

Abstract
The temporal response of pulsed nano-electrospray ionization mass spectrometry (nano-ESI-MS) was studied and its influence on ion formation and detection was characterized. Rise and decay times for the mass resolved ion current were determined to be 20 ± 3 msec and 61 ± 5 msec, respectively, which led to a maximum pulse rate of 12 Hz. Pulsed nano-ESI operation was demonstrated from a multi-sprayer source controlled by a high voltage pulsing circuit constructed in-house. The desired source mode of operation (e.g. pulsing or continuous) can be realized solely by controlling the voltage applied to each sprayer.
Keywords: Mass spectrometry, Nano-ESI, Pulsed ESI, ESI temporal response, Flared inlet capillary, Dual ion source
INTRODUCTION
Electrospray Ionization Mass Spectrometry (ESI-MS) has proven to be useful for the analysis of biologically relevant compounds due to its sensitivity, throughput, and ability to analyze large, non-volatile biological molecules from solution.[1] Nano-ESI is a low flow rate regime of ESI that has gained widespread use in the study of biopolymers.[2–9] The smaller sprayer tips and lower applied voltages used with nano-ESI reduce sample consumption and result in electrospray droplets of higher surface-to-volume ratios.[10–12] Higher surface-to-volume ratios allow more analyte molecules to be closer to the droplet surface and thus more easily desorbed into the gas phase, thereby improving sensitivity.[11,13]
Several researchers have utilized multiple conventional ESI sprayers on a variety of instruments to increase sample throughput. Throughput has been increased by a factor of four while mass measurement accuracies of below 5 ppm were maintained for a variety of metabolites by coupling an ESI source to each of four HPLC effluent streams and using a fifth channel to introduce a lock mass for calibration.[14] A dual sprayer setup on the front end of a TOF instrument was used to achieve mass accuracies of 3 ppm for ions below mass-to-charge (m/z) 1000 using sub-picomole amounts of sample.[15] Each sprayer was independently and sequentially sampled by alternately switching the high voltage (HV) applied to each sprayer.[15]
The use of HV switching as a means to sample each sprayer has several advantages. No longer is a mechanical barrier (e.g., a baffle) required as the switching mechanism.[16] By avoiding the use of a mechanical barrier, the sprayers can be positioned physically closer to the mass spectrometer; which is optimal for nano-ESI. The use of HV switching also allows for shorter transit times between sampling from different sprayers (~1 ms) as opposed to the use of a mechanical switching method (~100 ms); where the former is also advantageous to source stability, sample consumption, and selective sprayer sampling.[4,14,15]
In addition to increasing throughput, multiple ESI sources have been used in parallel arrangements to improve mass measurement accuracy, [4] reduce adverse affects from multiple solutions interacting prior to analysis, [4,17] and allow ion/ion reactions to be studied.[18–21] Work has been presented on the use of two nano-ESI sprayers operating continuously but at opposite polarities for ion/ion reactions.[22] Converting the continuous ESI process into a controlled pulsed mechanism alleviates the problem of sample loss. Pulsing a single nano-ESI source has been achieved by holding the sample solution at a high potential and using a cylindrical piezoelectric element to dispense 10 picoliter droplets with a drop-on-demand mechanism.[23] This method demonstrated detection limits comparable to non-pulsed nano-ESI but with lower total sample consumption and greater control of the amount of sample consumed over a given time period.[23] Recently multiple nano-ESI sprayers have been pulsed in experiments designed to explore ion-ion reactions.[20,21] Controlled pulsing of the ESI process can lead to more stable mass spectra because the sample flow rate can be matched with the reduced spraying rate. Pulsed ESI can be achieved by externally pulsing the voltage applied to the sprayer and has been successfully demonstrated at a reduced flow rate of 3 μL/min.[24] By modulating the spray voltage, nano-ESI has been pulsed at a maximum frequency of 350 kHz.[25] However, this frequency was determined by monitoring the current on a lens element immediately following the nano-ESI needle not by detecting ions passed through a mass spectrometer. At this point the reason for the discrepancy in pulsing rates is not clear, but understanding the limits of a pulsed nano-ESI system in a mass spectrometer is important in understanding the possible applications for such a setup.
The first section of the work presented here is the characterization of pulsed nano-ESI using mass spectrometric detection. The pulsed nano-ESI mechanism is studied to elucidate optimal operational limits and to explore the fundamental behavior associated with pulsing the spray high voltage. For the second section, a dual nano-ESI source has been developed that is controlled using applied voltages either in pulsed or continuous modes of operation. The dual source is used in conjunction with a flared inlet capillary that was built in-house and has been described elsewhere.[26,27] The source allows mass accuracy, sample throughput, and sprayer-to-sprayer reproducibility to be improved and was designed with the benefits associated with HV switching in mind. Furthermore, the source presented herein utilizes two nano-ESI sprayers yet does not require the instrument to be modified and consequently should be applicable to any mass spectrometer with an atmospheric sampling orifice.
EXPERIMENTAL
Samples
Peptides trialanine (AAA), leucine enkephalin (YGGFL), and Gramicidin S, polyethylene glycol 600 (PEG 600), and polypropylene glycol 425 (PPG 425) were purchased from Sigma (St. Louis, MO) and used without further purification. Unless noted otherwise, working solutions of each peptide were made at 100 μM in 75/20/5 by volume acetonitrilel/water/formic acid, except Gramicidin S solutions which were at 20 μM in 75/20/5 methanol/water/acetic acid. The PEG 600 solution was made to 100 μM in methanol and the PPG 425 solution was made to 50 μM in methanol with an equimolar amount of sodium acetate added for polymer cationization. Solutions for the solvent composition experiments were 100 μM YGGFL in methanol/water/acetic acid where the percent by volume composition of each component was parametrically adjusted. Methanol and water (100% by volume) solvent systems were also explored. HPLC grade acetonitrile, methanol, and water were purchased from Fisher Scientific (Fair Lawn, NJ). Certified A.C.S. formic and acetic (glacial) acids were also purchased from Fisher Scientific.
Instrumentation
Temporal Response Experiments
Time constant and solution composition experiments were performed on a home-built hybrid quadrupole time-of-flight mass spectrometer (Q-TOF).[28] The quadrupole is followed by an ion optics system which can deflect the ions toward a channeltron electron multiplier (Burle Industries, MA) or allow them to pass to the pusher of the TOF analyzer. For these experiments ions were deflected to the electron multiplier. The total distance from the tip of the nano-ESI needle (see description below) to the deflection electrodes is 0.552 m. The quadrupole was set to transmit ions of m/z 550–560; it has been experimentally determined in our lab that these ions have an average kinetic energy of 13.3 ± 0.2 eV.[28] Time constants were determined from the ion current measured at the electron multiplier, which was monitored with a 150 MHz digital oscilloscope (Tektronix, Richardson, TX).
The time constant experiments utilized a high voltage pulser (DEI PVX-4130, Fort Collins, CO) operated at various pulse widths to control the voltage applied to the nano-ESI sprayer. The high and low voltages for the pulser were supplied from a Spellman 4 kV power supply (Spellman, Hauppauge, NY) and an Ortec EG&G 3kV power supply (Ortec, Oak Ridge, TN). The high voltage pulser was triggered using a digital delay generator (Stanford Research Systems/DG535). Pulse widths from 15–125, 150–325, and 350–500 msec were generated from the digital delay generator operated with internal triggering rates of 5, 2, and 1 Hz respectively. The high voltage pulse was monitored on the digital oscilloscope with a 1000x oscilloscope probe (Tektronix, Richardson, TX) and the rise and fall times of the high voltage pulse were less than 1 μsec. The nano-ESI rise rime (time to initiate and generate steady state nano-ESI signal) and decay time (90% reduction in ion signal, where 100% ion signal was defined as the plateau achieved after the signal rise time) were measured by monitoring the ion current from the electron multiplier located immediately after the quadrupole. Data from the oscilloscope was downloaded to a PC using a standard GPIB connection and loaded into graphing software for processing.
Multi-Sprayer Pulsed Nano-ESI
The nano-ESI multi-sprayer experiments were performed on a Bruker Esquire quadrupole ion trap (Billerica, MA) with a flared inlet capillary developed in-house and described previously.[26,27] The flared capillary introduces a wider acceptance area for spray plume sampling which is beneficial for improving sensitivity when a single sprayer is utilized but also allows for the effective use of multiple sprayers. A metal mesh-cap comprised of 88% transmission metal mesh is attached to the flared end of the capillary to create a uniform electric field and allow the expanded acceptance area to be utilized. The incorporation of such a flared capillary requires minimal instrumental modifications and makes the alignment of nano-ESI sprayers with the instrumental sampling orifice much easier. The multi-sprayer nano-ESI experiments were conducted using a home-built source consisting of two nano-ESI sprayers secured to an X-Y-Z translational stage. The sprayers were each at an angle of ~ 5° with respect to and ~ 1.5 mm off of the axis of the flared inlet capillary. For clarity, the names left sprayer and right sprayer will be used throughout the paper to refer to the side of the transfer capillary that the sprayer is situated on when looking down onto the XZ-plane (See Figure 1). Both sprayers were immobilized on the translational stage such that any movement of the staging mechanism resulted in both sprayers being repositioned simultaneously. An updated design where each sprayer can be positioned independently is currently under development. The sprayers are constructed from Swagelok 1/4″ to 1/16″ reducing unions which accept 0.060″ O.D. × 0.045″ I.D. glass capillaries (Drummond Scientific Company, Broomall, PA) that were pulled at one end to ~ 4 μm using a Narishige model PP-830 dual stage glass electrode puller (Narishige International USA, Inc., Easy Meadow, NY). Nano-ESI solutions were injected into the pulled sprayer by direct infusion through the non-tapered end. Electrical contact is made with the nano-ESI solution via a platinum wire inserted into the open end of the sprayer which is in contact with the Swagelok body; spray is initiated and maintained through the applied voltage without any pneumatic assistance. Separate, independent voltage sources are connected to the Swagelok reducing unions such that the sprayers were electrically isolated from each other. Both sprayers were positioned 1–2 mm from the entrance of the flared inlet capillary and its metal mesh-cap.
Figure 1.

Schematic of the dual nano-ESI source and timing circuitry displaying the timing diagram for pulsed nano-ESI.
The multi-sprayer experiments used two separate EMCO (Sutter Creek, CA; model C25N) negative high voltage power supplies controlled by a timing circuit that was triggered by the start of the Esquire accumulation table within the scan function. The start of the accumulation table triggered two monostable multivibrators whose variable output pulse widths dictate the time duration when the voltage is applied to the sprayer of interest. The monostable output pulse width is determined by the user through control of an external resistor-capacitor network associated with each monostable. The amplitude of the applied high voltage was controlled by a variable resistor that modulates the amplitude of the TTL enable voltage on each EMCO supply. The sprayers could be operated on alternating scans or simultaneously on the same scan by varying the timing circuitry. In addition, but not demonstrated in this work, one sprayer can be operated continuously while the other sprayer is operated on alternating scans. Shown in Figure 1, the electronic control circuitry and a timing diagram indicate how the source operates in an alternating (pulsing) mode such that each sprayer operates on every other accumulation table.
RESULTS AND DISCUSSION
Every electrospray process involves some finite time following the initial application of the voltage to establish a steady flux of ions (rise-time). When the voltage is turned off there will also be some finite time for the flux of ions to decay (decay-time). In a pulsed experiment it is vital to understand the limits of these times as they will determine the maximum pulse rate for more complex experiments. The ion current of the protonated molecule of YGGFL (m/z 556) passing through a quadrupole mass filter was monitored on an electron multiplier as a function of the width of the pulsed voltage that was applied to the electrospray needle. The resulting ion signal from a 225 msec pulse width is shown in Figure 2a. The rise-time of the ion signal was measured to be 20 ± 3 msec, and was determined from the time that the ESI voltage was turned on until the time that the signal intensity plateaus, as illustrated in Figure 2a. In general there is a period of 10 msec after the voltage is pulsed on where no measurable ion signal is observed. The applied voltage should reach the requested value within 250 nsec of the initiation of the pulse and based on the average kinetic energy of the ions, the flight time of the ions through the mass spectrometer (250 μsec) is negligible compared to the 10 msec delay. Therefore, the majority of this 10 msec delay is most likely due to the generation of charged species and the formation of the Taylor cone. This delay is reasonable when considering the inherent wait time between voltage application and the formation of the requisite Taylor cone.[25] The average protonated molecule signal intensity achieved at the signal plateau was found to be independent of pulse width. The fluctuation at the top of the pulsed ion signal is believed to be from fluctuations inherent in the electrospray signal such as variations in ion desolvation and transmission efficiencies associated with an atmospheric ionization technique.
Figure 2.
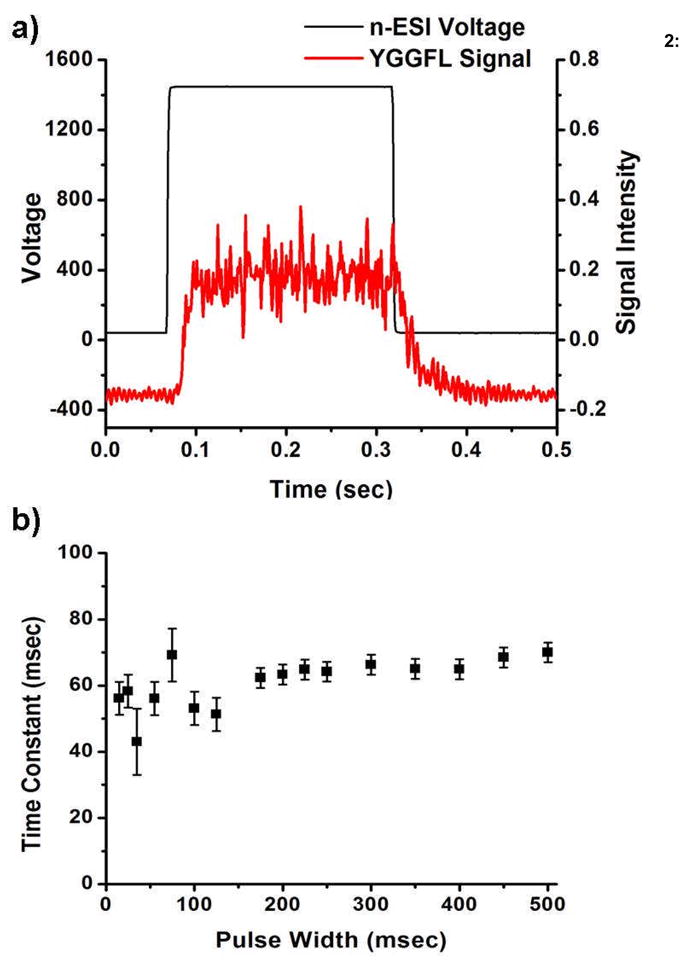
a) YGGFL signal from a 225 msec 1100 V pulse. b) Decay Time Constant as a function of pulse widths.
Immediately after turning off the electrospray voltage the ion signal begins to decrease, which is different than the observed lag in turning on the electrospray voltage. The decay-time in the ion signal, which is determined from the time the voltage is turned off, was 61 ± 5 msec. This decay-time should include the time it takes to break the Taylor cone, the time to stop the ion flux, and the flight time of the ions. The portion of the decay-time associated with stopping the ion flux includes the time where ions are still emitted from the liquid protruding from the sprayer tip even after the breaking of the Taylor cone.29 As noted above the ion flight time is approximately 250 μsec and the time it takes to break the Taylor cone is less than 50 μsec.[17,23,24] These results indicate that the majority of the decay time is in stopping the ion flux. The observed decay time is much longer than some reports found in the literature, which suggest decay times of less than 100 μsec.[24,25] However, these reports measured the current directly on an electrode that immediately follows the nano-ESI or ESI needle.[17,23,24] Additionally, the solutions that were used in determining decay times were pure solvents, high concentration salts, or contained glycol. The ion currents that are reported in the literature are in the 10 to 100 nA region which may be the result of solvent clusters and is much greater than the ion currents expected from desolvated analyte ions. Typical ion currents measured at the entrance of the quadrupole in our experiments are on the order of 10–100 pA. This suggests that the discrepancy between the decay times in other reports versus the 61 msec measured in our experiments is due to the nature of the analyte and the sensitivity of the detector, i.e., the ability of a lens, compared to an electron multiplier, to detect decreasing ion flux. Despite the large difference in decay times, reports that monitor the ion current in a pulsed electrospray system using a lens show that the trailing edge of the ion signal has a long decay time relative to the rise time.[17,23–25] In one report, time resolved photos show that ion signal is still present following the termination of the Taylor cone suggesting that charge in the spray solution takes some additional amount of time to dissipate and finally stop ion formation/desolvation.[25] In low flow rate systems, such as the ones in our experiments, the shape of the ion plume can become a mist which is possibly the environment that is created after the Taylor cone has dissipated but while there is still residual charge in the spray.[29] Following the ion decay a noisy signal was detected which lasted 75 ± 10 msec before the detector signal returned to baseline levels, i.e., background noise of the detector, which may be further evidence of this mist. As will be discussed later, the time that the noisy signal lasted was heavily dependant upon the solvent composition and the needle position.
As can be seen in Figure 2b at pulse widths above 125 msec the decay time is fairly reproducible. The pulsed voltage with widths below 125 msec was triggered at 5 Hz, meaning that the 25 msec pulse width has 175 msec between pulses. Longer pulse widths (recall that pulse widths from 150–325 and 350–500 msec were triggered at 2 and 1 Hz, respectively) all have greater than 175 msec between pulses; therefore, the time between pulses did not significantly impact the variability in the decay time. A possible explanation for the variability in the decay is that at longer pulse times the charge in the solution reaches a steady state which is comparable to a constant electrospray voltage. The literature suggests that at lower pulse widths the charge in the electrospray capillary is more localized and thus there can be more variability in the decay time.30 This affect may be exaggerated due to the use of a wire filament in the solution to generate the ESI voltage. The small dimension of the filament and the corresponding higher electric field lines in solution may make these experiments more susceptible to localized field imperfections than experiments with a metal, or metal coated, capillary.
The total time of 81 ± 6 msec, i.e. the sum of the rise and fall times, would lead to a maximum pulse rate of 12 ± 0.6 Hz or, including the full return to baseline, 6.4 ± 0.6 Hz. The solvent composition did have an effect on ESI response time and operation. The minimum decay time observed was in 20/5/75 methanol/water/acetic acid, although the signal-to-noise (S/N) ratio was much worse in these highly acidic solutions. It is believed that the lower decay times may be a result of the high ionic strength; however, additional experiments are required to probe this further. It has been shown in the literature that the sensitivity of an ESI experiment can be drastically reduced with an increased ionic strength, which due to the excess salts in these strongly acidic solutions may account for the poor signal-to-noise ratio.[31] Pure water and pure methanol solutions yielded decay times almost double those of all other solutions. In general, needle positioning relative to the instrumental sampling orifice also had an impact on the decay time. The shortest times were obtained when the needle was directly in front of the aperture and located ~ 1–2 mm axially from the aperture. The impact of needle position was significantly reduced in the source used for experiments on the Esquire due to the incorporation of a flared inlet capillary. At a constant needle position the rise time, regardless of solvent composition, was consistently ~ 20 msec.
Voltage Control of Sprayers
Independent Sprayer Operation and Optimization
The first concern when introducing multiple sprayers into the source region for a nano-ESI interface is the potential for a reduction in signal-to-noise (S/N) from each sprayer. Such S/N reduction could result from the requisite moving of each sprayer off axis to provide room for multiple sprayers and consequently using a less-than-optimal sample introduction scheme. To explore the extent of S/N loss by moving the sprayers off-axis an extreme sprayer geometry was examined. The intensity of the protonated molecule of AAA was measured in triplicate for a single, orthogonal, coaxial sprayer and a sprayer moved off-axis 1.5 mm and positioned ~ 30G° to the inlet capillary axis. The spectra (not shown) indicate that the average, acquired S/N was reduced by 27% by positioning the sprayer ~ 30° to the inlet capillary axis and ~ 1.5 mm off axis. This reduction in S/N is attributable to the use of a flared inlet capillary with a ~ 3 mm diameter opening such that an off-axis position of 1.5 mm (regardless of the sprayer angle) places the sprayer at the edge of the capillary acceptance region. To confirm this idea the orthogonal sprayer was moved from on-axis to 1.5 mm off axis; a S/N reduction of 22% was observed. Furthermore, going from the orthogonal sprayer positioned 1.5 mm off of the capillary axis to the ~ 30° sprayer positioned 1.5 mm off axis resulted in a 7% S/N reduction.
Improved mass measurement accuracy is beneficial in the spectral interpretation, elemental composition determination, and identification of unknowns. The introduction of an internal calibrant offers one method of improving mass accuracy, especially for trapping mass analyzers, because the analyte and calibrant can be exposed to the same trapping fields and space charge effects. To demonstrate the capability of this setup to address such a situation, both sprayers were operated simultaneously in a continuous fashion. Shown in Figure 3 is the resultant spectrum where 100 μM PEG 600 in methanol was sprayed from one sprayer to serve as the calibrant while the analyte, 100 μM YGGFL in 75/20/5 by volume acetonitrile/water/formic acid, was sprayed from the other sprayer. Utilizing this multi-sprayer configuration as shown in Figure 3 resulted in an improvement in mass measurement accuracy from 678 ppm to 119 ppm.
Figure 3.

PEG 600 solution (closed squares) and YGGFL solutions (open square) spraying simultaneously from their respective sprayers.
The spectrum in Figure 3 was obtained by lowering the voltage applied to the PEG sprayer from what gives the maximum ion signal, thereby suppressing the ESI process and consequently reducing the amount of ion formation. The suppression of the PEG signal in Figure 3 was necessary to avoid adversely affecting the MS signal for YGGFL due to the space charge capacity imposed by the ion trapping volume. This deliberate signal suppression further illustrates the ability of the setup to allow for independent control and optimization of the ionization processes by independently controlling and optimizing the ESI potentials applied to each sprayer. Without the ability to spray different sample solutions from their own, independent sprayers a mixture of the samples would have to be used. It is common that when multiple samples are in the same solution that one of the analytes will experience signal suppression. The spectrum in Figure 4a was obtained by spraying Gramicidin S and PPG from their own sprayer; both analytes are effectively detected. Separate sprayers allow each sample to be optimized in-terms of solvent and other additives (i.e. the addition of sodium to the PPG solution). If these two analytes are sprayed from the same solution the spectrum shown in Figure 4b is obtained. The PPG signal is significantly suppressed under these conditions which are more optimal for spraying the peptide.
Figure 4.

a) Mass spectrum of Gramicidin S (20 M) in one sprayer and PPG 425 (50 μM with 50 μM Na+) in the other sprayer. b) Mass spectrum from a single sprayer containing a mixture of the analytes from a). [M+2H]+2 Gramcidin S (open circle), [M+Na]+ PPG (closed squares).
Nano-ESI sprayers vary with regard to distributions of tip opening sizes and geometries as well as sprayer-counter electrode spacing and alignment. Exact tip-to-tip reproducibility can be problematic especially when trying to perform spectral comparisons for such applications as batch process monitoring or quantification studies. Sprayer tip alignment is often addressed through use of optical microscopes for positioning nano-ESI sprayers in front of an instrument’s sampling orifice. The novel design presented here and the use of a flared inlet capillary allow for signal optimization of multiple sprayers without the need for extensive sprayer re-alignment. To explore the accuracy of the voltage control, the same solution of YGGFL was put into both sprayers and the potential applied to each sprayer was optimized until the observed intensities from each sprayer were approximately identical, with only one sprayer operating at a time. For this experiment the voltage applied to the wire-mesh capillary cover was −1500 V. As a control, both sprayers were held at 0 V without any manipulation of their respective voltages and as a result, a 75 % difference between the signal intensity for the protonated molecule of YGGFL from each sprayer was observed (data not shown). The voltages on the left and right sprayers were then optimized to 200 V and 240 V, respectively. For the optimized case, an 8 % relative difference between the protonated molecule signal intensities was observed. In other experiments where the initial difference is not so great a 4% relative difference was readily achievable. It should also be noted that no extra precautions were taken to ensure both sprayers were equal distances from the instrumental sampling orifice, which is indicative of the ability to achieve comparable spectra without precise sprayer alignment.
Pulsed Operation
Another advantage of the proposed multiplexed source is the ability to introduce multiple analytes from separate solutions in an alternating fashion. The source can be pulsed by controlling the voltage applied to the desired nano-ESI sprayer. This pulsing can be synchronized with the ion accumulation and injection sequence, and ultimately the mass analysis step of the analyzer, to optimize the instrumental duty cycle. Such capabilities avoid the situation where a nano-ESI sprayer may be operating without being sampled by the mass spectrometer and allows lower sample flow rates to be realized, thus reducing sample consumption. To demonstrate this mode of operation, both sprayers were pulsed so each consecutive instrumental scan was sampling a different sprayer in an alternating manner where AAA and YGGFL solutions were directly infused into their respective sprayers and alternately pulsed for 30 minutes. In Figure 5, the two spectra (scans 3939 and 3940) show that on consecutive scans only one sprayer is being sampled at a time and alternating pulsing has been achieved. The bottom pane of the figure is a histogram representing the signal intensity for the protonated molecules of AAA and YGGFL over a series of instrumental scans where a scan was executed every 450 msec. As is evident from the histogram, alternating pulsing was achieved for the duration of the experiment, where scan number 3960 represents an elapsed experimental time of 29.7 minutes. The demonstrated stability of the pulsing mode of operation is promising for coupling this source to relevant separation techniques (e.g., HPLC).
Figure 5.

Consecutive mass spectra and histogram illustrating the dual pulsing mode of operation for AAA and YGGFL solutions.
CONCLUSIONS
Pulsed nano-ESI was performed and characterized by using ion detection after mass analysis. The average rise time and decay times were determined to be 20 ± 3 msec and 61 ± 5 msec respectively, which results in a maximum pulse rate of 12 Hz. Altering the solvent composition had minor effects on the decay time and no effect on the rise time, with the largest reduction in decay time resulting from the most acidic solutions.
As has been demonstrated for the multi-sprayer source, the desired mode of operation (i.e., both sprayers operating continuously or both sprayers being pulsed in an alternating manner) can be realized by controlling the voltage applied to each sprayer rather than mechanically switching the sprayers. The novel, multi-sprayer source design presented here should be compatible with most mass analyzers while requiring minimal instrumental modifications. The dual source provides a method to introduce analyte and internal calibrant to the mass spectrometer from independent nano-ESI sprayers for the purpose of improving mass measurement accuracy. By synchronizing the voltages applied to the sprayers with the ion accumulation event sample loss and consumption can be minimized. Both simultaneous and pulsing modes of operation can be achieved without having to re-position the nano-ESI sprayers and can be precisely controlled by means of regulating the voltages applied to the sprayers. Finally, the symmetry about the axis of the instrument sampling orifice should permit the installation of more sprayers on the same circumference as the existing two sprayers but on different planes with respect to the sampling orifice to increase the throughput realized with this system.
Acknowledgments
This work was supported in part by NIH grant GM49852.
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 2.Korner R, Wilm M, Morand K, Schubert M, Mann M. Nano Electrospray Combined with a Quadrupole Ion Trap for the Analysis of Peptides and Protein Digests. J Am Soc Mass Spectrom. 1996;7:150–156. doi: 10.1016/1044-0305(95)00626-5. [DOI] [PubMed] [Google Scholar]
- 3.Marina A, Carcia MA, Albar JP, Yague J, Castro JALd, Vazquez J. High-Sensitivity Analysis and Sequencing of Peptides and Proteins by Quadrupole Ion Trap Mass Spectrometry. J Mass Spectrom. 1999;34:17–27. doi: 10.1002/(SICI)1096-9888(199901)34:1<17::AID-JMS746>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Hannis JC, Muddiman DC. A Dual Electrospray Ionization Source Combined with Hexapole Accumulation to Achieve High Mass Accuracy of Biopolymers in Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J Am Soc Mass Spectrom. 2000;11:876–883. doi: 10.1016/S1044-0305(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 5.Emmett MR, Caprioli R. Micro-Electrospray Mass Spectrometry: Ultra-High Sensitivity Analysis of Peptides and Proteins. J Am Soc Mass Spectrom. 1994;5:605–613. doi: 10.1016/1044-0305(94)85001-1. [DOI] [PubMed] [Google Scholar]
- 6.Niessen WMA, Tinke AP. Liquid Chromatography-Mass Spectrometry General Principles and Instrumentation. J Chromatogr A. 1995;703:37–57. [Google Scholar]
- 7.Zeng L, Kassel DB. Developments of a Fully Automated Parallel HPLC/Mass Spectrometry System for the Analytical Characterization and Preparative Purification of Combinatorial Libraries. Anal Chem. 1998;70:4380–4388. [Google Scholar]
- 8.Bednar P, Papouskova B, Muller L, Bartak P, Stavek J, Pavlousek P, Lemr K. Utilization of Capillary Electrophoresis/Mass Spectrometry (CE/MSn) for the Study of Anthocyanin Dyes. J Sep Sci. 2005;28:1291–1299. doi: 10.1002/jssc.200500071. [DOI] [PubMed] [Google Scholar]
- 9.Soler C, Manes J, Pico Y. Comparison of Liquid Chromatography using Triple Quadrupole and Quadrupole Ion Trap Mass Analyzers to Determine Pesticide Residues in Oranges. J Chromatogr A. 2005;1067:115–125. doi: 10.1016/j.chroma.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 10.Fong KWY, Chan TWD. A Novel Nonmetalized Tip for Electrospray Mass Spectrometry at Nanoliter Flow Rate. J Am Soc Mass Spectrom. 1999;10:72–75. doi: 10.1016/S1044-0305(98)00127-5. [DOI] [PubMed] [Google Scholar]
- 11.Wilm M, Mann M. Analytical Properties of the Nanoelectrospray Ion Source. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 12.Glish GL, Vachet RW. The Basics of Mass Spectrometry in the Twenty–First Century. Nature Reviews. 2003;2:140–150. doi: 10.1038/nrd1011. [DOI] [PubMed] [Google Scholar]
- 13.Mann M. Electrospray: Its Potential and Limitations as an Ionization Method for Biomolecules. J Mass Spectrom. 1990;25:575–587. [Google Scholar]
- 14.Leclercq L, Delatour C, Hoes I, Brunelle F, Labrique X, Castro-Perez J. Use of a Five-Channel Mulitplexed Electrospray Quadrupole Time-of-Flight Hybrid Mass Spectrometer for Metabolite Identification. Rapid Commun Mass Spectrom. 2005;19:1611–1618. doi: 10.1002/rcm.1964. [DOI] [PubMed] [Google Scholar]
- 15.Satomi Y, Kudo Y, Sasaki K, Hase T, Takao T. Accurate Mass Measurement in nano-Electrospray Ionization Mass Spectrometry by Alternate Switching of High Voltage Between Sample and Reference Sprayers. Rapid Commun Mass Spectrom. 2005;19:540–546. doi: 10.1002/rcm.1813. [DOI] [PubMed] [Google Scholar]
- 16.Wolff J, Eckers C, Sage AB, Giles K, Bateman R. Accurate Mass Liquid Chromatography/Mass Spectrometry on Quadrupole Orthogonal Acceleration Time-of-Flight Mass Analyzers using Switching Between Separate Sample and Reference Sprays. 2. Applications using Dual-Electrospray Ion Source. Anal Chem. 2001;73:2605–2612. doi: 10.1021/ac001419a. [DOI] [PubMed] [Google Scholar]
- 17.Zhou F, Shui W, Lu Y, Yang P, Guo Y. High Accuracy Mass Measurement of Peptides with Internal Calibration using a Dual Electrospray Ionization Sprayer System for Protein Identification. Rapid Commun Mass Spectrom. 2002;16:505–511. doi: 10.1002/rcm.603. [DOI] [PubMed] [Google Scholar]
- 18.Ogorzalek Loo RR, Udseth HR, Smith RD. Evidence of Charge Inversion in the Reaction of Singly Charged Anions with Multiply Charged Macroions. J Phys Chem. 1991;95:6412–6415. [Google Scholar]
- 19.Ogorzalek Loo RR, Udseth HR, Smith RD. A New Approach for the Study of Gas-Phase Ion-Ion Reactions using Electrospray Ionization. J Am Soc Mass Spectrom. 1992;3:695–705. doi: 10.1016/1044-0305(92)87082-A. [DOI] [PubMed] [Google Scholar]
- 20.Xia Y, Liang X, McLuckey SA. Pulsed Dual Electrospray Ionization for Ion/Ion Reactions. J Am Soc Mass Spectrom. 2005;16:1750–1756. doi: 10.1016/j.jasms.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 21.Liang X, Han H, Xia Y, McLuckey SA. A Pulsed Triple Ionization Source for Sequential Ion/Ion Reactions in an Electrodynamic Ion Trap. J Am Soc Mass Spectrom. 2007;18:369–376. doi: 10.1016/j.jasms.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Danell RM, Myer MJ, Danell AS. Proc. 55th ASMS Conf. on Mass Spectrom. & Allied Topics; Indianapolis, Indiana. 2007. [Google Scholar]
- 23.Berggren WT, Westphall MS, Smith LM. Single-Pulse Nanoelectrospray Ionization. Anal Chem. 2002;74:3443–3448. doi: 10.1021/ac010975h. [DOI] [PubMed] [Google Scholar]
- 24.Wei J, Shui W, Zhou F, Lu Y, Chen K, Xu G, Yang P. Mass Spec Rev. 2002;21:148–162. doi: 10.1002/mas.10026. [DOI] [PubMed] [Google Scholar]
- 25.Alexander MS, Paine MD, Stark JPW. Naturally and Externally Pulsed Electrospray. Anal Chem. 2006;78:2658–2664. doi: 10.1021/ac0520036. [DOI] [PubMed] [Google Scholar]
- 26.Glish GL, Danell RM. Electrospray Ionization Device. 6,703,611. U.S. Patent. 2004
- 27.Danell RM, Glish GL. Doctoral Dissertation. The University of North Carolina; Chapel Hill: 2001. [Google Scholar]
- 28.Black D. PhD, Doctoral Dissertation. The University of North Carolina; Chapel Hill: 2005. [Google Scholar]
- 29.Koerner T, Turck K, Brown L, Oleschuk RD. Porous Polymer Monolith Assisted Electrospray. Anal Chem. 2004;76:6456–6460. doi: 10.1021/ac049438y. [DOI] [PubMed] [Google Scholar]
- 30.Pozniak BP, Cole RB. Proc. 54th ASMS Conf. Mass Spectrometry and Allied Topics; Seattle, Washington. 2006. [Google Scholar]
- 31.Wang G, Cole RB. Effect of Solution Ionic Strength on Analyte Charge State Distributions in Positive and Negative Ion Electrospray Mass Spectrometry. Anal Chem. 1994;66:3702–3708. [Google Scholar]