

Abstract
Cyclin-dependent kinase inhibitors (CKIs) are major contributors to the decision to enter or exit the cell cycle. The Caenorhabditis elegans genome encodes two CKIs belonging to the Cip/Kip family, cki-1 and cki-2. cki-1 has been shown to act as a canonical negative regulator of cell-cycle entry, while the role of cki-2 remains unclear. We identified cki-2 in a genome-wide RNAi screen to reveal genes essential for developmental cell-cycle quiescence. Examination of cki-2 knockout animals revealed extra rounds of cell divisions, verifying a role in establishing or maintaining the temporary cell-cycle arrest. Despite the overlapping defects, the pathways mediated by cki-1 and cki-2 are discrete since the extra cell phenotype conferred by a putative cki-2(null) mutation is enhanced upon additional loss of cki-1 activity. Moreover, the extra cell division defect of cki-2 is not increased with the additional loss of lin-35 Rb, as is seen with cki-1. Thus, both cki-1 and cki-2 mediate cell-cycle quiescence, but our genetic and phenotypic analyses demonstrate that they act within distinct pathways to exert control over the cell-cycle machinery.
Keywords: C. elegans, vulva development, cyclin-dependent kinase inhibitor, cell-cycle quiescence
Introduction
The developmental decision of cells to divide or arrest is generally determined during the G1 phase of the cell cycle. Studies in yeasts and cultured mammalian cells have identified a period, called START and the restriction point respectively,1–3 during G1 in which an irreversible commitment to the cell cycle is made. For example, animal cells that have progressed past the restriction point are refractory to serum withdrawal and can complete the cell cycle in the absence of further stimulation. Thus an elaborate system of checks and balances during G1 ensures that cell divisions are initiated appropriately during development. An important regulator of G1 progression, pRb, is encoded by the retinoblastoma tumor suppressor gene.4–8 One mechanism by which pRb inhibits G1/S progression is through transcriptional inhibition of genes required for S phase, such as cyclin E.9 In parallel, members of the Cip/Kip family of CKIs can interact with and inhibit the kinase activity of the Cdk2-cyclin E complex.10 By inactivating existing Cdk2-cyclin E complexes and inhibiting the production of new cyclin E, Cip/Kip and pRb act in concert to potentiate a G1 arrest.
We use the development of the C. elegans vulva as a model system to examine the regulatory mechanisms governing cell-cycle quiescence. The adult vulva originates from six vulva precursor cells (VPCs) that arise during the first larval stage (L1) and remain temporarily arrested in the G1 phase of the cell cycle until L3.11, 12 Since the vulva is not an organ that is required for viability of the animal, even defects that severely perturb its development or function can be studied. Using a lin-12 Notch gain-of-function mutation to direct the six VPCs to adopt inappropriate cell fates,13 we mark the VPCs as protrusions on the ventral surface of the adult known as pseudovulvae. Thus, in the lin-12(gf) genetic background, defects producing extra VPCs will result in easily observed extra pseudovulvae. We refer to the generation of greater than six pseudovulvae as the enhancer of lin-12(gf) multivulva (Elm) phenotype. elm genes identified in this manner act to limit the number of VPCs generated to six by controlling either cell-fate adoption or cell-cycle progression.14
A forward genetic screen for the Elm phenotype identified genes important in the regulation of cell cycles, such as cdc-14, lin-1, lin-31 and mdt-13.14, 15 We continued the Elm phenotype screen using RNAi feeding libraries to inhibit gene activities. Here, we describe the analysis of cki-2, a locus identified as an elm gene in the RNAi screen. The extra cell division defect of a putative null mutant indicates that cki-2 activity is required for the temporary cell-cycle arrest of the VPCs. In contrast to the related cki-1 locus, loss of cki-2 activity does not result in defects of multiple tissues nor are the defects enhanced by loss of lin-35 Rb function. These data suggest that both cki-1 and cki-2 perform functions during cell-cycle quiescence, but these functions are performed independently of each other.
Results
A genome-wide RNAi screen to discover genes necessary for normal VPC development identified the cyclin-dependent kinase inhibitor, cki-2
We previously performed a forward genetic screen for the enhancer of lin-12(gf) mutivulva (Elm) phenotype which identified genes required to direct a developmental cell-cycle arrest.14, 15 In ongoing screens for the Elm phenotype using RNAi, we identified a clone targeting cki-2, one of two C. elegans members of the Cip/Kip family of cyclin-dependent kinase inhibitors (CKIs).16–18 Unlike control-fed lin-12(gf) animals that generate six VPCs which in turn produce a maximum of six adult pseudovulvae, lin-12(gf) animals that ingested bacteria targeting cki-2 for RNAi-mediated inhibition (cki-2(RNAi)) can display greater than six pseudovulvae (Fig. 1A–B). The cki-2(RNAi); lin-12(gf) animals displayed an average of 5.1 pseudovulvae compared to 4.5 for control treated animals (Fig. 1C). The emergence of additional pseudovulvae following inhibition of cki-2 function by RNAi indicates the important role of cki-2 in restricting VPC production.
Figure 1.

The Elm phenotype of cki-2(RNAi). (A) An adult rrf-3(pk1426); lin-12(n950gf); lag-2(sa37) triple mutant tester animal treated with a control RNAi clone targeting gfp displays a maximum of six pseudovulvae (asterisks). Arrow indicates pseudovulva at normal vulva location. (B) Inactivation of cki-2 by RNAi results in the production of greater than six pseudovulvae on the tester strain. Scale bar indicates 50 μm. (C) Quantification of pseudovulvae displayed by the populations (n=50 each) represented in (A) and (B). cki-2(RNAi) significantly increased the average pseudovulvae number compared to the control population (P < 0.01).
cki-2 mutation results in extra VPCs through disruption of cell-cycle quiescence
To confirm the RNAi phenotype and to perform a careful genetic characterization of cki-2 function during development, we examined two deletion alleles of cki-2, tm3496 and ok2105 (Fig. 2A and Materials and Methods). The cki-2(tm3496) mutation deletes 362 base pairs to eliminate the weakly conserved C-terminus. In contrast, cki-2(ok2105) substitutes 1,606 base pairs, including the majority of the cki-2 coding sequence, with a duplication of the 9 base pair sequence immediately preceding the breakpoint. If expressed, the CKI-2(ok2105) truncated protein is predicted to be comprised of only 27 amino acids. Therefore, the cki-2(ok2105) mutation likely results in a null allele.
Figure 2.

cki-2(ok2105) mutant animals display cell-cycle quiescence defects. (A) Genomic organization of the cki-1 (black boxes) and cki-2 (grey boxes) coding sequences. Dashed lines designate regions deleted by the indicated mutations. Oligonucleotides used in PCR-based analyses are indicated as arrowheads. Break indicates approximately 4.8 kb separating cki-1 and cki-2. (B) PCR using genomic DNA template confirms cki-2(ok2105) deletion. Using conditions optimized for short products, PCR of wild-type DNA readily amplifies the 0.26 kb product of the A+B primer set (lane 1) while the 1.9 kb A+C product (lane 3) is not appreciable produced. Conversely, the cki-2(ok2105) mutant DNA does not support amplification using the A+B primer set, rather in combination with A+C generates a 0.3 kb product that spans the breakpoint of the mutation (lanes 2 and 4, respectively). (C) DAPI and anti-α-tubulin antibody staining of cki-2(ok2105) homozygous embryos visualizes the normal centrosome number, cells with greater than two centrosomes were not observed (n=30 embryos). (D) Nomarski image of wild-type L2 larvae shows three of six VPCs (arrows) temporarily arrested. (E) VPCs of cki-2(ok2105) L2 larva undergo extra cell divisions resulting from defective cell-cycle quiescence. Split arrows indicate sibling cells generated by extra rounds of cell divisions. Scale bars indicate 10 μm.
Since cki-2(RNAi) results in the production of extra pseudovulvae by the tester strain and the Cip/Kip family of CKIs control G1/S progression, we next examined the VPCs of the cki-2 mutant animals for extra rounds of cell divisions which would indicate defects in cell-cycle quiescence. The VPCs of both cki-2(tm3496) and cki-2(ok2105) mutant animals undergo extra cell divisions prior to the L2/L3 molt (Figs. 2E and 4), verifying a requirement for cki-2 activity in VPC cell-cycle quiescence. The cki-2(tm3496) extra VPC defect is less penetrant than cki-2(ok2105), supporting the notion that cki-2(ok2105) is a null allele. We confirmed the requirement for cki-2 by introducing a cki-2(wt) transgene into the cki-2(ok2105) mutant strain and observing rescue of the extra cell division defect (Fig. 4). Therefore, cki-2 activity contributes to the establishment or maintenance the L1-to-L3 cell-cycle quiescent period during VPC development.
Figure 4.

cki-2 is an important regulator of VPC cell-cycle quiescence. The percentage of VPCs observed to have undergone an extra round of cell division within the indicated genotype is presented. Except for the rescue experiment (n=7), at least 15 animals were examined just prior to the L2-to-L3 molt. Mean ± standard error values are indicated.
For subsequent genetic analyses of cki-2 function, we focused on the probable null allele, cki-2(ok2105). The cki-2(ok2105) animals appear superficially wild type despite the extra VPCs. We specifically note a lack of embryonic lethality and adult sterility, two phenotypes that can result from impaired cell cycles.19 The average brood size for cki-2(ok2105) hermaphrodites was reduced (195.1±101.1, 9 broods) compared to wild type (280.5±28.3, 11 broods). However, we determined that this decrease is the result of a partially penetrant egg-laying defect (Egl) that drastically reduced the number of offspring produced by the afflicted individual. Since non-Egl cki-2(ok2105) animals are otherwise capable of producing over 300 progeny (see Materials and Methods), cki-2 is not required for production of normal-sized broods. Importantly, the cki-2(ok2105) embryos exhibited neither defects in centrosome content (Fig. 2C) nor lethality (0.5%, n=1756) compared to wild-type embryos (0.7%, n=1411). Therefore, the cki-2(ok2105) mutation yields superficially normal animals that do not display cell cycle associated phenotypes, such as centrosome amplification or embryonic and larval lethality.
The cki-2 promoter is generally expressed during development
In order to determine the spatiotemporal expression pattern of cki-2, we generated transgenic worms expressing green fluorescent protein (GFP) under the control of the cki-2 promoter (Pcki-2::gfp). As previously reported,17 GFP is widely observed during embryogenesis. Similarly, GFP is also generally expressed during larval development, being observed within both dividing and terminally differentiated cells. For example, cells within the seam (Fig. 3A) and intestinal (Fig. 3B) lineages strongly express the Pcki-2::gfp reporter. The VPCs (Fig. 3C) and their descendent cells (Fig. 3D) also express GFP, further supporting a role for cki-2 in vulva development. While cki-2 activity may be regulated by additional post-transcriptional mechanisms that would not be discernible by the transcriptional reporter, the broad expression of GFP nevertheless indicates that cki-2 may act in the cell-cycle quiescence of a wide range of cell types.
Figure 3.

A Pcki-2::gfp reporter is widely expressed during larval development. During L1, GFP expression can be observed within cells of the intestine (A) and seam (B) lineages. Cells of the vulva lineage strongly express GFP at both the L2 (C) and L4 (D) stages. All images depict Nomarski (left panels) and GFP expression (right panels) of larvae harboring a Pcki-2::gfp extrachromosomal array. Arrows indicate VPCs and scale bars represent 10 μm.
cki-2 mutation does not result in a general disruption of cell-cycle quiescence
Since cki-2 is widely expressed and loss of cki-1 activity disrupts the cell-cycle quiescence of multiple developing tissues, we examined cell types other than the VPCs in the cki-2(ok2105) animals for similar extra cell division defects. In prior studies, cki-1(RNAi) resulted in the production of extra intestinal nuclei, seam cells and distal tip cells.16, 20 In addition, loss of cdc-14, a regulator of cki-1, produced extra cells within the M-lineage at a low frequency.15 Accordingly, we examined intestinal nuclei, distal tip cells, seam cells and cells within the M-lineage of cki-2(ok2105) mutant animals and found no evidence for defects in control of cell cycles (Table 1). Therefore, for these non-vulval cell types that express the cki-2 promoter during development, cki-2 activity is not required for normal cell-cycle quiescence.
Table 1.
Tissue-restricted defects of cki-2(ok2105) mutant animals
| tissue1, age | wild-type | n | cki-2(ok2105) | n |
|---|---|---|---|---|
| intestinal nuclei, hatching | 19.8±0.5 | 20 | 20.0±0.2 | 20 |
| intestinal nuclei, L4 | 32.6±1.0 | 20 | 32.2±1.2 | 20 |
| M-Lineage, hatching | 1±0 | 20 | 1±0 | 20 |
| M-Lineage, L2 | 16.2±0.2 | 19 | 16.4±0.2 | 20 |
| Distal Tip cell, L4 | 2±0 | 50 | 2±0 | 50 |
| seam cell, young adult | 16±0.4 | 20 | 15.9±0.5 | 20 |
| VPC, L2 | 6±0 | 30 | 8.6 ±0.8 | 30 |
Examination of intestine, M-lineage, Distal Tip and seam cells was aided using the elt-2, hlh-8, lag-2 and scm tissue-specific GFP markers, respectively.
cki-2 controls cell-cycle quiescence through a cyclin E-dependent pathway
To establish a requirement for cki-2 activity within the regulatory hierarchy controlling cell-cycle quiescence, we examined the cki-2(ok2105) phenotype in combination with mutations of cye-1 and lin-35, the sole C. elegans homologs of cyclin E and Rb, respectively.21, 22 As predicted for a cyclin-dependent kinase inhibitor, the extra VPC divisions in cki-2(ok2105) animals were strongly suppressed in combination with even heterozygous mutation of the cye-1 locus (Fig. 4). Intriguingly, the cki-2(ok2105) extra VPC phenotype was not enhanced in the lin-35(n745); cki-2(ok2105) double mutant animals (Fig 4), in contrast to the strong synergy displayed between cki-1 and lin-35 mutations.20 These data indicate that both cki-1 and cki-2 depend on cye-1 activity to promote the ectopic cell divisions but only the cki-1 activity is functionally redundant with the role played by lin-35 Rb. Thus, cki-1 and cki-2 perform partially overlapping roles in controlling VPC cell-cycle quiescence that are genetically distinguishable.
Genetic interactions suggest that cki-1 and cki-2 act in parallel pathways
Since cki-1 and cki-2 both regulate cell-cycle quiescence of the VPCs, we examined the genetic relationship between the two loci. We examined cki-1(gk132), the only identified mutation believed to specifically disrupt cki-1 activity. Since cki-1(gk132) homozygous animals die during embryogenesis, our analyses were limited to evaluation of cki-1(gk132) heterozygotes. Unlike the cki-1(gk132) mutation that displays weak haploinsufficiency for control of VPC cell-cycle quiescence,15 the cki-2(ok2105) mutation appears completely recessive (Fig. 4). To assess the effect of decreasing both cki-1 and cki-2 activities simultaneously, we first examined mnDf100, a chromosome II deficiency that deletes both cki-1 and cki-2 loci.17, 23 Animals heterozygous for the mnDf100 mutation present an extra VPC defect that is indistinguishable from cki-1(gk132) heterozygotes. Unexpectedly, upon analysis of the cki-1(gk132)/cki-2(ok2105) double-heterozygous combination, VPC cell-cycle quiescence was strongly disrupted as demonstrated by the highly penetrant extra VPC divisions (Fig. 4). The enhanced extra cell division phenotype is surprising since both the mnDf100/+ and the cki-1(gk132)/cki-2(ok2105) animals are predicted to be hemizygous for both cki-1 and cki-2 loci. The most likely explanation for the greater than expected extra cell division defect of cki-1(gk132)/cki-2(ok2105) animals is that the cki-1(gk132) mutation also disrupts the activity of the adjacent cki-2 locus. The equally strong extra VPC defect seen in cki-2(ok2105)/mnDf100 animals is consistent with the hypothesis that the gk132 mutation, like mnDf100, disrupts both cki-1 and cki-2 functions. Overall, these data indicate that cki-1 and cki-2 likely act in parallel pathways to control cell-cycle quiescence since a decrease in cki-1 activity enhances the extra VPC defect of the putative cki-2 null allele.
Given the strong enhancement of the cki-2 cell-cycle defect in combination with loss of cki-1 in the VPCs, we re-investigated a role for cki-2 in regulation of cell cycles outside the VPCs. As described above, intestines of cki-2(ok2105) mutants develop with the wild type nuclear complement, therefore we examined if loss of cki-2 function in combination with mutations of known cell-cycle regulators, such as cki-1, could reveal a weak role. The cki-1(gk132)/cki-2(ok2105) trans-heterozygous combination that results in a highly penetrant extra VPC defect yields an average nuclei number that is identical to heterozygous cki-1(gk132) alone (Fig. 5). Similarly, inhibition of cki-1 by RNAi in either wild-type or cki-2(ok2105) mutant animals results in an indistinguishable extra intestinal nuclei phenotype. Moreover, we examined the combination of cki-2(RNAi) and fzr-1(ku298), a mutation of a C. elegans Cdc20/Cdh1 homolog that functions redundantly with lin-35.24 Again, loss of cki-2 activity did not enhance the extra intestinal nuclei defect of fzr-1(ku298) mutant animals (Fig. 5). Thus, the inability of cki-2 loss of function to enhance the extra intestinal nuclei phenotype of either cki-1 or fzr-1 supports that cki-2 plays no role in regulation of cell-cycle quiescence during development of the intestine.
Figure 5.

cki-2 is not required for control of intestinal divisions. Young adult animals were analyzed for each of the indicated genotypes to determine the average number of intestinal nuclei (n≥20). Bars indicate mean ± standard deviation. * indicates that P > 0.2.
cki-2 expression is impaired by the cki-1(gk132) mutation
In order to test the hypothesis that the cki-1(gk132) mutation inhibits expression of the cki-2 locus, we used quantitative real-time PCR to measure cki-2 mRNA levels. Since homozygous cki-1(gk132) mutation results in embryonic lethality, measurement of cki-2 expression in the cki-1(gk132) homozygotes during larval development is not possible. Thus, we examined cki-1(gk132)/cki-2(ok2105) trans-heterozygotes. This mutant combination ensures that only mRNA expressed from the cki-2 locus located on the cki-1(gk132) chromosome is measured since the cki-2(ok2105) deletion eliminates sequences necessary for PCR amplification (Fig. 2B). mRNA expression of the cki-2 locus adjacent to the cki-1(gk132) mutation is decreased 8-fold when compared to expression from cki-2 with a cki-1(wt) neighbor (Fig. 6). These data demonstrate that the cki-1(gk132) mutation inhibits expression of the cki-2 locus that is approximately 5 kb downstream, thus providing a molecular explanation for the unexpected genetic interaction.
Figure 6.
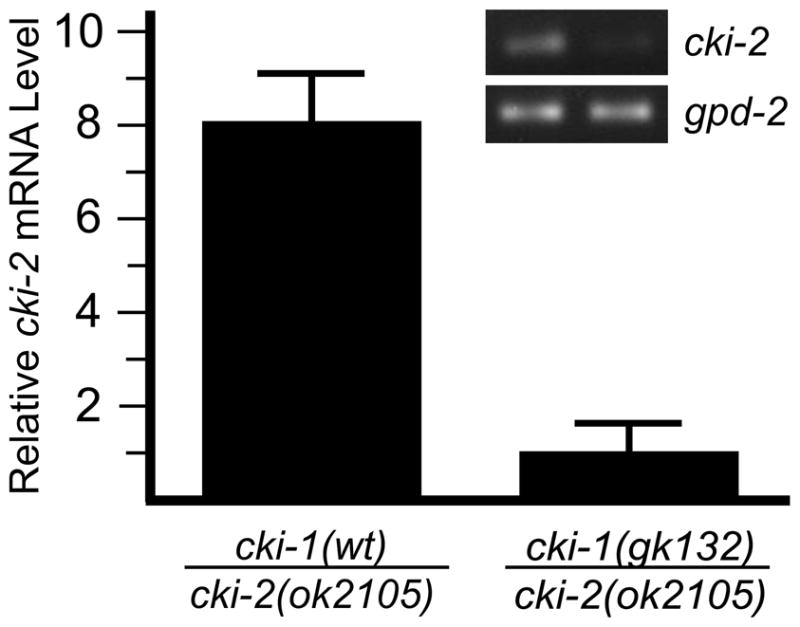
The cki-1(gk132) mutation disrupts expression of the adjacent cki-2 gene. Comparison of quantitative real-time PCR analysis of steady-state mRNA indicates that cki-2 expression from the cki-1(gk132) chromosome is decreased approximately 8-fold compared to the wild-type control. Inset image illustrates a representative result of a semi-quantitative, reverse-transcription PCR to visualize cki-2 mRNA from wild-type (left lane) and cki-1(gk132) (right lane) animals. GPDH (gpd-2) is used as the normalization control for both experiments.
Discussion
Our studies demonstrate that cki-2 regulates cell-cycle quiescence, likely through the control of the core components of the cell-cycle machinery. While our conclusions are based upon the characterization of phenotypes resulting from a putative cki-2 null allele, our attention to cki-2 was initially directed by RNAi data. RNAi and the related transgene-mediated co-suppression techniques provide convenient methods to inhibit gene activity; however, their expedience is counter-balanced by the potential for inconsistent or indirect effects.25–27 For example, a previous report using co-suppression to inhibit cki-2 activity (cki-2cs 28) found that cki-2cs germ lines were unable to properly expel centrioles, resulting in multipolar mitotic spindles and embryonic lethality. While there is little doubt that centrosome amplification and death was observed, the cki-2(ok2105) mutant animals clearly demonstrate that these previously reported defects were not the direct result of cki-2 loss of activity. We conclude that cki-2 is not required for centriolar expulsion during oogenesis since cki-2(ok2105) animals are deficient in cki-2 activity but display no evidence of impaired centrosome function, fertility nor viability.
The data presented here reveal a rate-limiting role for cki-2 as a canonical CKI, similar to the previously characterized role of cki-1. Several prior studies determined that cki-1(RNAi) causes defects in cell-cycle quiescence and results in obvious developmental disruptions.16–18 In contrast, cki-2(RNAi) did not yield an obvious phenotype16–18 and only upon closer experimental scrutiny was a subtle effect previously observed.20 Interestingly, overexpression of either CKI-1 or CKI-2 are sufficient to induce cell-cycle arrest16, 17; although only transgenes expressing cki-1(wt), and not cki-2(wt), were able to provide limited rescuing activity to mnDf100 homozygous embryos.17 In the present study, we examined the putative null allele, cki-2(ok2105) and conclude that cki-2 plays an important role in cell-cycle quiescence of the VPCs. As observed in our RNAi screen, cki-2 loss of function results in the production of extra VPCs through additional rounds of cell divisions. The cell-cycle quiescence defect conferred by cki-2(ok2105) is sensitive to the expression of cye-1, supporting that cki-2 functions as a CKI to regulate the core cell-cycle machinery. Since the cki-2(ok2105) mutation disrupts quiescence without affecting cki-1 expression (data not shown), cki-1 does not provide sufficient activity to compensate for loss of cki-2 activity. Therefore, cki-1 and cki-2 provide independent functions during cell-cycle quiescence.
Our data reveal two important distinctions between the roles played by cki-1 and cki-2. First, despite the general expression of the cki-2 reporter, the cki-2(ok2105) extra cell phenotype is cell-type restricted. In contrast, cki-1 loss-of-function produces defects in multiple tissues. The phenotypic differences between cki-1 and cki-2 mutant animals likely reflect differences in post-transcriptional regulation, cell-type specific dependence or protein function between the two genes. Although overexpression of either CKI-1 or CKI-2 is sufficient to arrest cell divisions, it is currently not possible to unequivocally demonstrate that the proteins perform unique functions, such as initiation vs. maintenance of quiescence. Second, we observe that cki-2 activity, unlike cki-1, is not functionally redundant with lin-35. A prior study determined that the lin-35-mediated process acted in parallel to cki-1 and cki-2 based on increased cell divisions in lin-35 mutant animals following concurrent RNAi inactivation of cki-1 and cki-2.20 In fact, our cki-2 data suggest that the previously observed lin-35 enhancement was due solely to the inactivation of cki-1. These data suggest that either cki-1, cki-2 and lin-35 act within three partially overlapping pathways or that cki-2 and lin-35 act together within with a pathway that is parallel to a cki-1-mediated process. Recent data obtained from replacing the p57 locus with p27 indicate that these two related mammalian CKIs perform both specific and shared roles.29 As technologies emerge that would allow high efficiency homologous recombination in C. elegans,30 it will be possible to uncover the specific molecular functions that differentiate cki-1 and cki-2.
Materials and Methods
Strains
All strains are N2 Bristol derived and grown under standard conditions at 20°C.31 The following mutant alleles were used in these studies: cye-1(eh10), lin-35(n745), mnDf100 unc-4(e120), cki-1(gk132), cki-2(ok2105), cki-2(tm3496), rrf-3(pk1426), lin-12(n950), lag-2(sa37), ayIs7[hlh-8::GFP], wIs78[ajm-1::gfp; scm-1::gfp; unc-119(+); F58E10(+)], rtIs14[elt-2::GFP] and qIs19[Plag-2::GFP].
RNAi screen
The Elm phenotype screen14 was modified to accommodate the reverse genetic approach. The rrf-3(pk1426) mutation32, 33 was incorporated into the lin-12(n950); lag-2(sa37) tester strain to enhance sensitivity to RNAi. The screening of the genome-wide library34 was performed essentially as described previously,35 with the exception that our screen searched only for the Elm phenotype using the triple-mutant tester strain. The feeding-RNAi clone, II-5H10, was found to induce the Elm phenotype. The clone was sequenced and verified as targeting T05A6.2, also known as cki-2.
Analysis of cki-2
The FX3496 strain was obtained from the National BioResource Project. RB1692, containing the cki-2(ok2105) deletion allele, was obtained from the C. elegans Genetics Center. The strain was back-crossed at least four generations before phenotypic analysis. PCR was used to localize the gene disruption and to generate a product that was sequenced to reveal the molecular lesion. Approximate positions of oligonucleotide primers used in reactions are shown in Figure 2A. Primer combination A+C amplified a 1.9 kb product from wild-type genomic DNA template, while a 0.3 kb product was generated using the cki-2(ok2105) deletion template. Primer combination A+B amplified 0.26 kb product from wild-type genomic DNA and was used to confirm the absence of full-length cki-2 sequences in the homozygous mutant animals. Oligonucleotide primer sequences are as follows:
Primer A: 5′-CTCGAGGATCATGGCGGCAACAACAGCCG-3′
Primer B: 5′-TCGGAAGCAGAAATCGACTC-3′
Primer C: 5′-AAACAAGAGCGAAGGTCGAA-3′
The sequence of the cki-2(ok2105) deletion/insertion mutation was determined to be:
…ATCTGATATTCATGATATTCATGCAAATGGT…
The underlined bases indicate the nine base pair duplication replacing the 1,606 bp normally found in wild type.
The Pcki-2::gfp transcriptional reporter was created by insertion of the 3.1 kb region upstream of the cki-2 coding sequence into pPD114.108, a kind gift from A. Fire. The reporter was co-injected with the dominant Rol-marker, pRF4, and four stable transgenic lines were isolated. Mosaic maintenance of the extrachromosomal transgenic array allowed visualization of GFP expression within specific tissues.
cDNA production and Real-time PCR analysis
cki-1(gk132)/mIn1 hermaphrodites and cki-2(ok2105) homozygous males were mated and cross-progeny were collected based on GFP expression. Approximately 100 L2-aged GFP-negative and GFP-positive individuals corresponding to cki-1(gk132)/cki-2(ok2105) and mIn1/(cki-2(ok2105), respectively, were pooled and their RNA isolated with the RNeasy kit (Qiagen) and reverse transcribed using Superscript III (Invitrogen). Relative expression levels were determined using the ΔΔCt method 36 with data from triplicate multiplexed reactions normalized to gpd-2 mRNA. Real-time PCR was performed with SYBR Green PCR Mix (Applied Biosystems) on a 7900HT machine using the 9600 emulation setting (Applied Biosystems). cki-2 was amplified using Primers A and B (above). Amplification of gpd-2 used the following primers: 5′-ACCGGAGTCTTCACCACCATC-3′ and 5′-TTCCTGATGGTCCGTCAACAG-3′.
Microscopy
To assay cell-cycle quiescence during development, synchronously developing populations were examined for extra cell divisions at 0, 18, 24 or 48 hours post-feeding. VPCs were visualized using Nomarski optics. Seam cells, distal tip cells, cells of the M-lineage and intestinal nuclei were visualized with the aid of the scm37, lag-238, hlh-839 and elt-240 GFP markers, respectively. Embryos were processed for immunohistochemistry using standard methanol/formaldehyde fixation protocols.41 Anti-α-tubulin monoclonal antibody (DM1A; Sigma-Aldrich) was used at 1:2000 dilution. Alexa Fluor 488 labeled goat anti-mouse secondary antibody (A11017; Invitrogen) was used at 1:500 dilution. Nomarski and epifluorescent images were collected using a Zeiss AxioImager microscope, AxioCam camera and Axiovision software. Image cropping and annotations were performed using Adobe Photoshop and Illustrator software.
Determination of viability and brood size
L4-aged hermaphrodites were individually placed on fresh plates. The parental animals were transferred at least once daily for at least three days. After two days, larvae and unhatched eggs were counted. Eleven N2 animals produced broods of 252, 252, 254, 260, 261, 278, 282, 286, 313, 317 and 330 total progeny. A subset of plates was analyzed to determine embryonic lethality, ten unhatched eggs were found among 1,411 viable siblings. Nine cki-2(ok2105) broods were enumerated as described. Egg-laying defective cki-2(ok2105) mutants produced broods of 34, 86,127 and 152 progeny, while cki-2(ok2105) mutants able to lay eggs for the duration of the experiment produced broods of 203, 249, 286, 309 and 310 offspring. Embryonic lethality was determined to be 0.51% in these broods with nine unhatched eggs found among the 1,756 total viable siblings.
Acknowledgments
We are grateful to Mike Boxem and Patricia Ernst for critical reading of the manuscript and Erika Artinger for help with the real-time PCR analyses. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the NIH National Center for Research Resources, and the National BioResource Project (NBRP) of Japan. This work was supported by grants to R.M.S. from the National Institutes of Health (GM077031) and the American Cancer Society (IRG-82-003-21).
Abreviations
- CKI
cyclin-dependent kinase inhibitor
- Elm
enhancer of lin-12(gf) multivulva
- RNAi
RNA interference
- VPC
vulva precursor cell
Contributor Information
Sarah H. Buck, Email: Sarah.H.Buck@dartmouth.edu.
Daniel Chiu, Email: Daniel.Chiu@dartmouth.edu.
R. Mako Saito, Email: Richard.M.Saito@dartmouth.edu.
References
- 1.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A. 1974;71:1286–90. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Temin HM. Stimulation by serum of multiplication of stationary chicken cells. J Cell Physiol. 1971;78:161–70. doi: 10.1002/jcp.1040780202. [DOI] [PubMed] [Google Scholar]
- 3.Woollard A, Nurse P. G1 regulation and checkpoints operating around START in fission yeast. Bioessays. 1995;17:481–90. doi: 10.1002/bies.950170604. [DOI] [PubMed] [Google Scholar]
- 4.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–9. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 5.DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–83. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 6.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 7.Friend SH, Horowitz JM, Gerber MR, Wang XF, Bogenmann E, Li FP, et al. Deletions of a DNA sequence in retinoblastomas and mesenchymal tumors: organization of the sequence and its encoded protein. Proc Natl Acad Sci U S A. 1987;84:9059–63. doi: 10.1073/pnas.84.24.9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harbour JW, Lai SL, Whang-Peng J, Gazdar AF, Minna JD, Kaye FJ. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science. 1988;241:353–7. doi: 10.1126/science.2838909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–62. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 10.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 11.Euling S, Ambros V. Heterochronic genes control cell cycle progress and developmental competence of C.elegans vulva precursor cells. Cell. 1996;84:667–76. doi: 10.1016/s0092-8674(00)81045-4. [DOI] [PubMed] [Google Scholar]
- 12.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–56. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 13.Greenwald IS, Sternberg PW, Horvitz HR. The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell. 1983;34:435–44. doi: 10.1016/0092-8674(83)90377-x. [DOI] [PubMed] [Google Scholar]
- 14.Clayton JE, van den Heuvel SJ, Saito RM. Transcriptional control of cell-cycle quiescence during C.elegans development. Dev Biol. 2008;313:603–13. doi: 10.1016/j.ydbio.2007.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saito RM, Perreault A, Peach B, Satterlee JS, van den Heuvel S. The CDC-14 phosphatase controls developmental cell-cycle arrest in C. elegans. Nat Cell Biol. 2004;6:777–83. doi: 10.1038/ncb1154. [DOI] [PubMed] [Google Scholar]
- 16.Hong Y, Roy R, Ambros V. Developmental regulation of a cyclin-dependent kinase inhibitor controls postembryonic cell cycle progression in Caenorhabditis elegans. Development. 1998;125:3585–97. doi: 10.1242/dev.125.18.3585. [DOI] [PubMed] [Google Scholar]
- 17.Fukuyama M, Gendreau SB, Derry WB, Rothman JH. Essential embryonic roles of the CKI-1 cyclin-dependent kinase inhibitor in cell-cycle exit and morphogenesis in C elegans. Dev Biol. 2003;260:273–86. doi: 10.1016/s0012-1606(03)00239-2. [DOI] [PubMed] [Google Scholar]
- 18.Feng H, Zhong W, Punkosdy G, Gu S, Zhou L, Seabolt EK, et al. CUL-2 is required for the G1-to-S-phase transition and mitotic chromosome condensation in Caenorhabditis elegans. Nat Cell Biol. 1999;1:486–92. doi: 10.1038/70272. [DOI] [PubMed] [Google Scholar]
- 19.O’Connell KF, Leys CM, White JG. A genetic screen for temperature-sensitive cell-division mutants of Caenorhabditis elegans. Genetics. 1998;149:1303–21. doi: 10.1093/genetics/149.3.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boxem M, van den Heuvel S. lin-35 Rb and cki-1 Cip/Kip cooperate in developmental regulation of G1 progression in C. elegans. Development. 2001;128:4349–59. doi: 10.1242/dev.128.21.4349. [DOI] [PubMed] [Google Scholar]
- 21.Fay DS, Han M. Mutations in cye-1, a Caenorhabditis elegans cyclin E homolog, reveal coordination between cell-cycle control and vulval development. Development. 2000;127:4049–60. doi: 10.1242/dev.127.18.4049. [DOI] [PubMed] [Google Scholar]
- 22.Lu X, Horvitz HR. lin-35 and lin-53, two genes that antagonize a C.elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell. 1998;95:981–91. doi: 10.1016/s0092-8674(00)81722-5. [DOI] [PubMed] [Google Scholar]
- 23.Sigurdson DC, Spanier GJ, Herman RK. Caenorhabditis elegans deficiency mapping. Genetics. 1984;108:331–45. doi: 10.1093/genetics/108.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fay DS, Keenan S, Han M. fzr-1 and lin-35/Rb function redundantly to control cell proliferation in C. elegans as revealed by a nonbiased synthetic screen. Genes Dev. 2002;16:503–17. doi: 10.1101/gad.952302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Captan VV, Goszczynski B, McGhee JD. Neither maternal nor zygotic med-1/med-2 genes play a major role in specifying the Caenorhabditis elegans endoderm. Genetics. 2007;175:969–74. doi: 10.1534/genetics.106.066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 27.Ahringer J. WormBook. The C. elegans Research Community. 2006. Reverse Genetics. [Google Scholar]
- 28.Kim DY, Roy R. Cell cycle regulators control centrosome elimination during oogenesis in Caenorhabditis elegans. J Cell Biol. 2006;174:751–7. doi: 10.1083/jcb.200512160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Susaki E, Nakayama K, Yamasaki L, Nakayama KI. Common and specific roles of the related CDK inhibitors p27 and p57 revealed by a knock-in mouse model. Proc Natl Acad Sci U S A. 2009;106:5192–7. doi: 10.1073/pnas.0811712106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robert VJ, Bessereau JL. Manipulating the Caenorhabditis elegans genome using mariner transposons. Genetica. 2009 doi: 10.1007/s10709-009-9362-2. [DOI] [PubMed] [Google Scholar]
- 31.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simmer F, Moorman C, van der Linden AM, Kuijk E, van den Berghe PVE, Kamath RS, et al. Genome-Wide RNAi of C. elegans Using the Hypersensitive rrf-3 Strain Reveals Novel Gene Functions. PLoS Biology. 2003;1:e12. doi: 10.1371/journal.pbio.0000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, et al. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol. 2002;12:1317–9. doi: 10.1016/s0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
- 34.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–7. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 35.Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, et al. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 2004;14:2162–8. doi: 10.1101/gr.2505604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Abrahante JE, Daul AL, Li M, Volk ML, Tennessen JM, Miller EA, et al. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell. 2003;4:625–37. doi: 10.1016/s1534-5807(03)00127-8. [DOI] [PubMed] [Google Scholar]
- 38.Blelloch R, Anna-Arriola SS, Gao D, Li Y, Hodgkin J, Kimble J. The gon-1 gene is required for gonadal morphogenesis in Caenorhabditis elegans. Dev Biol. 1999;216:382–93. doi: 10.1006/dbio.1999.9491. [DOI] [PubMed] [Google Scholar]
- 39.Corsi AK, Kostas SA, Fire A, Krause M. Caenorhabditis elegans twist plays an essential role in non-striated muscle development. Development. 2000;127:2041–51. doi: 10.1242/dev.127.10.2041. [DOI] [PubMed] [Google Scholar]
- 40.Fukushige T, Hawkins MG, McGhee JD. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev Biol. 1998;198:286–302. [PubMed] [Google Scholar]
- 41.Duerr JS. WormBook. The C. elegans Research Community. 2006. Immunohistochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]