

Abstract
A method of performing collision induced dissociation (CID) on the charge-reduced parent ion as it is formed during electron capture dissociation (ECD), called ECD+CID, is described. In ECD+CID, the charge-reduced parent ion is selectively activated using resonant excitation and collisions with the helium bath gas inside a linear quadrupole ion trap ECD device (ECDLIT). It has been observed that ECD+CID can improve the sequence coverage for melittin over performing ECD alone (i.e., from 76 % to 88 %). Perhaps just as important, ECD+CID can be used to reduce the extent of multiple electron capture events observed when performing ECD in the ECDLIT. Consequently, the abundance of mass-to-charge ratios corresponding to ECD product ions that contain neutralized protons is decreased, simplifying the interpretation of the product ion spectrum.
Introduction
Electron capture dissociation (ECD) has become an important tool for tandem mass spectrometry (MS/MS) analyses of multiply charged parent ions.1-3 The successful application of ECD to the study of post-translational modifications (PTMs),4-7 peptide/protein sequencing,1-3, 8-10 and other biologically relevant molecules6, 11, 12 has led to a growing importance of mass spectrometry in such fields. The wide-spread use of ECD and its increase in popularity is evidenced by several review articles that have been published on this topic.13-17
Since 2004, it has become possible to interact low energy electrons with multiply charged cations in mass spectrometers other than Fourier transform ion cyclotron resonance (FTICR-MS) instruments.18-25 Recently a practicable hybrid linear quadrupole ion trap (LIT)-time of flight (TOF) mass spectrometer where ECD is performed in a modified LIT (ECDLIT) has been developed.26 The ECDLIT is housed within a magnetic field that is generated by a neodymium magnet and orientated parallel to the LIT central axis. The magnetic field helps radially confine electrons to the central axis and aid in their axial transmission through the ECDLIT. In addition, the helium bath gas present at 10-3 mbar and the radial restoring force applied to the ions by the quadrupolar field inherent with the ECDLIT ensure that the parent ions for ECD are focused to the same central axis as the electrons. The combination of the static magnetic field convoluted with the electrodynamic and collisional ion focusing within the ECDLIT provides the ion-electron overlap necessary for efficient ECD.
The MS/MS efficiency (i.e, the percentage of isolated parent ion converted to and detected as product ions) for doubly charged substance P on the ECDLIT is nine percent.27 This efficiency measurement is in the range of what has been reported for ECD performed on FTICR-MS instruments where the conversion efficiency of parent ions to product ions is between 5 and 30 %.15 Activated ion ECD (AI-ECD) has been used to improve the extent of dissociation observed with respect to performing ECD alone.28-35 The supplemental activation is required in ECD to help disrupt non-covalent, intramolecular interactions28 or to increase the amount of parent ion internal energy due to the minimal amount of energy imparted by ion-electron inelastic collisions.36 Because most ECD experiments to-date have been performed under the ultra-high vacuum conditions of FTICR-MS, infrared (IR) radiation has been the ion activation method of choice. IR radiation is more convenient to use in FTICR-MS than collisional activation because the latter requires a target gas to be introduced into the ICR cell which must be pumped away prior to mass analysis.37, 38
Because collisional activation is a resonant process in ion trapping instruments, it can be performed mass-selectively. Therefore, the charge-reduced ion resulting from non-dissociative electron capture can be excited exclusively without having to be isolated. The ability to do mass-selective activation is not possible with IR activation where the parent ion of interest must be isolated first or else any ion that enters the path of the laser could be activated and dissociated.28, 31, 39 Furthermore, the energetics of ion-neutral collisional activation can be controlled by modulating the amplitude of the supplemental ac waveform used to achieve resonance excitation. Consequently, the [M+nH](n-1)+• ion can be made to undergo collision induced dissociation (CID) rather than simply being ejected from the ECDLIT as with the double resonance (DR) ECD technique.40, 41 Due to the constant ∼10-3 mbar helium bath gas pressure present in the ECDLIT, CID is readily implemented. In the case of ECD+CID, the parent ion of interest to resonantly excite for CID is the first charge-reduced species, [M+nH](n-1)+•. The mass-to-charge of this ion is obviously known a priori because the ion isolated for ECD is [M+nH]n+.
By using CID to simultaneously aid ECD in the ECDLIT, a unique form of AI-ECD can be implemented. We are referring to this process of simultaneously applying ECD and CID, as ECD+CID to differentiate it from experiments where the CID occurs after ECD (e.g. ECD/CID). This is an important distinction as will be discussed below. The ability to mass-selectively activate the charge-reduced species during ECD in the ECDLIT makes it possible to improve the extent of parent ion dissociation, and thus peptide sequence coverage, compared to performing ECD alone.
In addition to increasing the sequence coverage another feature of ECD+CID, and the main reason it is desirable to perform the CID simultaneous with, rather than subsequent to, the ECD, is related to the ECDLIT electron capture rate. Due to the high rate of electron capture in the ECDLIT, performing ECD on multiply charged parent ions results in multiple electron capture events under typical operating conditions. When a parent ion ([M+nH]n+) captures a low kinetic energy electron the charge-reduced species ([M+nH](n-1)+•) is formed which then ideally dissociates into product ions. However, a significant percentage of this charge-reduced species remains undissociated. If the charge state of the initial, even-electron parent ion is greater than two, the charge-reduced peak will be multiply charged; therefore a second electron can be captured which would produce [M+nH](n-2)+. With successive non-dissociative electron capture events, the charge is decreasing but the number of hydrogens remains constant. With each electron captured, a proton is being neutralized, resulting in the intact species containing one or more hydrogens than if that same charge state would have been formed directly from electrospray ionization (ESI). When the charge-reduced species containing additional hydrogens dissociates, its product ions would be observed at mass-to-charge ratios that differ from their theoretical values for c and z product ions, making spectral interpretation and sample identification complicated.
Scheme 1 depicts the process of multiple electron capture events. EC1 and EC2 represent first and second electron capture (but no dissociation) events and D1 and D2 indicate the dissociation channels that could follow each electron capture during ECD. If the parent ion ([M+nH]n+) were to capture one electron but not undergo dissociation, it would form the odd-electron charge-reduced capture product ([M+nH](n-1)+•). Due to the aforementioned electron beam density and concomitant rate of electron capture in the ECDLIT, the [M+nH](n-1)+• ion could then capture a second electron to form [M+nH](n-2)+ (EC2), and so on. If the [M+nH](n-1)+• ion undergoes dissociation to form product ions via the D1 channel, product ions corresponding to typical ECD experiments would be observed. However, product ions formed from the D2 pathway would contain neutralized protons that could result in mass-to-charge ratios corresponding to the presence of additional hydrogen(s). To limit the amount of EC2 (and D2), in the normal ECD experiment, the electron flux has to be reduced, which reduces the fragmentation and MS/MS efficiency. In ECD+CID (denoted by dashed rectangle in Scheme 1) the charge-reduced ion ([M+nH](n-1)+•) is resonantly excited and dissociated at the same time the parent ion ([M+nH]n+) is being irradiated with electrons. As a result, EC1D1 becomes the dominant reaction pathway, to the detriment of EC2D2, improving the fragmentation and MS/MS efficiencies.
Scheme 1.

In this manuscript, the effect of simultaneously performing CID on the charge-reduced parent ion during ECD (i.e., ECD+CID) to improve peptide sequence coverage over performing ECD alone is demonstrated. In addition, the ability of ECD+CID to prevent multiple electron capture events in the ECDLIT instrument and to aid in de novo peptide sequencing is presented. The effects of the convoluted electrodynamic and static magnetic fields on ion motion in the ECDLIT are also discussed.
Experimental
Samples
The peptides melittin (GIGAVLKVLTTGLPALISWIKRKRQQ-NH2) and β-endorphin (YGGFMTSEKSQTPLVTLFKNAIIKNAYKKGE) were purchased from Sigma-Aldrich, Inc. (St. Louis, MO) and used without further purification. A solution was made to a concentration of 5 μM in 50:50 v% methanol/water. Acetic acid (1% by volume) was added to the final sample mixture to aid in the electrospray process.
Instrumentation
Mass spectrometry experiments were performed on a NanoFrontier LIT-TOF (Hitachi High Technologies).42 Ions were generated using nanoelectrospray ionization. The basic operation of this mass spectrometer has been described previously.42 For the ECD+CID experiments, the ECDLIT was modified so a supplemental ac waveform can be applied to one pair of rods of the ECDLIT quadrupole rod set. The supplemental ac waveform allows an ion of interest to be resonantly excited and thus undergo activation through collisions with the 10-3 mbar pressure of helium bath gas present in the ECDLIT.
Simultaneous ECD+CID
In ECD+CID experiments, the parent ion [M+nH]n+ is selected in the LIT and transferred to the ECDLIT. Then the parent ion is irradiated with electrons while a supplementary ac waveform is simultaneously applied at a resonant frequency of the odd-electron, charge-reduced ion, [M+nH](n-1)+•, to effect CID. Because of the magnetic field, there are two resonance frequencies for each mass-to-charge ratio as described by Equation 1:
| (1) |
where, ω is the observed frequency of radial motion for a given ion and ωq and ωc are the ion's secular and cyclotron frequencies, respectively. ωq can be expressed in terms of experimental parameters following the procedure outlined by Douglas et. al.43 The voltages applied to the ECDLIT rods create the quadrupolar potential, φ(x,y,t):
| (2). |
In Equation 2, x and y refer to the radial dimensions of the LIT; r0 is the radius of the inscribed circle of the rod array; Vrf is the zero-to-peak amplitude of the drive rf voltage applied to the LIT, U is the dc potential applied to the LIT rod set, which is typically 0 V, and Ω is the angular frequency of Vrf. From Equation 2 it can be shown that if U = 0 V, ωq can be expressed as:
| (3). |
Independently, the effect of the magnetic field on the ion motion is described by the cyclotron equation:44
| (4). |
In Equation 4, B is the magnetic field strength present in the ECDLIT in units of Tesla. Substituting Equations 3 and 4 into Equation 1 and solving for m/z results in:
| (5). |
where ω0 is the frequency of the applied supplemental dipolar waveform. From Equation 5 it can be seen that for a constant set of experimental parameters (i.e., B, Ω, ω0, and r0), the zero-to-peak drive rf voltage amplitude (Vrf) is proportional to the mass-to-charge that is in resonance. The relationship between mass-to-charge and the drive rf amplitude to an ion's resonance points is shown for the [M+4H]4+ charge state of melittin in Figure 1 under the conditions where B = 150 mT, Ω = 467.3 kHz, ω0 = 47.2 kHz, r0 = 6.0 mm. The extracted ion current for [M+4H]4+ (m/z 712.2) is plotted as a function of Vrf in Figure 1A. Two resonance points are observed, at Vrf values of 133 and 141 V0-p, respectively. The reduction in [M+4H]4+ abundance at Vrf 133 and 141 V0-p is indicative of CID occurring as the parent ion is brought into resonance with the supplemental ac waveform. A supplemental ac waveform amplitude of 6.0 Vp-p was sufficient to cause dissociation. The difference between being on- and off-resonance with the supplemental ac waveform is demonstrated by the mass spectra in Figure 1B where the top (*) spectrum corresponds to the off-resonance condition indicated in Figure 1A. No dissociation is observed when the parent ion is not in resonance with the supplemental waveform. Conversely, the bottom (**) spectrum in Figure 1B was acquired when the parent ion was in resonance with the supplemental ac waveform as indicated in Figure 1A. In such a case, the [M+4H]4+ parent ion is activated through collisions with the helium bath gas and dissociates into product ions. The CID spectra acquired at each resonance point (i.e. Vrf = 133 and 141 V0-p) were the same suggesting that the choice of which resonance point to use is not critical.
Figure 1.

A) Plot of the extracted ion current for [M+4H]4+ melittin (m/z 712.638) as a function of rf amplitude (V0-p). Two resonance points are observed (at 133 and 141 V0-p) due to the convoluted magnetic and electrodynamic fields in the ECDLIT. B) Mass spectra acquired when the [M+4H]4+ parent ion was off-(*), top spectrum) and on- (**), bottom spectrum) resonance.
Instrumental Parameters
Two different ECD+CID experiments were performed. First, ECD+CID was used to improve the extent of dissociation over that realized using ECD alone. For ECD alone, melittin [M+4H]4+ was irradiated for 10 ms with 1.2 eV electrons while for ECD+CID, melittin [M+4H]4+ was irradiated for 190 ms with 1.2 eV electrons while [M+4H]3+• (m/z 949.595, Vrf = 221 V0-p) was resonantly excited with an 8.0 Vp-p supplemental ac waveform. The longer electron irradiation time in the ECD+CID experiment is necessary because the electrons are heated somewhat by the supplemental ac waveform and thus electron density along the axis of the ECDLIT is reduced. However, data is acquired for the same length of time in both experiments (i.e. few scans are averaged in the ECD+CID experiment).
A different set ECD+CID experimental parameters were used to demonstrate the ability to prevent multiple electron capture events during ECD in the ECDLIT. For this experiment the instrumental parameters were varied until the ECD and ECD+CID experiments displayed similar mass spectra. During ECD alone, melittin [M+4H]4+ was irradiated for 7.0 ms with 1.0 eV electrons. Under ECD+CID conditions, [M+4H]4+ was irradiated for 80 ms with 1.2 eV electrons while [M+4H]3+• (m/z 949.595, Vrf = 221 V0-p) was resonantly excited with a 6.0 Vp-p supplemental ac waveform.
Results and Discussion
ECD+CID for Improved Sequence Coverage
As with FTICR-MS instruments the use of ion activation with ECD does improve parent ion dissociation with the ECDLIT. However, unlike FTICR-MS systems, the ECDLIT is capable of using mass-to-charge selective CID for parent ion activation during the electron irradiation portion of an ECD experiment. In Figure 2A the result of using ECD+CID (upward spectrum) is compared to performing ECD alone (downward spectrum). When ECD is performed by itself under typical conditions, the most abundant product ion is the charge-reduced species (e.g., [M+4H]3+•). Conversely, in ECD+CID the [M+4H]3+• ion is collisionally activated and made to undergo dissociation, thereby improving the peptide sequence coverage. The amino acid sequence of melittin given at the bottom of Figure 2 indicates the sites of backbone cleavage. A wide solid arrow represents cleavage observed under both ECD+CID and ECD conditions; a narrow arrow indicates cleavage only observed in ECD experiment; a wide open arrow represents cleavage from ECD+CID only. All charge states of a product ion associated with the same N-Cα bond cleavage are represented by one arrow. The sequence coverage achieved under ECD+CID was 88 % versus 77 % with ECD alone. The ability to mass-selectively excite only the [M+4H]3+• ion during the ECD+CID experiment, rather than all first generation product ions as would be done using IR, ensures that additional product ions are from the CID of the charge-reduced species. As shown in Figure 2, examples of cleavage unique to ECD+CID of both the N-Cα bond (c and x ions) and the peptide bond (b and y ions) are observed. However, the only peptide bond dissociation that contributed to the improved sequence coverage achieved with ECD+CID was the cleavage N-terminal to Pro14 to generate the y132+ ion. Unlike ECD, with CID the amide bond N-terminal to proline is preferentially cleaved, which allows ECD+CID to provide complementary information to ECD alone.
Figure 2.

(A) ECD+CID spectrum (upward) and ECD spectrum (downward) of [M+4H]4+ of melittin. The amino acid sequence for melittin is shown at the bottom of the figure and indicates sites of backbone cleavage. Wide solid arrows indicate product ions (c or z) that were observed in both dissociation experiments. Wide open arrows represent backbone cleavages unique to the ECD experiment by itself, while the narrow arrow indicates cleavage exclusive to the ECD+CID experiment. (B) Expanded mass-to-charge regions around melittin [M+4H]3+• from (2A) The top spectrum is the theoretical isotopic distribution expected if the [M+3H]3+ charge state were formed directly from ESI. The middle spectrum is mass-to-charge region for [M+4H]3+• from the ECD+CID spectrum demonstrating its removal due to resonance excitation. The bottom spectrum is the mass-to-charge region for [M+4H]3+• from the ECD spectrum.
The data shown in Figure 2B is a blow-up of the charged reduced region from the spectra in Figure 2A. This shows that the [M+4H]3+• ion was resonantly excited and eliminated from the spectrum (either by dissociation, or ejection). The theoretical isotopic distribution for the even electron [M+3H]3+ species when it is formed directly from ESI is given in the top of Figure 2B. Under ECD+CID conditions (middle of Figure 2B) the major isotopomers associated with the odd-electron, charge-reduce species are no longer observed because the [M+4H]3+• ion was resonantly excited to effect dissociation. The experimentally observed isotopic distribution for the [M+4H]3+• ion under ECD conditions without CID is shown in Figure 3C. It can be seen that compared to the theoretical distribution of the even electron [M+3H]3+ species in the top spectrum of Figure 2B, the distribution in the bottom spectrum is shifted to higher mass-to-charge values indicative of the additional hydrogen on [M+4H]3+• that results from the neutralization of a proton in [M+4H]4+.
Figure 3.

Expanded mass-to-charge regions around z6-z9 ions unique to ECD+CID in Figure 2. A) z7+2 and z8+2 ions are present in ECD+CID (top) experiment but absent under ECD alone conditions (bottom). B) z9+2 and z6+ ions are present in ECD+CID (top) experiment but absent under ECD alone conditions (bottom).
Considering the amino acid sequence from Figure 2, it can be seen that the improved sequence coverage observed with ECD+CID is due to cleavage C-terminally to Pro14 and the formation of z6—z9 product ions. Shown in Figure 3 are the mass-to-charge regions associated with these z-ions. For each product ion the theoretical, monoisotopic mass-to-charge value is given, and the corresponding isotopomer is indicated with an arrow. The data in Figure 3 clearly show that the z ions in question are observed under ECD+CID conditions and not formed with ECD alone.
The increase in sequence coverage for melittin is not that great because ECD itself is quite efficient in the ECDLIT for this species. As in FTICR-MS instruments, the efficiency in the ECDLIT decreases with increasing peptide length or decreasing charge. Figure 4 shows the results obtained with β-endorphin (32 residues versus 26 for melittin) using ECD+CID and ECD alone. In this case the sequence coverage improves from 72% to 97%, with the only cleavage not observed for ECD+CID being on the N-terminal side of proline. Like melittin the improved sequence coverage for β-endorphin is mainly from the C-terminal side of the proline. These results suggests that proline has an inhibitory effect on cleave to its C-terminal side in ECD. Further studies of this are in progress.
Figure 4.

ECD+CID spectrum (upward) and ECD spectrum (downward) of [M+4H]4+ of β-endorphin. The amino acid sequence for β-endorphin is shown at the bottom of the figure and indicates sites of backbone cleavage, annotated the same as in Figure 2.
ECD+CID for Improved de novo Sequencing
Ion-electron reactions are an attractive set of MS/MS methods for peptide analyses because they can provide extensive sequence coverage and allow labile bonds (e.g., PTMs) to remain intact while only requiring the electron kinetic energy and flux to be tuned.13 The data in Figure 2, where only ECD was performed (bottom spectrum), used electrons with kinetic energies optimized for maximum electron capture cross-section. However, the relative intensity of the product ions from dissociation is quite small so there might be benefit to increasing the electron flux under such conditions in an attempt to improve the extent of dissociation in ECD. To explore the possibility of higher electron flux improving peptide sequence coverage with the ECDLIT, the current passed through the electron filament was increased so the electron current passing through the ECDLIT and measured on the quadrupole ions guide changed from 0.33 to 0.80 μA.26 Increasing the electron flux introduces a larger number of electrons to the ECDLIT. When parent ions contain more than two protons, their charge-reduced species would still be multiply-charged and thus capable of capturing another electron. As described in Scheme 1, the second electron capture can lead to product ions with mass-to-charge values that differ from theory, making spectral interpretation more difficult. By using ECD+CID the first charge-reduced species ([M+nH](n-1)+•) can be resonantly excited and dissociated, thereby preventing the second electron capture event. Thus, while ECD alone may result in more extensive backbone cleavage when a higher flux of incident electrons (i.e., 0.80 μA versus 0.33 μA) is used, interpretation of the MS/MS spectrum would be difficult without a priori knowledge of the peptide sequence. ECD+CID should allow the ambiguity introduced by multiple electron capture events (i.e., sequential proton neutralization) to be avoided.
The result of increasing the electron flux to a measured electron current of 0.85 μA and using only ECD is shown in the spectrum with the peaks pointing down in Figure 5A. The parent ion was the [M+4H]4+ charge state of melittin. ECD+CID with a measured electron current of 0.80 μA is shown in the spectrum pointing up in Figure 5A where the parent ions for ECD and CID were the [M+4H]4+ and [M+4H]3+• species, respectively. The two spectra appear similar which is due to the majority of the peptide backbone cleavage sites being the same in ECD+CID and ECD. The amino acid sequence in the bottom of Figure 5 shows the locations of c/z ion formation; as with Figure 2, the arrows are used to represent which MS/MS method is responsible for each cleavage. Under the present experimental conditions, a larger number of N-Cα bonds are cleaved with just ECD compared to ECD+CID (84 % vs. 76 %). However, this improved sequence coverage is due only to the ability to annotate product ions which have mass-to-charge values different than what is theoretically expected because the amino acid sequence was known a priori.
Figure 5.
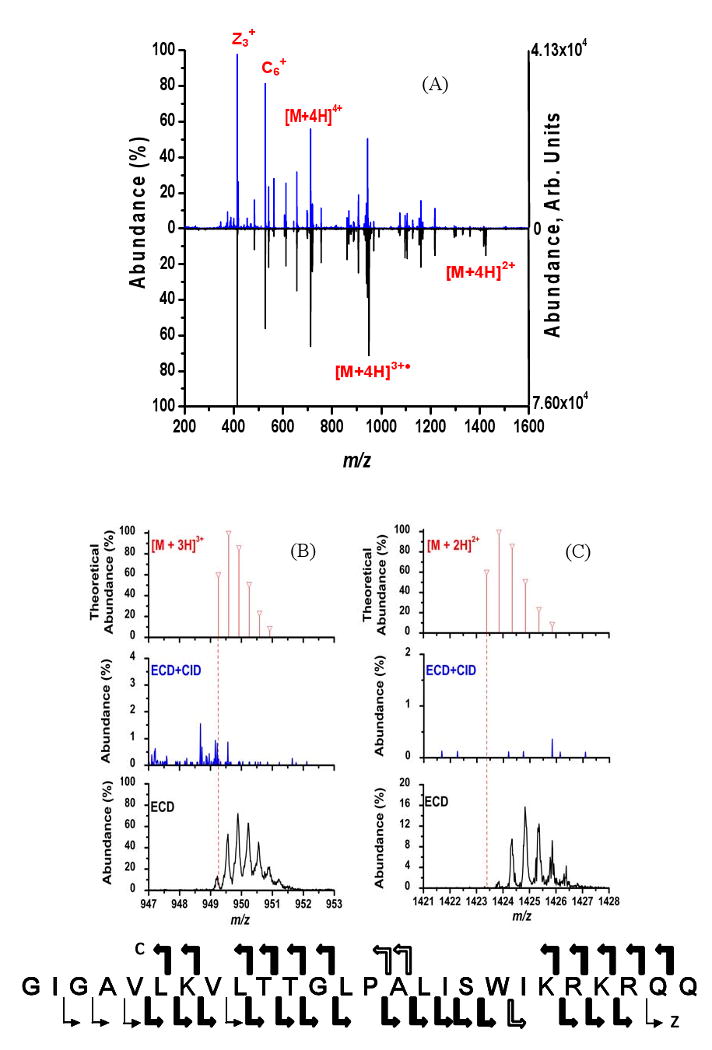
(A) ECD+CID spectrum (upward) and ECD spectrum (downward) of [M+4H]4+ of melittin. The amino acid sequence for melittin at the bottom of the figure indicates the cleavage sites and is annotated the same as in Figure 2. (B and C) Expanded mass-to-charge regions around melittin [M+4H]3+• and [M+4H]2+, respectively, from 5A. The theoretical isotopic distributions expected if the charge states were formed directly from ESI are given for [M+3H]3+ and [M+2H]2+ ion the top spectra. The middle spectra are these mass-to-charge regions for the ECD+CID experiment and the bottom spectra are the same mass-to-charge regions for just the ECD spectra.
Focusing on various regions of the spectra shown in Figure 5A reveals some important differences between the ECD and ECD+CID spectra. Shown in Figure 5B and 5C are the theoretical isotopic distributions for the [M+3H]3+ and [M+2H]2+ melittin charge states generated directly from ESI and the corresponding mass-to-charge regions of the experimental ECD+CID and ECD spectra from Figure 5A. The vertical, dashed line in the 5B and 5C represents the monoisotopic mass-to-charge value for each charge state that would be expected in the absence of any neutralized protons. The mass-to-charge region around [M+4H]3+•, shown in middle spectra in 5B and 5C, provides evidence for the successful application of ECD+CID. Using ECD+CID the [M+4H]3+• ion is effectively removed from the ECDLIT and thus its ability to participate in any multiple electron capture events has been essentially eliminated. In contrast, the results from performing just ECD, bottom spectrum in Figure 5B, show that the [M+4H]3+• isotopic distribution is present with a significant abundance. From Figure 5B, it can be seen that the capture of one electron by [M+4H]4+ does result in a small amount of the even-electron [M+3H]3+ being formed; most likely due to a loss of H• from [M+4H]3+•.
The result of resonantly exciting [M+4H]3+• is demonstrated in Figure 5C. Under ECD+CID conditions (middle spectrum, Figure 5C), the [M+4H]2+ ion is not observed due to the resonance excitation of the [M+4H]3+• intermediate. Conversely, the [M+4H]2+ ion is present when just ECD is performed (bottom spectrum, Figure 5C), where the mass-to-charge values of the isotopic distribution indicate the presence of two neutralized protons with respect to [M+2H]2+ formed directly from ESI. The distribution for [M+4H]2+ in Figure 5C does show a small amount of [M+3H]2+•, most likely due to a loss of H• but this time from [M+4H]2+. Because [M+4H]2+ is formed readily under typical ECD conditions in the ECDLIT, the ability to perform ECD+CID is beneficial. The electron densities for the ECD+CID and ECD experiments were 1.0 and 1.1 μA/mm2, respectively. By using approximately the same electron densities for both experiments, the effectiveness of ECD+CID can be observed directly from Figure 5. With an electron irradiation time of only 7 ms for the ECD control experiment, the high electron densities readily achievable within the ECDLIT allow multiple electron capture events to occur. Therefore, even at short (i.e., 7 ms) irradiation times, it is necessary to minimize EC2D2 (Scheme 1) when higher electron fluxes (i.e., 0.80 μA vs 0.33 μA) are used.
The ability of ECD+CID to reduce the amount of ambiguity in product ion identification resulting from multiple electron capture events is demonstrated in Figure 6. The mass-to-charge values for the z23+2 and z24+2 melittin product ions from Figure 5 are shown in Figure 6A and 6B, respectively. The data in Figure 6 are presented in the same format as Figure 5B and C, where the theoretical isotopic distributions are shown in top spectra, the ECD+CID are shown in middle spectra and ECD results in the bottom spectra, respectively. For both product ions under just ECD conditions it was observed that their experimental isotopic distributions were shifted corresponding to the presence of one neutralized proton (bottom spectra). Under ECD+CID conditions, the abundances of both product ions were significantly reduced (middle spectra). Therefore, the formation of the z23+2 and z24+2 product ions can be attributed to multiple electron capture events. The data from Figure 6 demonstrate the utility of ECD+CID for simplifying the spectra when multiple electron capture events can occur because the abundance of the mass-shifted product ions have been significantly reduced in the MS/MS spectrum.
Figure 6.
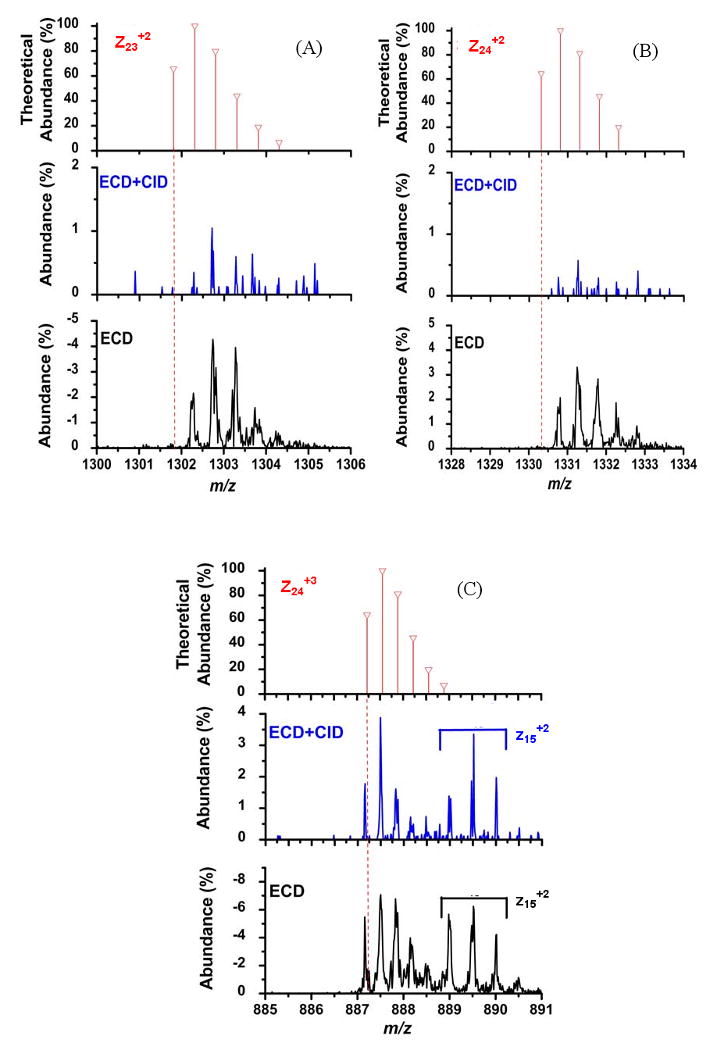
Mass-to-charge regions around the (A) z23+2, (B) z24+2, and (C) z24+3 and z15+2 melittin product ions from Figure 5A. The theoretical isotopic distributions are shown in the top spectra. The middle spectra are these mass-to-charge regions for the ECD+CID spectra and the bottom spectra are the same mass-to-charge regions for just the ECD spectra.
The spectra in Figure 6C serve as experimental evidence that product ions without neutralized protons are retained during ECD+CID and are observed at their expected mass-to-charge value. Shown in Figure 6C are the mass-to-charge regions encompassing the z24+3 and z15+2 product ions from the results in Figure 5. The theoretical isotopic distribution for the z24+3 product ion is shown in top spectrum of Figure 6C. The z24+3 product ion serves as an internal control experiment since it is not expected to undergo any mass shifts because it has retained three protons. Under both ECD+CID and ECD conditions, the z24+3 product ion is detected at its theoretical mass-to-charge value. The results shown in Figure 6C thus support the previous discussion regarding Figures 5A and 5B that the z23+2 and z24+2 ions, respectively, were reduced in abundance due to ECD+CID which prevented the [M+4H]3+• ions from capturing another electron.
In Figure 6C the z15+2 product ion was also detected at its theoretical mass-to-charge value under both experimental conditions. Since the z15+2 product ion only has two protons remaining, it could contain at least one neutralized proton similar to the z23+2 and z24+2 product ions from Figure 6A and 6B. However, the z15+2 product ion was not affected by the ECD+CID experiment and therefore it does not contain any neutralized protons and thus it comes directly from the [M+4H]4+ parent ion.
These results indicate another use for the ECD+CID experiment, determining the location of the neutralized proton in the charge reduced species. From the amino acid sequence of melittin provided in Figure 5 and assuming the protons reside at basic residues, the results presented in Figures 6 suggest that the neutralized proton observed under ECD conditions is located on Lys7. There is an extra proton in the z23+2 and z24+2 product ions, but not in the z15+2 product ion. Lys7 is in the only basic residue that is in the z23+2 and z24+2 product ions but not in the z15+2 product ion. Thus, if the parent ion sequence is known, ECD+CID could be used to further study characteristics of non-dissociative electron capture such as factors that dictate the location of electron capture and charge reduction for peptides and proteins.
Conclusions
A new technique entitled simultaneous ECD, CID (ECD+CID) has been implemented on a recently developed ECDLIT instrument. The ability to perform ECD+CID is unique to this ECDLIT instrument; a single mass-to-charge of interest can be activated through collisions with the ∼10-3 mbar helium bath gas. The activation is achieved by applying a supplemental ac waveform to the ECDLIT rod set to resonantly excite ions of mass-to-charge of interest.
The ability of ECD+CID to aid in peptide sequencing by increasing the number of sequence specific product ion and eliminating multiple electron capture events was demonstrated using melittin. By dissociating the charge-reduced species ([M+4H]3+•) as it is formed from [M+4H]4+ undergoing a non-dissociative electron capture event, the amount of peptide sequence coverage observed was increased from 76 % to 88 %. The abundance of product ions observed at mass-to-charge ratios that are shifted from their expected values under typical ECD conditions, due to multiple electron capture are reduced in the MS/MS spectrum via ECD+CID. Product ions mass-to-charge ratios can be shifted when multiple electron capture events occur because neutralized protons remain in the ion. Eliminating these peaks simplifies MS/MS spectral interpretation and allows increased electron flux to be used. It has also been demonstrated that information about the location of the neutralized proton inherent with electron capture can be obtained by comparing ECD+CID and ECD results.
Acknowledgments
This work was supported in part by NIH grant 5R21RR020207
References
- 1.Zubarev RA, Kelleher NL, McLafferty FW. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 2.Kruger NA, Zubarev RA, Horn DM, McLafferty FW. Int J Mass Spectrom. 1999;185/186/187:787–793. [Google Scholar]
- 3.Kruger NA, Zubarev RA, Carpenter BK, Kelleher NL, Horn DM, McLafferty FW. Int J Mass Spectrom. 1999;1999:1–5. [Google Scholar]
- 4.Bakhtiar R, Guan Z. Biochem Biophys Res Commun. 2005;334:1–8. doi: 10.1016/j.bbrc.2005.05.138. [DOI] [PubMed] [Google Scholar]
- 5.Shi SDH, Hemling ME, Carr SA, Horn DM, Lindh I, McLafferty FW. Anal Chem. 2001;73:19–22. doi: 10.1021/ac000703z. [DOI] [PubMed] [Google Scholar]
- 6.Håkansson K, Cooper HJ, Emmett MR, Costello CE, Marshall AG, Nilsson CL. Anal Chem. 2001;73:4530–4536. doi: 10.1021/ac0103470. [DOI] [PubMed] [Google Scholar]
- 7.Adamson JT, Håkansson K. J Proteome Res. 2006;5:493–501. doi: 10.1021/pr0504081. [DOI] [PubMed] [Google Scholar]
- 8.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Anal Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 9.Fung YME, Liu H, Chan TWD. J Am Soc Mass Spectrom. 2006;17:757–771. doi: 10.1016/j.jasms.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Leymarie N, Costello CE, O'Connor PB. J Am Chem Soc. 2003;125:8949–8958. doi: 10.1021/ja028831n. [DOI] [PubMed] [Google Scholar]
- 11.Håkansson K, Hudgins RR, Marshall AG. J Am Soc Mass Spectrom. 2003;14:23–41. doi: 10.1016/S1044-0305(02)00708-0. [DOI] [PubMed] [Google Scholar]
- 12.Adamson JT, Hakansson K. Anal Chem. 2007;79:2901–2910. doi: 10.1021/ac0621423. [DOI] [PubMed] [Google Scholar]
- 13.Zubarev RA. Mass Spectrom Rev. 2003;22:57–77. doi: 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- 14.Zubarev RA. Curr Opin Biotechnol. 2004;15:12–16. doi: 10.1016/j.copbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Cooper HJ, Håkansson K, Marshall AG. Mass Spectrom Rev. 2005;24:201–222. doi: 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- 16.Bakhtiar R, Guan Z. Biotech Lett. 2006;28:1047–1059. doi: 10.1007/s10529-006-9065-z. [DOI] [PubMed] [Google Scholar]
- 17.Tsybin YO, Quinn JP, Tsybin OY, Hendrickson CL, Marshall AG. J Am Soc Mass Spectrom. 2008;19:762–771. doi: 10.1016/j.jasms.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Baba T, Hashimoto Y, Hasegawa H, Hirabayashi A, Waki I. Analytical Chemistry. 2004;76:4263–4266. doi: 10.1021/ac049309h. [DOI] [PubMed] [Google Scholar]
- 19.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Proc Natl Acad Sci USA. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. J Proteome Res. 2005;4:628–632. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silivra OA, Kjeldsen F, Ivonin IA, Zubarev RA. J Am Soc Mass Spectrom. 2005;16:22–27. doi: 10.1016/j.jasms.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Ding L, Brancia FL. Anal Chem. 2006;78:1995–2000. doi: 10.1021/ac0519007. [DOI] [PubMed] [Google Scholar]
- 24.Liang X, Hager JW, McLuckey SA. Anal Chem. 2007;79:3363–3370. doi: 10.1021/ac062295q. [DOI] [PubMed] [Google Scholar]
- 25.Kaplan DA, Hartmer R, Speir JP, Stoermer C, Gumerov D, Easterling ML, Brekenfeld A, Kim T, Laukien F, Park MA. Rapid Commun Mass Spectrom. 2008;22:271–278. doi: 10.1002/rcm.3356. [DOI] [PubMed] [Google Scholar]
- 26.Satake H, Hasegawa H, Hirabayashi A, Hashimoto Y, Baba T. Analytical Chemistry. 2007;79:8755–8761. doi: 10.1021/ac071462z. [DOI] [PubMed] [Google Scholar]
- 27.Bushey JM, Baba T, Glish GL. The 56th ASMS Conference on Mass Spectrometry and Allied Topics; 2008. [Google Scholar]
- 28.Horn DM, Ge Y, McLafferty FW. Anal Chem. 2000;72:4778–4784. doi: 10.1021/ac000494i. [DOI] [PubMed] [Google Scholar]
- 29.Sze SK, Ge Y, Oh HB, McLafferty FW. Anal Chem. 2003;75:1599–1603. doi: 10.1021/ac020446t. [DOI] [PubMed] [Google Scholar]
- 30.Tsybin YO, Witt M, Baykut G, Kjeldsen F, Håkansson P. Rapid Commun Mass Spectrom. 2003;17:1759–1768. doi: 10.1002/rcm.1118. [DOI] [PubMed] [Google Scholar]
- 31.Oh HB, McLafferty FW. Bull Korean Chem Soc. 2006;27:389–394. [Google Scholar]
- 32.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JEP, Coon JJ. Anal Chem. 2007;79:477–485. doi: 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia Y, Han H, McLuckey SA. Anal Chem. 2008;80:1111–1117. doi: 10.1021/ac702188q. [DOI] [PubMed] [Google Scholar]
- 34.Tsybin YO, He H, Emmett MR, Hendrickson CL, Marshall AG. Anal Chem. 2007;79:7596–7602. doi: 10.1021/ac071165u. [DOI] [PubMed] [Google Scholar]
- 35.Mihalca R, van der Burgt YEM, McDonnell LA, Duursma M, Cerjak I, Heck AJR, Heeren RMA. Rapd Commun Mass Spectrom. 2006;20:1838–1844. doi: 10.1002/rcm.2520. [DOI] [PubMed] [Google Scholar]
- 36.O'Brien JT, Prell JS, Holm AIS, Williams ER. J Am Soc Mass Spectrom. 2008;19:772–779. doi: 10.1016/j.jasms.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laskin J, Futrell JH. Mass Spectrometry Reviews. 2005;24:135–167. doi: 10.1002/mas.20012. [DOI] [PubMed] [Google Scholar]
- 38.Amster IJ. Journal of Mass Spectrometry. 1996;31:1325–1337. [Google Scholar]
- 39.Håkansson K, Chalmers MJ, Quinn JP, McFarland MA, Hendrickson CL, Marshall AG. Anal Chem. 2003;75:3256–3262. doi: 10.1021/ac030015q. [DOI] [PubMed] [Google Scholar]
- 40.Lin C, Cournoyer JJ, O'Connor PB. J Am Soc Mass Spectrom. 2006;17:1605–1615. doi: 10.1016/j.jasms.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Lin C, Cournoyer JC, O'Connor PB. J Am Soc Mass Spectrom. 2008;19:780–789. doi: 10.1016/j.jasms.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satake H, Hasegawa H, Hirabayashi A, Hashimoto Y, Baba T. Anal Chem. 2007;79:8755–8761. doi: 10.1021/ac071462z. [DOI] [PubMed] [Google Scholar]
- 43.Douglas DJ, Frank AJ, Mao DM. Mass Spec Rev. 2005;24:1–29. doi: 10.1002/mas.20004. [DOI] [PubMed] [Google Scholar]
- 44.Marshall AG, Hendrickson CL, Jackson GS. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]