidative stress.
It is interesting that relaxin is able to fully restore NOx excretion during ANGII hypertension despite the known action of ANGII to increase oxidative stress via activation of NADPH oxidase (31-32). Increased superoxide production directly reduces NO bioavailability by binding with NO to produce peroxynitrite and by reducing the availability of tetrahydrobiopterin, an essential NOS cofactor, which reduces NO production and causes the NOS enzymes to switch to superoxide generation (33). Indeed, we observed that both chronic ANGII and L-NAME models exhibited significant oxidative stress and that relaxin reduced urinary excretion of hydrogen peroxide and TBARS and renal cortex nitrotyrosine content in the rats treated with ANGII. This is consistent with work by Masini et al which demonstrated that relaxin decreases lipid peroxidation in myocardial tissue following ischemia reperfusion injury (34) and lipid peroxidation and 8-hydroxy-2′-deoxyguanosine levels in ileal tissue from animals subjected to splanchnic artery occlusion (35). However, we did not observe any decrease in oxidative stress markers during L-NAME hypertension suggesting that the anti-oxidant effects of relaxin require stimulation of endogenous NO.
Our findings indicate that relaxin can also prevent ANGII induced renal injury but is not protective during chronic NOS inhibition. The protective effect observed during ANGII treatment may be predominantly due to the blood pressure lowering effect of relaxin in this model. However, others have demonstrated renoprotective effects of relaxin in experimental models of renal disease with normal blood pressure (9, 36) and it is likely that with established injury relaxin can directly remodel the extracellular matrix via reduced collagen synthesis, increased activity of matrix metalloproteinases, and antagonism of pro-fibrotic factors (11). In cultured kidney cells, relaxin has been shown to promote fibronectin degradation via the ubiquitin-proteasomal pathway (37) and to increase the activity of matrix metalloproteinases (38). Most likely, a combination of hemodynamic and direct actions of relaxin account for the remarkable ability of relaxin to preserve renal function and prevent / reverse renal injury in the male relaxin knockout mouse (39), and in the aging rat (40).
Perspectives
Over half of the people who are treated for hypertension do not have adequate blood pressure control. New, effective treatments for hypertension are desperately needed, and our data indicate that relaxin is a potential therapeutic agent for the treatment of hypertension and the resulting end organ damage. However, this study suggests that an assessment of the endogenous NO system is critical before use of relaxin to treat cardiovascular disease, since in states where the endogenous NO system cannot be stimulated, relaxin may not be an effective treatment. This work also suggests a potential role for relaxin as an antioxidant and highlights the need for future studies to investigate the impact of relaxin on pro- and anti-oxidant pathways.
Supplementary Material
Acknowledgments
We thank Elaine Unemori, Dennis Stewart and Corthera, Inc. for providing the relaxin, Kirk Conrad for helpful discussions, and Harold Snellen for technical assistance
Sources of Funding: This work was supported by a Postdoctoral Fellowship from the American Heart Association and the University of Florida Multidisciplinary Training Program in Hypertension T32HL083810 (to J.M.S.) and a grant from the National Institutes of Health (DK056843 to C.B.).
Footnotes
Disclosures: The authors have no relationships to disclose.
References
- 1.Sherwood OD. Relaxin's physiological roles and other diverse actions. Endocrine Rev. 2004;25:205–234. doi: 10.1210/er.2003-0013. [DOI] [PubMed] [Google Scholar]
- 2.McGuane JT, Parry LJ. Relaxin and the extracellular matrix: molecular mechanisms of action and implications for cardiovascular disease. Expert Rev Mol Med. 2005;7:1–18. doi: 10.1017/S1462399405009944. [DOI] [PubMed] [Google Scholar]
- 3.Conrad KP. Unveiling the vasodilatory actions and mechanisms of relaxin. Hypertension. 2010;56:2–9. doi: 10.1161/HYPERTENSIONAHA.109.133926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher C, MacLean M, Morecroft I, Seed A, Johnston F, Hillier C, McMurray J. Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? Circulation. 2002;106:292–295. doi: 10.1161/01.cir.0000025630.05387.45. [DOI] [PubMed] [Google Scholar]
- 5.Debrah DO, Conrad KP, Jeyabalan A, Danielson LA, Shroff SG. Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension. 2005;46:745–750. doi: 10.1161/01.HYP.0000184230.52059.33. [DOI] [PubMed] [Google Scholar]
- 6.Danielson LA, Sherwood OD, Conrad KP. Relaxin is a potent renal vasodilator in conscious rats. J Clin Invest. 1999;103:525–533. doi: 10.1172/JCI5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samuel CS, Unemori EN, Mookerjee I, Bathgate RA, Layfield SL, Mak J, Tregear GW, Du XJ. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–4133. doi: 10.1210/en.2004-0209. [DOI] [PubMed] [Google Scholar]
- 8.St-Louis J, Massicotte G. Chronic decrease of blood pressure by rat relaxin in spontaneously hypertensive rats. Life Sci. 1985;37:1351–1357. doi: 10.1016/0024-3205(85)90251-6. [DOI] [PubMed] [Google Scholar]
- 9.Garber SL, Mirochnik Y, Brecklin C, Slobodskoy L, Arruda JA, Dunea G. Effect of relaxin in two models of renal mass reduction. Am J Nephrol. 2003;23:8–12. doi: 10.1159/000066302. [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Chakravorty A, Bathgate RA, Dart AM, Du XJ. Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. Hypertension. 2010;55:1260–1266. doi: 10.1161/HYPERTENSIONAHA.109.149369. [DOI] [PubMed] [Google Scholar]
- 11.Lekgabe ED, Kiriazis H, Zhao C, Xu Q, Moore XL, Su Y, Bathgate RA, Du XJ, Samuel CS. Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension. 2005;46:412–418. doi: 10.1161/01.HYP.0000171930.00697.2f. [DOI] [PubMed] [Google Scholar]
- 12.Nistri S, Bani D. Relaxin receptors and nitric oxide synthases: search for the missing link. Reprod Biol Endocrinol. 2003;1:5. doi: 10.1186/1477-7827-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bani-Sacchi T, Bigazzi M, Bani D, Mannaioni PF, Masini E. Relaxin-induced increased coronary flow through stimulation of nitric oxide production. Br J Pharmacol. 1995;116:1589–1594. doi: 10.1111/j.1476-5381.1995.tb16377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baccari MC, Bani D. Relaxin and nitric oxide signalling. Curr Protein Pept Sci. 2008;9:638–645. doi: 10.2174/138920308786733921. [DOI] [PubMed] [Google Scholar]
- 15.Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol. 2006;176:213–254. doi: 10.1007/3-540-32967-6_7. [DOI] [PubMed] [Google Scholar]
- 16.Portaluppi F, Boari B, Manfredini R. Oxidative stress in essential hypertension. Curr Pharm Des. 2004;10:1695–1698. doi: 10.2174/1381612043384619. [DOI] [PubMed] [Google Scholar]
- 17.Baylis C. Nitric oxide deficiency in chronic kidney disease. Am J Physiol. 2008;294:F1–F9. doi: 10.1152/ajprenal.00424.2007. [DOI] [PubMed] [Google Scholar]
- 18.Novak J, Ramirez RJ, Gandley RE, Sherwood OD, Conrad KP. Myogenic reactivity is reduced in small renal arteries isolated from relaxin-treated rats. Am J Physiol. 2002;283:R349–R355. doi: 10.1152/ajpregu.00635.2001. [DOI] [PubMed] [Google Scholar]
- 19.Quattrone S, Chiappini L, Scapagnini G, Bigazzi B, Bani D. Relaxin potentiates the expression of inducible nitric oxide synthase by endothelial cells from human umbilical vein in in vitro culture. Mol Hum Reprod. 2004;10:325–330. doi: 10.1093/molehr/gah040. [DOI] [PubMed] [Google Scholar]
- 20.Failli P, Nistri S, Quattrone S, Mazzetti L, Bigazzi M, Sacchi TB, Bani D. Relaxin up-regulates inducible nitric oxide synthase expression and nitric oxide generation in rat coronary endothelial cells. FASEB J. 2002;16:252–254. doi: 10.1096/fj.01-0569fje. [DOI] [PubMed] [Google Scholar]
- 21.Bani D, Failli P, Bello MG, Thiemermann C, Bani Sacchi T, Bigazzi M, Masini E. Relaxin activates the L-arginine-nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension. 1998;31:1240–1247. doi: 10.1161/01.hyp.31.6.1240. [DOI] [PubMed] [Google Scholar]
- 22.Nistri S, Chiappini L, Sassoli C, Bani D. Relaxin inhibits lipopolysaccharide-induced adhesion of neutrophils to coronary endothelial cells by a nitric oxide-mediated mechanism. FASEB J. 2003;17:2109–2111. doi: 10.1096/fj.03-0216fje. [DOI] [PubMed] [Google Scholar]
- 23.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int. 1984;26:137–143. doi: 10.1038/ki.1984.147. [DOI] [PubMed] [Google Scholar]
- 24.Xiao S, Erdely A, Wagner L, Baylis C. Uremic levels of BUN do not cause nitric oxide deficiency in rats with normal renal function. Am J Physiol Renal Physiol. 2001;280:F996–F1000. doi: 10.1152/ajprenal.2001.280.6.F996. [DOI] [PubMed] [Google Scholar]
- 25.Suto T, Losonczy G, Qiu C, Hill C, Samsell L, Ruby J, Charon N, Venuto R, Baylis C. Acute changes in urinary excretion of nitrite-nitrate (UNOXV) do not predict renal vascular NO production. Kidney Int. 1995;48:1272–1277. doi: 10.1038/ki.1995.411. [DOI] [PubMed] [Google Scholar]
- 26.Sasser JM, Moningka NC, Cunningham MW, Jr, Croker B, Baylis C. Asymmetric dimethylarginine in angiotensin II-induced hypertension. Am J Physiol. 2010;298:R740–R746. doi: 10.1152/ajpregu.90875.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conrad KP, Debrah DO, Novak J, Danielson LA, Shroff SG. Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology. 2004;145:3289–3296. doi: 10.1210/en.2003-1612. [DOI] [PubMed] [Google Scholar]
- 28.Bani D, Baccari MC, Nistri S, Calamai F, Bigazzi M, Sacchi TB. Relaxin up-regulates the nitric oxide biosynthetic pathway in the mouse uterus: involvement in the inhibition of myometrial contractility. Endocrinology. 1999;140:4434–4441. doi: 10.1210/endo.140.10.7055. [DOI] [PubMed] [Google Scholar]
- 29.Mookerjee I, Hewitson TD, Halls ML, Summers RJ, Mathai ML, Bathgate RA, Tregear GW, Samuel CS. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 2009;23:1219–1229. doi: 10.1096/fj.08-120857. [DOI] [PubMed] [Google Scholar]
- 30.Danielson LA, Kercher LJ, Conrad KP. Impact of gender and endothelin on renal vasodilation and hyperfiltration induced by relaxin in conscious rats. Am J Physiol. 2000;279:R1298–R1304. doi: 10.1152/ajpregu.2000.279.4.R1298. [DOI] [PubMed] [Google Scholar]
- 31.Fukui T, Ishizaka N, Rajagopalan S, Laursen JB, Capers Q, 4th, Taylor WR, Harrison DG, de Leon H, Wilcox JN, Griendling KK. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ Res. 1997;80:45–51. doi: 10.1161/01.res.80.1.45. [DOI] [PubMed] [Google Scholar]
- 32.Welch WJ, Blau J, Xie H, Chabrashvili T, Wilcox CS. Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am J Physiol. 2004;288:H22–H28. doi: 10.1152/ajpheart.00626.2004. [DOI] [PubMed] [Google Scholar]
- 33.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, Sacchi TB. Relaxin counteracts myocardial damage induced by ischemia-reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology. 1997;138:4713–4720. doi: 10.1210/endo.138.11.5520. [DOI] [PubMed] [Google Scholar]
- 35.Masini E, Cuzzocrea S, Mazzon E, Muia C, Vannacci A, Fabrizi F, Bani D. Protective effects of relaxin in ischemia/reperfusion-induced intestinal injury due to splanchnic artery occlusion. Br J Pharmacol. 2006;148:1124–1132. doi: 10.1038/sj.bjp.0706811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garber SL, Mirochnik Y, Brecklin CS, Unemori EN, Singh AK, Slobodskoy L, Grove BH, Arruda JA, Dunea G. Relaxin decreases renal interstitial fibrosis and slows progression of renal disease. Kidney Int. 2001;59:876–882. doi: 10.1046/j.1523-1755.2001.059003876.x. [DOI] [PubMed] [Google Scholar]
- 37.McDonald GA, Sarkar P, Rennke H, Unemori E, Kalluri R, Sukhatme VP. Relaxin increases ubiquitin-dependent degradation of fibronectin in vitro and ameliorates renal fibrosis in vivo. Am J Physiol Renal Physiol. 2003;285:F59–F67. doi: 10.1152/ajprenal.00157.2002. [DOI] [PubMed] [Google Scholar]
- 38.Heeg MH, Koziolek MJ, Vasko R, Schaefer L, Sharma K, Müller GA, Strutz F. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 2005;68:96–109. doi: 10.1111/j.1523-1755.2005.00384.x. [DOI] [PubMed] [Google Scholar]
- 39.Samuel CS, Zhao C, Bond CP, Hewitson TD, Amento EP, Summers RJ. Relaxin-1-deficient mice develop an age-related progression of renal fibrosis. Kidney Int. 2004;65:2054–2064. doi: 10.1111/j.1523-1755.2004.00628.x. [DOI] [PubMed] [Google Scholar]
- 40.Danielson LA, Welford A, Harris A. Relaxin improves renal function and histology in aging Munich Wistar rats. J Am Soc Nephrol. 2006;17:1325–1333. doi: 10.1681/ASN.2005121307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.