

Abstract
Activation of nuclear factor (NF)-κB, one of the most investigated transcription factors, has been found to control multiple cellular processes in cancer including inflammation, transformation, proliferation, angiogenesis, invasion, metastasis, chemoresistance and radioresistance. NF-κB is constitutively active in most tumor cells, and its suppression inhibits the growth of tumor cells, leading to the concept of ‘NF-κB addiction’ in cancer cells. Why NF-κB is constitutively and persistently active in cancer cells is not fully understood, but multiple mechanisms have been delineated including agents that activate NF-κB (such as viruses, viral proteins, bacteria and cytokines), signaling intermediates (such as mutant receptors, overexpression of kinases, mutant oncoproteins, degradation of IκBα, histone deacetylase, overexpression of transglutaminase and iNOS) and cross talk between NF-κB and other transcription factors (such as STAT3, HIF-1α, AP1, SP, p53, PPARγ, β-catenin, AR, GR and ER). As NF-κB is ‘preactive’ in cancer cells through unrelated mechanisms, classic inhibitors of NF-κB (for example, bortezomib) are unlikely to mediate their anticancer effects through suppression of NF-κB. This review discusses multiple mechanisms of NF-κB activation and their regulation by multitargeted agents in contrast to monotargeted agents, thus ‘one size does not fit all’ cancers.
Keywords: NF-κB, constitutive activation, cancer, inflammation, multitargeted therapy
Introduction
In the previous century, many research initiatives were focused on understanding the genetic etiology of human tumorigenesis. However, cancer remains a major health problem and is responsible for one in eight deaths worldwide (Garcia et al., 2007). Also, an estimated 55% increase in cancer incidence is expected by the year 2020 (Warren et al., 2008).
Cancer is an extremely complex disease (Roukos, 2010). Large-scale analysis of genes has shown that the types of mutations that occur in various cancers are highly heterogeneous (Hudson et al., 2010). Only a minority of cancers are caused by germline mutations, whereas the vast majority (~90%) are linked to somatic mutations and environmental factors (Aggarwal et al., 2009). The mutations found in the cancer cell genome accumulate over the lifetime of the cancer patient. Mutation rate increases in the presence of substantial exogenous mutagenic exposures, such as tobacco smoke carcinogens, naturally occurring toxic chemicals present in some foods or various forms of radiation including ultraviolet light. These exposures are associated with increased rates of lung, liver and skin cancer, respectively (Grivennikov et al., 2010). Moreover, these agents induce inflammatory responses, suggesting a strong association between inflammation, nuclear factor (NF)-κB and cancer (Mantovani et al., 2008; Aggarwal and Gehlot, 2009; Grivennikov et al., 2010).
Currently, 18 485 genes and 464 139 tumors have been sequenced, resulting in a cataloge of 20 090 unique mutations, which have been curated in the Catalogue of Somatic Mutations in Cancer (http://www.sanger.ac.uk/cosmic, v47 release, 24 May, 2010). Studies of the genetic complexities of breast, colorectal, pancreatic and brain cancers have revealed that cancer genomes are highly complex (Bignell et al., 2010; Ding et al., 2010; Pleasance et al., 2010a), with a range of 48–101 somatic alterations in each tumor, depending on the cancer type (Jones et al., 2008; Parsons et al., 2008; Bell, 2010). Within a given cancer type, there is considerable intertumor heterogeneity, resulting in large number of altered genes. However, this complexity is reduced significantly by considering the biological pathways, rather than the altered gene themselves (Wood et al., 2007; Verhaak et al., 2010). For example, 12 core biological processes or pathways appear to be deregulated in most pancreatic tumors, although precisely how this deregulation is achieved varies from tumor to tumor (Jones et al., 2008). This suggests alterations in the complex network of signaling pathways (Ledford, 2010), and has very clear practical implications for the development of targeted therapeutics, as it is less likely that a drug targeting just one mutated gene or one particular pathway alone could be effective for treating any type of cancer.
The area of research involving NF-κB has grown tremendously in the past decade. This is evident from the fact that although NF-κB was discovered only 25 years ago (Sen and Baltimore, 1986) and is one of ~2000 estimated transcription factors in humans (Lander et al., 2001; GuhaThakurta, 2006), ~10% of research articles listed in PubMed on the subject of transcription factors are associated with NF-κB. Furthermore, of the more than 39 000 articles published about NF-κB, about 19 000 are associated with tumors and cancers underscoring the importance of this transcription factor in cancer studies.
As NF-κB is the key transcription factor involved in the inflammatory pathway, NF-κB is constitutively active in most cancers (Table 1), and many of the signaling pathways implicated in cancer are likely to be networked to the activation of NF-κB (Figure 1) (Karin, 2009; Grivennikov et al., 2010; Grivennikov and Karin, 2010). Mammalian NF-κB is a family of transcription factors that includes five members: RelA/p65, c-Rel, RelB, NF-κB1 (p50) and NF-κB2 (p52) (Ghosh and Karin, 2002; Vallabhapurapu and Karin, 2009). The primary regulation of the NF-κB pathway is through the association of NF-κB complexes with their inhibitor, IκB proteins. There are multiple human IκB proteins, including IκBα, IκBβ, IκBε and IκBζ. In addition, the precursors p50 and p52 and the full-length proteins p105 and p100 also function as IκB proteins. The principle inactive form of the NF-κB complex is a p50–p65 (RelA)–IκBα trimer, primarily located in the cytoplasm. In the classic or canonical pathway, in response to various external stimuli, IκBα is phosphorylated at Ser 32 and Ser 36 by the IκBα kinase (IKK). This promotes K-48 ubiquitination of IκBα by the SCF–βTrCP complex and its degradation by the proteasome. The released NF-κB dimer (p50–p65, which is also phosphorylated by IKK) is translocated in the nucleus, where it binds to its cognate response elements in promoters to activate the transcription of responsive genes (Vallabhapurapu and Karin, 2009). At the NF-κB responsive promoters, the p65 subunit of NF-κB is further modified by acetylation and methylation, and it interacts with additional coactivators (Werner et al., 2005). A second, non-canonical or alternative pathway involves activation of the p100–RelB complex to p52–RelB in response to specific extracellular signals (Senftleben et al., 2001). Unlike its response to IκBα, p100, after phosphorylation at Ser 866 and Ser 870, undergoes limited processing to generate p52, also regulated by SUMOylation (Vatsyayan et al., 2008).
Table 1.
Different mechanisms of constitutive activation of NF-κB in cancers
| Cancer type | Mutated gene | Pathway | Reference |
|---|---|---|---|
| Cancers | |||
| Acute myeloid leukemia | |||
| c-KIT | PI3K–Akt | (Lennartsson et al., 2005; Linnekin, 1999) | |
| FLT3 | PI3K–Akt | (Choudhary et al., 2005; Grosjean-Raillard et al., 2008) | |
| ERK–MAPK | (Takahashi et al., 2005) | ||
| Ras | PI3K-PKB–Akt-IKK | (Birkenkamp et al., 2004) | |
| Chronic myeloid leukemia | |||
| Bcr–Abl | MEKK1 | (Nawata et al., 2003) | |
| Pancreatic cancer | |||
| K-Ras | PI3K-RacEGF-Rac | (Almoguera et al., 1988) | |
| PI3K–Akt-IKK | (Liptay et al., 2003) | ||
| Prostate cancer | |||
| AR | PI3K–Akt | (Newmark et al., 1992; Sun et al., 2010) | |
| Small cell lung cancer | |||
| K-Ras | TBK1–IKKβ | (Basseres et al., 2010) | |
| Head and neck cancer | |||
| EGFR | CK2–IKK | (Molinolo et al., 2009) | |
| Glioma | |||
| EGFR/PTEN | PI3K–Akt-IKK | (Wang et al., 2004) | |
| PDGF | PI3K–Akt-IKK | (Smith et al., 2008) | |
| Non-small cell lung cancer | |||
| K-Ras/EGFR | PI3K–Akt-IKK | (Tang et al., 2006) | |
| Breast cancer | |||
| HER2 | PI3K–Akt-IKK | (Cao et al., 2007) | |
| TAB1 | TAK1–IKKβ | (Neil and Schiemann, 2008) | |
| Multiple myeloma | |||
| NFKB2 | Alternative | (Demchenko et al., 2010) | |
| CYLD, NFKB1, and TACI | Classical | (Demchenko et al., 2010) | |
| NIK, TRAF2, TRAF3, cIAP1 and 2, and CD40 | Both alternative | (Demchenko et al., 2010) | |
| Others | |||
| B-cell line | Enhanced IκBα degradation | (Miyamoto et al., 1994) | |
Abbreviations: AR, androgen receptor; cIAP, cellular inhibitor of apoptosis; CK2, casein kinase 2; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; FLT3, FMS-like tyrosine kinase 3; IKK, IκB kinase; MAPK, mitogen-activated protein kinase; MEKK, MAPK kinase; NIK, NF-κB inducing kinase; PKB, protein kinase B; PDGF, platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog; TAB1, TAK1 binding protein 1; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; TAK1, transforming growth factor (TGF)-β activated kinase 1; TBK1, TNF receptor-associated NF-κB kinase (TANK)-binding kinase 1; TRAF, TNF receptor-associated factor.
Figure 1.

Signaling network of NF-κB activation in cancer. Various pathways of NF-κB activation in cancers are shown. The sites of action of some phytochemicals are also indicated in the boxes. The network converges at three major sites (IKK kinase such as TAK1, IKK itself and the p50–p65 heterodimer). ASK1, apoptosis signal-regulating kinase 1; BAFF-R, TNF family member B cell-activating factor-receptor; CK II, casein kinase II; HTLV, human T-lymphotropic virus; LTβR, lymphotoxin-β receptor; MyD88, myeloid differentiation primary response gene 88; RalB, RAS-like protein B; RANK, receptor activator for nuclear factor κB; RIP, receptor interacting protein; Syk, spleen tyrosine kinase; TAK1, transforming growth factor (TGF)-β activating kinase 1; TBK, TRAF family member-associated NF-κB activator (TANK)-binding kinase; TLR, toll-like receptor; TNFR, tumor necrosis factor receptor; TRADD, TNFR1-associated death domain; TRAF, TNF-receptor-associated factor; β-TrCP, β-transducin repeat-containing protein; TWEAK-R, TNF-related weak inducer of apoptosis-receptor; UV, ultra violet.
For activation of NF-κB by canonical and non-canonical pathways, the regulatory kinase is IKK, which is a complex of three proteins, two catalytic (IKKα and IKKβ) and one regulatory (IKKγ, also known as the NF-κB essential modulator (NEMO)). The IKK complex is believed to be activated by a wide variety of stimuli that finally modify IKKγ by K-63 ubiquitinylation (Bhoj and Chen, 2009; Chen and Sun, 2009) and by phosphorylation (Perkins, 2006; Scheidereit, 2006). However, pathways that do not involve IKK have been described for NF-κB activation (Kato et al., 2003; O’Dea et al., 2008; Ge et al., 2009). Even the IKK family, the key kinases that phosphorylate IκBα, is known to phosphorylate proteins other than IκBα (Chariot, 2009; Hutti et al., 2009). The p65 transactivator subunit has been shown to be phosphorylated by IKK (Figure 2). In addition to phosphorylation, it is also acetylated and methylated that are catalyzed by different set of enzymes (discussed later). Moreover, the activated NF-κB interacts and cooperates with other transcription factors such as STAT3, HIF-1α and AP1 to up- or down-modulate gene expression (Figure 3).
Figure 2.

Sites of modification in p65 (RelA) subunit of NF-κB in cancer. Locations of various modification sites in the Rel homology domain and transactivation domains (TAD1 and 2) of p65 are shown. The possible effects are shown in the boxes. Ac, actylation; K, lysine residues; M, methylation; P, phosphorylation; S, serine residues; Ub, ubiquitination.
Figure 3.

NF-κB interactions in cancer. NF-κB interacts with many transcription factors and transcriptional regulators. The interactions could be direct at the promoter, such as in AP1, HIF-1α, Notch1, JunD, CREB, SP1 or off the promoter, such as STAT3, p53 or both. The single-head arrow indicates activation, whereas the double-head arrow indicates direct and indirect activation. The blunt-end arrow indicates inhibition and negative regulation. For PPARs, see (Delerive et al., 1999; Chung et al., 2000; Planavila et al., 2005); for SIRT1 (Yeung et al., 2004); for Sir2 (Yang et al., 2007); for RXR (Na et al., 1999); for Daxx (Park et al., 2007); for GR (Ray and Prefontaine, 1994); for ER (Galien and Garcia, 1997); for SMRT (Lee et al., 2000); for Egr-1 (Chapman and Perkins, 2000); for HDACs (Ashburner et al., 2001; Liu et al., 2005); for PIAS (Chen et al., 2001); for FoxO4 (Zhou et al., 2009); for κB-ras (Tago et al., 2010); for β-catenin (Deng et al., 2002); for Egr-3 & 4 (Wieland et al., 2005); for TFIIB (Schmitz et al., 1995); for p300 (Morimoto et al., 2008); for BRCA1 (Benezra et al., 2003); for E2F1 (Lim et al., 2007); for IRF-1 (Sgarbanti et al., 2008); for SP-1 (Perkins et al., 1993); for C/EBPβ (Zwergal et al., 2006); for JunD (Toualbi-Abed et al., 2008); for Notch-1 (Cheng et al., 2001); for SRF (Franzoso et al., 1996); for HIF-1α (Scortegagna et al., 2008); for MEF2 (Kumar et al., 2005); for p53 (Jeong et al., 2004); for STAT3 (Yoshida et al., 2004; Yu and Kone, 2004); for AP-1 (Stein et al., 1993); for CDX2 (Kim et al., 2004); for PR (Kovalenko et al., 2003); and for AR (Palvimo et al., 1996).
In the present review, we will discuss the ramifications and networking of these diverse pathways in the etiology and treatment of cancer in an attempt to show that targeting just one step or one pathway would not be a successful strategy for the prevention or treatment of cancer.
NF-κB mutation and cancer
The p65 transactivator subunit of NF-κB, RelA, was recognized earlier as the potential oncogene (Gilmore, 2003). However, the mutations that confer Rel-A, c-Rel, or other NF-κB proteins with oncogenic potentialities are not abundant and are mainly limited to lymphoid malignancies. REL gene amplifications have been detected frequently in many types of human B-cell, and to a lesser extent T cell, lymphoma (reviewed in (Gilmore et al., 2004; Courtois and Gilmore, 2006)). Chronic lymphocytic leukemia, the most common adult leukemia, is currently incurable with conventional chemotherapeutic agents. Rel-A is a prognostic marker and a therapeutic target in this disease (Pepper et al., 2009; Lopez-Guerra and Colomer, 2010).
Similarly, the NFKB2 gene undergoes structural alterations in certain T-cell lymphomas, chronic lymphocytic leukemias, myelomas and B-cell lymphomas (Courtois and Gilmore, 2006). In Hodgkin’s lymphoma, mutations or deletions in the IκBα gene have been described (Cabannes et al., 1999). The Bcl-3 gene, another family member of IκBα, is overexpressed and translocated in B-cell leukemia (Martin-Subero et al., 2007; Chapiro et al., 2008). Similarly, amplification of the c-Rel gene is reported in several types of B-cell lymphoma (Pileri et al., 2003). Mutations in other NF-κB genes, such as NEMO, result more frequently in immunological disorders (Courtois and Gilmore, 2006).
In lymphoid malignancies, the dysregulation of NF-κB activation results in aberrant expression of target gene proteins such as cyclin D1, cyclin D2, c-myc, c-myb, BCL2 and BCL-XL that regulate cell proliferation or survival, as well as cytokines such as interleukin (IL)-2, IL-6 and CD40-L that regulate growth and proliferation of lymphocytes. Thus, constitutively active NF-κB has been implicated in various lymphoid malignancies (Aggarwal and Gehlot, 2009). Acute myeloid leukemia involves the activation of RTK Flt3, N-Ras and K-Ras, which activate NF-κB through the Akt pathway (Stirewalt and Radich, 2003; Tenen, 2003). As can be seen in Table 1, although NF-κB genes are not directly mutated in most cases, mutations in key genes such as RAS, phosphatidylinositol 3-kinase (PI3K)/Akt1, TP53 and EGFR affect the cellular processes that are known to involve the activation of NF-κB in some capacity (Grivennikov et al., 2010).
NF-κB as a tumor promoter and tumor suppressor
In recent years, in vitro studies have established strong support for the critical role of NF-κB in cancer. Abnormally high NF-κB activity is a clinical hallmark of chronic inflammation and has been found in many types of cancer cells. Therefore, drugs that inhibit NF-κB activity have been found to be useful additions to the chemotherapy regimens of a variety of cancers (Hoffmann et al., 2007). However, some recent findings have suggested that this generalization should be viewed with caution (Luedde et al., 2007; Bettermann et al., 2010). The role of the NF-κB signaling pathway in the development of hepatocellular carcinoma, for example, continues to remain controversial (Greten et al., 2004; Pikarsky et al., 2004; Vainer et al., 2008). Using a conditional knockout approach for IKKγ (NEMO), Luedde et al. (2007) showed that elimination of NF-κB activity in hepatocytes surprisingly resulted in elevated inflammatory cytokine expression and spontaneous carcinogenesis in every animal within a year, suggesting a tumor suppressor role for NF-κB (Luedde et al., 2007). They further showed that the conditional knockout of TAK1, the kinase that phosphorylates IKK, gives a similar phenotype. In an earlier study, deletion of IKKβ was also shown to induce hepatocellular carcinoma (Maeda et al., 2005). However, the pathway through which hepatocellular carcinoma is generated does not involve NF-κB (Bettermann et al., 2010).
Recent experimental data from several laboratories have revealed that IKKα also functions as a tumor suppressor in human squamous cell carcinomas of the skin, lungs, and head and neck (Maeda et al., 2007; Van Waes et al., 2007). Chemical carcinogenesis studies in mice have shown that reduction in IKKα expression increased the number and size of Ras-initiated skin tumors and promoted their progression, indicating that reduced IKKα expression provides a selective growth advantage that cooperates with Ras activity to promote skin carcinogenesis (Zhu et al., 2009). Of interest, IKKα kinase activity is not required for the development of mouse embryonic skin. Further mechanistic study revealed that IKKα interacts with histone H3 in nucleosomes and blocks the access of histone methyl transferase SUV39h1 to H3 (Zhu et al., 2007).
Two other tumor suppressor genes that regulate NF-κB activation, CYLD and A20, belong to the deubiquitinase family. Mutations in the CYLD gene that encodes for a tumor suppressor protein negatively regulate NF-κB activation. CYLD codes for a deubiquitinase (Brummelkamp et al., 2003; Kovalenko et al., 2003; Trompouki et al., 2003) that catalyses removal of K-63 polyubiquitin groups required for the activation of TAK1 and NEMO (Bhoj and Chen, 2009; Chen and Sun, 2009). A20 (Vereecke et al., 2009; Hymowitz and Wertz, 2010) also acts as a tumor suppressor gene (Shembade et al., 2010; Sriskantharajah and Ley, 2010).
In contrast to these findings, however, a recent study showed that conditional ablation of IKKβ inhibited melanoma tumor development in mice (Yang et al., 2010a). In addition, the non-canonical IKK family member IKKε, a recently discovered breast cancer oncoprotein, was shown to be essential for regulating antiviral signaling pathways. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKε promoted cell transformation (Hutti et al., 2009). Using integrative genomic approaches, Boehm et al. identified IKKε as a breast cancer oncogene (Boehm et al., 2007).
The above discussion clearly indicates that the IKK complex that was once believed to be responsible only for activating the canonical pathway of NF-κB may have many diverse functions. The roles of IKKs and their regulators vary with tissue types and with the context in which they act. Some of these points will be further elaborated in the following sections.
Constitutive activation of NF-κB in cancers (NF-κB addiction)
It has become increasingly clear that besides having a role in regulating adaptive immune response, NF-κB signaling also has a critical role in cancer development and progression (Aggarwal, 2004; Basseres and Baldwin, 2006; Karin, 2006; Mantovani et al., 2008; Prasad et al., 2010). The major tumor-promoting mechanism is the production of tumor-promoting cytokines by immune/inflammatory cells that activate transcription factors such as NF-κB, STAT3 and AP1, which induce genes responsible for cell proliferation, survival, angiogenesis and metastasis (Grivennikov et al., 2010). Constitutive activation of NF-κB has been reported in a wide variety of malignancies such as hematological, gastrointestinal, genitourinary, gynecological, thoracic, head and neck, and breast tumors and in melanoma and fibrosarcoma (Table 1) (reviewed in (Prasad et al., 2010)). Though the activated NF-κB was initially described in lymphoid cancers, it is also detected in most solid tumors as well (Karin et al., 2002). Recently, activated NF-κB was shown to be a prognostic factor in metastatic serous ovarian carcinoma. Immunoblotting showed NF-κB p65 phosphorylation in 72 (96%) of 75 effusions (Kleinberg et al., 2009).
Diverse mechanisms have been ascribed to the constitutive activation of NF-κB in cancers. Autocrine secretion of inflammatory mediators (chemokines and cytokines such as tumor necrosis factor α (TNF-α) and IL-1β) have been shown to activate NF-κB constitutively in head and neck cancer cells, acute myeloid leukemia, T-cell lymphoma, breast cancer (Giri and Aggarwal, 1998; Wolf et al., 2001; Estrov et al., 2003; Jackson-Bernitsas et al., 2007; Braunstein et al., 2008) and acute myeloid leukemia. Another mechanism that is implicated in the constitutive activation of NF-κB is mutations and/or overexpression of ligands and receptors such as epidermal growth factor (Sethi et al., 2007), HER-2/Neu (Pianetti et al., 2001; Le Page et al., 2005), hepatocyte growth factors (Fan et al., 2005) and integrins (Guo and Giancotti, 2004; Nikolopoulos et al., 2004). Constitutive activation of NF-κB is also due to aberrant expression/activation of kinases such as IKK in brain cancer (Politi et al., 2008) and in liver cancer (Jiang et al., 2010), NIK in melanoma (Dhawan et al., 2008), GSK-3β in pancreatic cancer (Wilson and Baldwin, 2008), Akt in breast cancer (Pianetti et al., 2001), Raf in multiple myeloma (Keats et al., 2007), and Bcr–abl in acute lymphoblastic leukemia and chronic myeloid leukemia (Reuther et al., 1998). Mutations in signaling intermediates such as MUC1-C (Ahmad et al., 2007, 2009) and CARD11 (Lenz et al., 2008) are also reported to activate NF-κB in an IKK-independent and IKK-dependent manner, respectively. In addition, in many types of cancer, chemotherapy and radiotherapy induce constitutive activation of NF-κB, thereby making the tumor non-responsive to the treatment. Additional mechanisms for activation of NF-κB in cancers have been discussed in the following sections.
NF-κB network
There are essentially three sites at which NF-κB signaling has been shown to be networked: IKK, IκBα and NF-κB heterodimer. As the IKK complex is the most extensively investigated site in canonical pathway of activation by TNF-α and Toll-like receptors, it is the major site wherein most of the diverse stimuli converge for activation of NF-κB. IκBα is also activated by many stimuli in an IKK-independent manner. Finally, the p50–p65 heterodimer interacts with many other transcription factors that are regulated by epigenetic modifications at the promoters of responsive genes, as summarized in Figures 2 and 3. The other pathways that interact and network with NF-κB pathways mainly in cancer are epidermal growth factor receptor (EGFR), RAS, TP53, PI3K–AKt and mTOR. These will be discussed in detail in the following section.
Signaling interactions
In the canonical pathway, IKKβ is the kinase that phosphorylates IκBα, and IKKγ is the regulator that is activated by upstream signaling emanating from ligand–receptor interaction. Although IKKα is the part of the complex, it is dispensable. TNF-α interacts with TNFR1-TRADD-TRAF2-RIP-TAK1-TAB1, which activates the IKK complex. The formation of this complex requires K-63 polyubiquitination as an important step, wherein the RING domain of TRAF acts as K-63 ubiquitin ligase. All TRAFs except TRAF1 have a RING domain; however, the RING domain of TRAF2 does not have active ligase activity. Recently, sphingosine-1-phosphate was shown to act as a cofactor for TRAF2 ubiquitin ligase activity (Alvarez et al., 2010). Whether the kinase that phosphorylates IKKβ is TAK1 has not been unequivocally established because MEKK3 is another potential kinase that has been shown to have a role in activating IKKβ. NIK is another kinase that may have a role in this pathway. TAK1/TAB1 is an established kinase in Toll-like receptor and IL-1-mediated activation of NF-κB, in which TRAF6 was the first ubiquitin ligase shown to activate TAK1/TAB1 by K-63 polyubiquitinylation (Deng et al., 2000). In contrast, the non-canonical NF-κB pathway proceeds primarily through activation of IKKα. The non-canonical pathway is activated mainly through members of the TNF receptor superfamily, such as the lymphotoxin-β receptor (Hacker and Karin, 2006; Scheidereit, 2006).
The cell signaling intermediates that are frequently mutated in human cancers have been directly/indirectly networked with NF-κB pathway. Several G-protein-coupled receptors were also recently identified and shown to contribute to the malignant phenotype in head and neck squamous cell carcinoma, including overexpression of H- and K-RAS (Vitale-Cross et al., 2004; Hunter et al., 2005; Lu et al., 2006; Thomas et al., 2006; Patel et al., 2007). Aberrant function of the PI3K, phosphatase and tensin homolog (PTEN), AKT and mTOR signaling networks is a frequent event in head and neck squamous cell carcinoma (Molinolo et al., 2009). G-protein-coupled receptor-coupled protein kinase A (PKA) phosphorylates p65 at Ser 276 and is responsible for constitutive activation of NF-κB and for modulation of its responsive genes implicated in the pathogenesis of head and neck squamous cell carcinoma (Arun et al., 2009). In lung cancer, activating mutations of the protein kinases EGFR, ERBB2 and BRAF and inactivating mutations of STK11 have been reported (Brose et al., 2002; Naoki et al., 2002; Sanchez-Cespedes et al., 2002; Paez et al., 2004; Stephens et al., 2004; Davies et al., 2005). In small cell lung cancer, several tumor suppressor genes are inactivated, including TP53 (80–90% of cases), RB1 (60–90% of cases) and PTEN (13% of cases). Infrequent activating mutations have been found in PIK3CA, EGFR and KRAS (all 10% or lower), and MYC is amplified in 20% of cases (Sher et al., 2008) (http://www.sanger.ac.uk/genetics/CGP/cosmic/).
Recent sequencing of a small cell lung cancer genome (Pleasance et al., 2010b) has shown a complex signature of tobacco exposure, a potent NF-κB activator (Shishodia and Aggarwal, 2004; Ahn and Aggarwal, 2005) (Tang et al., 2006). Epidermal growth factor and EGFR pathways (Ras–Raf–MEK) (Duffy and Kummar, 2009) have been shown to activate NF-κB through Akt activation as well as through direct tyrosine phosphorylation of IκBα (Sethi et al., 2007). The PI3K–Akt signaling pathway, induced by EGFR and Her-2, is involved in the constitutive activation of NF-κB in prostate cancer cell lines (Koumakpayi et al., 2010). GSK3β is another kinase shown to regulate activation of NF-κB because genetic disruption of GSK3β abrogates TNF-α- or IL-1β-induced NF-κB activation (Hoeflich et al., 2000). GSK3β activates a subset of NF-κB responsive genes by phosphorylating p65 at Ser 468 (Steinbrecher et al., 2005).
With use of lung cell lines expressing oncogenic K-Ras, NF-κB has been shown to be activated in these cells in a K-Ras-dependent manner, and this activation by K-Ras has been shown to require IKKβ kinase activity. Together, these results reveal the importance of the NF-κB subunit p65–RelA in K-Ras-induced lung transformation and identify IKKβ as a potential therapeutic target for K-Ras-induced lung cancer (Basseres et al., 2010). Interaction between K-Ras, TP53 and NF-κB signaling was demonstrated by Meylan et al. in mouse model of lung adenocarcinoma (Meylan et al., 2009). TGF-β also activates NF-κB by acetylation of p65 by activating PKA (Ishinaga et al., 2009). Furthermore, TGF-β downregulates PTEN by activating NF-κB in pancreatic cancer cells (Chow et al., 2010), suggesting a linkage between NF-κB and PTEN in TGF-β signaling.
Altered TAB1–IKK interaction promotes TGF-β-mediated NF-κB activation during breast cancer progression (Neil and Schiemann, 2008). Another pathway that has been linked with many pathophysiological conditions is the Akt–mTOR pathway. The mTOR downstream from Akt controls NF-κB activity in PTEN-null/inactive prostate cancer cells by interaction with and stimulation of IKK. Akt requires the mTOR-associated protein Raptor to induce NF-κB activity. Correspondingly, the mTOR inhibitor rapamycin has been shown to suppress IKK activity in PTEN-deficient prostate cancer cells through a mechanism that may involve dissociation of Raptor from mTOR (Dan et al., 2008).
IKK complexes and their regulators
Initially, IKKα and IKKβ were characterized as part of a large complex of molecular weights ranging from 700 to 900 kDa (Chen et al., 1996; DiDonato et al., 1997). The IKKγ (NEMO) has subsequently been identified through genetic complementation of an NF-κB activation-defective cell line (Yamaoka et al., 1998). Not every component of the high-molecular weight IKK complex has been characterized, and the exact stoichiometry of the IKK complex has not been unambiguously determined. As IKK activation is involved in diverse modes of activating NF-κB, it is not surprising that the complex is dynamic and that there may be many as yet uncharacterized transient components associated with it. A recent review by Israel, 2010 contains a detailed discussion about the properties and characteristics of these subunits.
It now seems clear that TAK1 functions as an IKK kinase, at least in response to certain signals (Wang et al., 2001; Bhoj and Chen, 2009). TAK1 complexes with TAB1 and TAB2 (or TAB3) and phosphorylates IKKβ. Another kinase, MEKK3, has also been suggested to act upstream of the IKK complex, as cells lacking MEKK3 are partially defective in NF-κB activation in response to certain stimuli (Huang et al., 2004). NIK is the upstream kinase for activation of the IKKα subunit, and it requires neither IKKβ nor NEMO (Park et al., 2005). In another study, the oncoprotein MUC1 interacted with the highmolecular weight IKK complex (IKKβ and γ) and was associated with constitutive activation of NF-κB p65 in many human cancer cell lines (Ahmad et al., 2007, 2009).
The role of IKKs has not been restricted to the NF-κB activation pathway (Chariot, 2009). The kinase activity of IKKβ is also required in TNF-α-induced mTOR pathway. The IKKβ physically interacts with and phosphorylates TSC1, resulting in its suppression, which activates the mTOR pathway, enhances angiogenesis and results in tumor development. Furthermore, expression of activated IKKβ is associated with TSC1 Ser 511 phosphorylation and VEGF production in multiple tumor types, and correlates with poor clinical outcome in breast cancer patients (Lee et al., 2007). IKKβ has also been shown to phosphorylate and induce the degradation of transcription factor FOXO3a, which has an important role in controlling cell proliferation and survival, therefore, promoting tumorigenesis (Hu et al., 2004). IKKα, free from IKKβ and γ, can also be found in the nucleus, where it phosphorylates histone H3 on Ser 10, triggering its subsequent CREBbinding protein-mediated acetylation on K14, a crucial step in modulating chromatin accessibility (Gloire et al., 2006). IKKα also phosphorylates the SMRT repressor, which is recruited by p50 and p52 homodimers, and induces its nuclear export (together with histone deacetylase 3) and degradation. Then it phosphorylates chromatin-bound p65 on Ser 536, leading to the displacement of SMRT-histone deacetylase 3 repressor activity and allowing p300 to acetylate p65 at K310, an event necessary for full transcription (Hoberg et al., 2006).
The role of IKKγ (NEMO) has also been reported in pathways other than NF-κB. In interferon regulatory factor signaling pathways, the interferon regulatory factor 3/interferon regulatory factor 7 pathway and two IKK-related kinases (TANK-binding kinase-1 and IKKε) are activated by NEMO through its interaction with TANK (Zhao et al., 2007). In the DNA damage-induced ATM activation pathway, SUMOylation of NEMO and IKKε is essential for NF-κB activation (Wu and Miyamoto, 2007; Renner et al., 2010). Chaperones such as hsp90 and hsp70 have also been described as components of the IKK complex. Hsp70 seems to behave as a NEMO-interacting inhibitor of NF-κB signaling, whereas hsp90 has been associated with its cochaperone cdc37 and behaves as a stabilizing factor of IKK through its interaction with cdc37 and the IKKα and IKKβ kinase domains (Salminen et al., 2008).
Modification of p65
The NF-κB transactivator subunit has been shown to undergo extensive and diverse post-translational modifications, such as phosphorylation, acetylation and methylation (Figure 2). These modifications regulate the DNA-binding and oligomerization properties of p65, a principle target for phosphorylation by various kinases. These kinases function both in the cytoplasm and in the nucleus, and are differentially induced by various stimuli. Six different serine phosphoacceptor sites have been identified in Rel-A (p65): serines (S) 236, S276, S311, S468, S529 and S536. Phosphorylation of S276 is mediated by the catalytic subunit of protein kinase A (PKAc) (Mosialos and Gilmore, 1993) and mitogen- and stress-activated kinase-1 (Vermeulen et al., 2003; Jamaluddin et al., 2009; Reber et al., 2009). S311 is phosphorylated by protein kinase C-ζ (Duran et al., 2003; Kai et al., 2009). Activated casein kinase II catalyzes the phosphorylation of p65 at S529 (Wang et al., 2000). S536 is targeted for phosphorylation either by the IKKs (Sakurai et al., 2003) or by ribosomalsubunit kinase-1 (Bohuslav et al., 2004). Of interest, PKA phosphorylates p65 in the cytoplasm, whereas MASK-1 phosphorylates p65 in the nucleus. However, PKA-interacting protein 1 (AKIP1) facilitates the nuclear translocation of PKA and retention of phosphorylated p65 in the nucleus (Gao et al., 2008). Another kinase, PKCζ, phosphorylates p65 at Ser 311. p65 is also subject to phosphorylation by a number of other kinases including GSK3β at S468 (Gong et al., 2008), AKT/PI3K and NF-κB-activating kinase (also known as TANK-binding kinase-1 and TRAF2-associated kinase (reviewed in (Chen and Greene, 2004)). NIK and Cot have been shown to cooperate to trigger p65 phosphorylation (Wittwer and Schmitz, 2008).
Ubiquitination of p65 is required for its degradation. As part of the negative regulation of NF-κB, the p65 in cytoplasm is phosphorylated at Ser 236 by IKKα (Lawrence et al., 2005), which accelerates its proteasomal degradation in the nucleus. Ubiquitinylation of p65 at lysine 195 (Fan et al., 2009) is mediated by COMMD1 (Maine et al., 2007) and GCN5 (Mao et al., 2009). Another altogether different mode of processing of p65 has been reported that requires copine-1-dependent endoproteloysis of the N-terminus of p65 (Ramsey et al., 2008).
Phosphorylation of p65 is primarily reported at serine residues. However, there is a report of tyrosine phosphorylation of p65 by Syk that is activated by PKCδ and is responsible for thrombin-induced ICAM-1 expression in endothelial cells (Bijli et al., 2008). Thrombin and collagen also induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of PKA from an NF-κB–IκBα complex (Gambaryan et al., 2010). Recently, WIP1 phosphatase has been shown to dephosphorylate p65 at Ser 536, which is required for p300 interaction and transactivation. Thus, WIP1 negatively regulates NF-κB signaling (Chew et al., 2009).
There are three main sites of acetylation within RELA (p65): lysines 218, 221 and 310 (Chen et al., 2002), and these modifications have different consequences on the function of NF-κB (Chen and Greene, 2004). Recently, it was shown that the acetylation of lysine 310 of p65 impairs the Set9-mediated methylation of lysines 314 and 315, which is important for the ubiquitinylation and degradation of chromatin-associated p65 (Yang et al., 2010b). Reversible lysine methylation of the p65 subunit is carried out by a lysine methylase, the nuclear receptorbinding SET domain-containing protein 1, and a lysine demethylase, F-box and leucine-rich repeat protein 11. Overexpression of F-box and leucine-rich repeat protein 11 inhibits NF-κB activity, and a high level of nuclear receptor-binding SET domain-containing protein 1 activates NF-κB and reverses the inhibitory effect of F-box and leucine-rich repeat protein 11, whereas reduced expression of nuclear receptor-binding SET domain-containing protein 1 decreases NF-κB activation. The targets are K218 and K221 of p65, which are methylated in cells with activated NF-κB. This shows that reversible lysine methylation of p65 is also an important element in the complex regulation of NF-κB (Lu et al., 2010).
Multiple post-translational modifications of NF-κB can, therefore, affect DNA binding, interactions with coactivators and corepressors, and the termination of the NF-κB response. These modifications might regulate each other and/or might create specific marks for the recruitment/docking of different effectors to control the temporal and spatial activation of NF-κB (Chen and Greene, 2004; Perkins, 2006).
Multiple pathways of activation of NF-κB and targeting NF-κB for cancer prevention and therapy
As alluded to in previous sections, although the NF-κB pathway constitutes the central pathway regulating inflammation and cancer, multiple signaling pathways are operating at any given time in any given cancer (for an elaborate review, refer to Staudt (2010)). First, the NF-κB pathway is activated by diverse stimuli that are networked and regulated by many other pathways (for example, EGFR/HER2-PI3K-Akt-IKKα, TP53, PTEN, Akt-mTOR, G-protein-coupled receptor-RAS-RAF-Akt and Wnt–β-catenin), including canonical and non-canonical pathways (Figure 1). Another altogether different mechanism was shown for the activation of IκBα involving tyrosine phosphorylation (Imbert et al., 1996; Singh et al., 1996), brought about by syk kinase induced by H2O2 (Takada et al., 2003). In another study, EGFR tyrosine kinase activity induced Tyr 42 phosphorylation of IκBα (Sethi et al., 2007). H2O2-induced Tyr phosphorylation does not require degradation of IκBα for activation of NF-κB (Imbert et al., 1996; Tang et al., 2006); however, pervanadate-induced Tyr-phosphorylated IκBα does undergo degradation (Mukhopadhyay et al., 2000). Of interest, hypoxiainduced activation of NF-κB in fetal lung fibroblasts also involved phosphorylation of IκBα at Tyr 42 residues (Wright et al., 2009). Therefore, it is likely that a hypoxic tumor environment may activate NF-κB through such a mechanism.
Second, the activated NF-κB (p50–p65) cooperates and interacts with many other transcription factors to form on and off promoter complexes, such as STAT3 (Grivennikov and Karin, 2010), SP1(Perkins et al., 1993) HIF-1α (Scortegagna et al., 2008) and so on (Figure 1). Because of the networking of the pathways inhibiting one step or one the pathway, activates the alternative route of activation. This can be exemplified by the fact that as IκBα is degraded by proteasomal pathway, the proteasomal inhibitors are developed to inhibit the NF-κB pathway. Velcade (bortezomib) is such a drug already approved by Food and Drug Administration for the treatment of multiple myeloma. However, recent reports indicate that velcade now induces NF-κB, instead of inhibiting as was thought by inducing calpane (Li et al., 2010) and caspasedependent (Hideshima et al., 2009b) mechanisms. In a similar way, IKKβ inhibitor MLN120B blocks the canonical pathway and growth of MM cell lines but does not inhibit the non-canonical NF-κB pathway (Hideshima et al., 2009a). Therefore, inhibitors that block more than one step in the pathway or inhibit multiple pathways would be more effective for the treatment of cancer. Figure 4 shows cancer drugs approved by Food and Drug Administration, although not conceived as NF-κB inhibitors, these drugs can suppress NF-κB activation. For instance, EGFR kinase inhibitors have been shown to suppress NF-κB activation induced by epidermal growth factor (Sethi et al, 2007).
Figure 4.
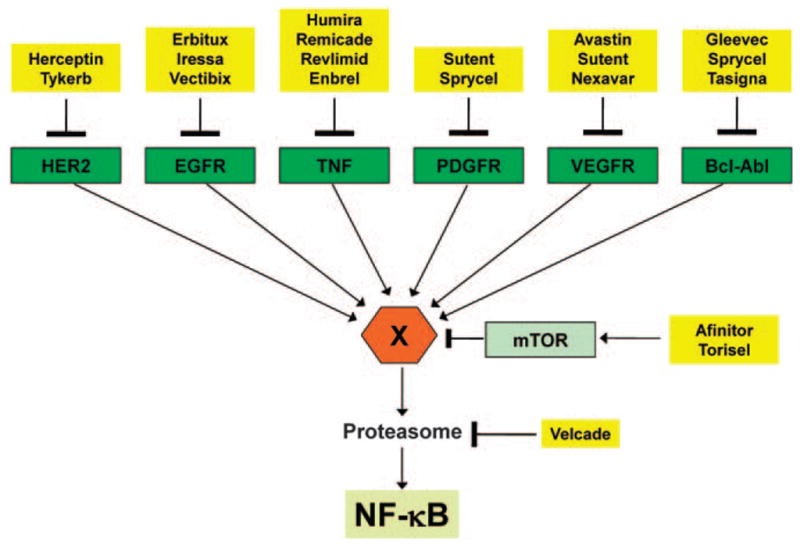
Suppression of NF-kB activation by the Food and Drug Administration-approved drugs for cancer therapy. Molecular targets for many Food and Drug Administration-approved drugs for treatment of cancer are shown. Although these drugs act through their defined molecular targets, they inhibit NF-κB via pathway(s) that are not well defined. The X indicates an intermediate that could be either IKK or its upstream activators, which may be different for different pathways. EGFR, epidermal growth factor receptor; mTOR, mammalian target of rapamycin; PDGF, platelet-derived growth factor receptor; TNF, tumor necrosis factor; VEGFR, vascular endothelial cell growth factor receptor.
Almost 15 years ago, we showed that nutraceuticals such as curcumin effectively inhibits TNF-α-induced activation of NF-κB through inhibition of IκBα phosphorylation (Singh and Aggarwal, 1995). Subsequently, curcumin was shown to inhibit not only IKK kinase activity but also p65 phosphorylation at serine 536, and p65 acetylation (Aggarwal et al., 2006); and p300–HAT (Balasubramanyam et al., 2004). Like curcumin, anacardic acid was found to suppress NF-κB activation by inhibition of HAT (Sung et al., 2008). Interestingly, several nutraceuticals such as berberine, butein, piceatannol, xanthohumol and wedelolactone, have been shown to directly bind to IKKβ through cysteine 179 (Kobori et al., 2004; Pandey et al., 2007, 2008; Harikumar et al., 2009; Son et al., 2010). Other nutraceuticals such as picroliv, thymoquinone, xanthohumol and plumbagin were found to inhibit NF-κB activity through directly binding to Cys 38 in p65 (Anand et al., 2008; Sethi et al., 2008; Harikumar et al., 2009). Sesquiterpene lactones were also found to suppress NF-κB through direct interaction with Cys 38 in p65 (Garcia-Pineres et al., 2001). Recently, we showed that crotepoxide inhibits the NF-κB pathway by inhibiting TAK1 directly, thus chemosensitizing tumor cells through inhibition of the proinflammatory pathway (Prasad et al., 2010). Thus, it is clear that various nutraceuticals can suppress NF-κB-mediated inflammatory pathways and this may be linked to their chemopreventive potential.
Conclusions
Extensive sequence-based analysis of the genome of various cancers has revealed that the driver mutations in a wide variety of genes fall into pathways of multiple signal transduction networks. Therefore, it is less likely that targeting just one mutated gene or one particular pathway could be effective alone for treating any type of cancer. Of interest, the inflammatory linkage and constitutive activation of NF-κB has emerged as one of the attractive targets for intervention and treatment of cancer. However, NF-κB itself is activated by diverse stimuli and by highly networked pathways, suggesting the need for a multitargeted approach. Nutraceuticals derived from fruits, vegetables and spices that target multiple steps in the NF-κB pathways are emerging as promising agents for the prevention and treatment of cancers.
Acknowledgments
We thank Tamara Locke for carefully editing the manuscript. Dr Aggarwal is the Ransom Horne, Jr, Professor of Cancer Research. This work was supported by a grant from the Clayton Foundation for Research (B.B.A.), a core grant from the National Institutes of Health (CA-16 672), a program project grant from National Institutes of Health (NIH CA-124787-01A2), and a grant from the Center for Targeted Therapy of MD Anderson Cancer Center. M.M.C. thanks the University of Delhi, Department of Science & Technology, India-PURSE grant Dean(R)/2010/1142 and MD Anderson Cancer Center for financial support.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Gehlot P. Inflammation and cancer: how friendly is the relationship for cancer patients? Curr Opin Pharmacol. 2009;9:351–369. doi: 10.1016/j.coph.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15:425–430. doi: 10.1158/1078-0432.CCR-08-0149. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Ichikawa H, Takada Y, Sandur SK, Shishodia S, Aggarwal BB. Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol Pharmacol. 2006;69:195–206. doi: 10.1124/mol.105.017400. [DOI] [PubMed] [Google Scholar]
- Ahmad R, Raina D, Joshi MD, Kawano T, Ren J, Kharbanda S, et al. MUC1-C oncoprotein functions as a direct activator of the nuclear factor-kappaB p65 transcription factor. Cancer Res. 2009;69:7013–7021. doi: 10.1158/0008-5472.CAN-09-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad R, Raina D, Trivedi V, Ren J, Rajabi H, Kharbanda S, et al. MUC1 oncoprotein activates the IkappaB kinase beta complex and constitutive NF-kappaB signalling. Nat Cell Biol. 2007;9:1419–1427. doi: 10.1038/ncb1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–233. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P, Kunnumakkara AB, Harikumar KB, Ahn KS, Badmaev V, Aggarwal BB. Modification of cysteine residue in p65 subunit of nuclear factor-kappaB (NF-kappaB) by picroliv suppresses NF-kappaB-regulated gene products and potentiates apoptosis. Cancer Res. 2008;68:8861–8870. doi: 10.1158/0008-5472.CAN-08-1902. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Arun P, Brown MS, Ehsanian R, Chen Z, Van Waes C. Nuclear NF-kappaB p65 phosphorylation at serine 276 by protein kinase A contributes to the malignant phenotype of head and neck cancer. Clin Cancer Res. 2009;15:5974–5984. doi: 10.1158/1078-0432.CCR-09-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- Basseres DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–3546. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DW. Our changing view of the genomic landscape of cancer. J Pathol. 2010;220:231–243. doi: 10.1002/path.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra M, Chevallier N, Morrison DJ, MacLachlan TK, El-Deiry WS, Licht JD. BRCA1 augments transcription by the NF-kappaB transcription factor by binding to the Rel domain of the p65/RelA subunit. J Biol Chem. 2003;278:26333–26341. doi: 10.1074/jbc.M303076200. [DOI] [PubMed] [Google Scholar]
- Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, Heymann F, et al. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell. 2010;17:481–496. doi: 10.1016/j.ccr.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijli KM, Fazal F, Minhajuddin M, Rahman A. Activation of Syk by protein kinase C-delta regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via tyrosine phosphorylation of RelA/p65. J Biol Chem. 2008;283:14674–14684. doi: 10.1074/jbc.M802094200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenkamp KU, Geugien M, Schepers H, Westra J, Lemmink HH, Vellenga E. Constitutive NF-kappaB DNA-binding activity in AML is frequently mediated by a Ras/PI3-K/PKB-dependent pathway. Leukemia. 2004;18:103–112. doi: 10.1038/sj.leu.2403145. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC. p53 induces NF-kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem. 2004;279:26115–26125. doi: 10.1074/jbc.M313509200. [DOI] [PubMed] [Google Scholar]
- Braunstein S, Formenti SC, Schneider RJ. Acquisition of stable inducible up-regulation of nuclear factor-kappaB by tumor necrosis factor exposure confers increased radiation resistance without increased transformation in breast cancer cells. Mol Cancer Res. 2008;6:78–88. doi: 10.1158/1541-7786.MCR-07-0339. [DOI] [PubMed] [Google Scholar]
- Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene. 1999;18:3063–3070. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci USA. 2007;104:15852–15857. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapiro E, Radford-Weiss I, Bastard C, Luquet I, Lefebvre C, Callet-Bauchu E, et al. The most frequent t(14;19)(q32;q13)-positive B-cell malignancy corresponds to an aggressive subgroup of atypical chronic lymphocytic leukemia. Leukemia. 2008;22:2123–2127. doi: 10.1038/leu.2008.102. [DOI] [PubMed] [Google Scholar]
- Chapman NR, Perkins ND. Inhibition of the RelA(p65) NF-kappaB subunit by Egr-1. J Biol Chem. 2000;275:4719–4725. doi: 10.1074/jbc.275.7.4719. [DOI] [PubMed] [Google Scholar]
- Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Cheng P, Zlobin A, Volgina V, Gottipati S, Osborne B, Simel EJ, et al. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J Immunol. 2001;167:4458–4467. doi: 10.4049/jimmunol.167.8.4458. [DOI] [PubMed] [Google Scholar]
- Chew J, Biswas S, Shreeram S, Humaidi M, Wong ET, Dhillion MK, et al. WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat Cell Biol. 2009;11:659–666. doi: 10.1038/ncb1873. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Muller-Tidow C, Berdel WE, Serve H. Signal transduction of oncogenic Flt3. Int J Hematol. 2005;82:93–99. doi: 10.1532/IJH97.05090. [DOI] [PubMed] [Google Scholar]
- Chow JY, Ban M, Wu HL, Nguyen F, Huang M, Chung H, et al. TGF-beta downregulates PTEN via activation of NF-kappaB in pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol. 2010;298:G275–G282. doi: 10.1152/ajpgi.00344.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, et al. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J Biol Chem. 2000;275:32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor i. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Hunter C, Smith R, Stephens P, Greenman C, Bignell G, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010;115:3541–3552. doi: 10.1182/blood-2009-09-243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, et al. beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell. 2002;2:323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Dhawan P, Su Y, Thu YM, Yu Y, Baugher P, Ellis DL, et al. The lymphotoxin-beta receptor is an upstream activator of NF-kappaB-mediated transcription in melanoma cells. J Biol Chem. 2008;283:15399–15408. doi: 10.1074/jbc.M708272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy A, Kummar S. Targeting mitogen-activated protein kinase kinase (MEK) in solid tumors. Target Oncol. 2009;4:267–273. doi: 10.1007/s11523-009-0125-x. [DOI] [PubMed] [Google Scholar]
- Duran A, Diaz-Meco MT, Moscat J. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J. 2003;22:3910–3918. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrov Z, Shishodia S, Faderl S, Harris D, Van Q, Kantarjian HM, et al. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood. 2003;102:987–995. doi: 10.1182/blood-2002-11-3550. [DOI] [PubMed] [Google Scholar]
- Fan S, Gao M, Meng Q, Laterra JJ, Symons MH, Coniglio S, et al. Role of NF-kappaB signaling in hepatocyte growth factor/scatter factor-mediated cell protection. Oncogene. 2005;24:1749–1766. doi: 10.1038/sj.onc.1208327. [DOI] [PubMed] [Google Scholar]
- Fan Y, Mao R, Zhao Y, Yu Y, Sun W, Song P, et al. Tumor necrosis factor-alpha induces RelA degradation via ubiquitination at lysine 195 to prevent excessive nuclear factor-kappaB activation. J Biol Chem. 2009;284:29290–29297. doi: 10.1074/jbc.M109.018994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Brown K, Daucher MB, Bressler P, Siebenlist U. Activation of the serum response factor by p65/NF-kappaB. EMBO J. 1996;15:3403–3412. [PMC free article] [PubMed] [Google Scholar]
- Galien R, Garcia T. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res. 1997;25:2424–2429. doi: 10.1093/nar/25.12.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem. 2010;285:18352–18363. doi: 10.1074/jbc.M109.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, Asamitsu K, Hibi Y, Ueno T, Okamoto T. AKIP1 enhances NF-kappaB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J Biol Chem. 2008;283:7834–7843. doi: 10.1074/jbc.M710285200. [DOI] [PubMed] [Google Scholar]
- Garcia M, Jemal A, Ward EM, Center MM, Hao Y, Siegel RL, et al. Global Cancer Facts & Figures 2007. American Cancer Society; 2007. pp. 1–48. ( http://www.cancer.org) [Google Scholar]
- Garcia-Pineres AJ, Castro V, Mora G, Schmidt TJ, Strunck E, Pahl HL, et al. Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J Biol Chem. 2001;276:39713–39720. doi: 10.1074/jbc.M101985200. [DOI] [PubMed] [Google Scholar]
- Ge J, Xu H, Li T, Zhou Y, Zhang Z, Li S, et al. A Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc Natl Acad Sci USA. 2009;106:13725–13730. doi: 10.1073/pnas.0907200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Gilmore TD. The Re1/NF-kappa B/I kappa B signal transduction pathway and cancer. Cancer Treat Res. 2003;115:241–265. [PubMed] [Google Scholar]
- Gilmore TD, Kalaitzidis D, Liang MC, Starczynowski DT. The c-Rel transcription factor and B-cell proliferation: a deal with the devil. Oncogene. 2004;23:2275–2286. doi: 10.1038/sj.onc.1207410. [DOI] [PubMed] [Google Scholar]
- Giri DK, Aggarwal BB. Constitutive activation of NF-kappaB causes resistance to apoptosis in human cutaneous T cell lymphoma HuT-78 cells. Autocrine role of tumor necrosis factor and reactive oxygen intermediates. J Biol Chem. 1998;273:14008–14014. doi: 10.1074/jbc.273.22.14008. [DOI] [PubMed] [Google Scholar]
- Gloire G, Dejardin E, Piette J. Extending the nuclear roles of IkappaB kinase subunits. Biochem Pharmacol. 2006;72:1081–1089. doi: 10.1016/j.bcp.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Gong R, Rifai A, Ge Y, Chen S, Dworkin LD. Hepatocyte growth factor suppresses proinflammatory NFkappaB activation through GSK3beta inactivation in renal tubular epithelial cells. J Biol Chem. 2008;283:7401–7410. doi: 10.1074/jbc.M710396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean-Raillard J, Ades L, Boehrer S, Tailler M, Fabre C, Braun T, et al. Flt3 receptor inhibition reduces constitutive NFkappaB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Apoptosis. 2008;13:1148–1161. doi: 10.1007/s10495-008-0243-4. [DOI] [PubMed] [Google Scholar]
- GuhaThakurta D. Computational identification of transcriptional regulatory elements in DNA sequence. Nucleic Acids Res. 2006;34:3585–3598. doi: 10.1093/nar/gkl372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Harikumar KB, Kunnumakkara AB, Ahn KS, Anand P, Krishnan S, Guha S, et al. Modification of the cysteine residues in IkappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood. 2009;113:2003–2013. doi: 10.1182/blood-2008-04-151944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Chauhan D, Kiziltepe T, Ikeda H, Okawa Y, Podar K, et al. Biologic sequelae of I{kappa}B kinase (IKK) inhibition in multiple myeloma: therapeutic implications. Blood. 2009a;113:5228–5236. doi: 10.1182/blood-2008-06-161505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood. 2009b;114:1046–1052. doi: 10.1182/blood-2009-01-199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Xia Y, Verma IM. Inflammatory tales of liver cancer. Cancer Cell. 2007;11:99–101. doi: 10.1016/j.ccr.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Huang Q, Yang J, Lin Y, Walker C, Cheng J, Liu ZG, et al. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004;5:98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nat Rev Cancer. 2005;5:127–135. doi: 10.1038/nrc1549. [DOI] [PubMed] [Google Scholar]
- Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM, et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell. 2009;34:461–472. doi: 10.1016/j.molcel.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nat Rev Cancer. 2010;10:332–341. doi: 10.1038/nrc2775. [DOI] [PubMed] [Google Scholar]
- Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, et al. Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabé RR, et al. International Cancer Genome Consortium . International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishinaga H, Jono H, Lim JH, Komatsu K, Xu X, Lee J, et al. Synergistic induction of nuclear factor-kappaB by transforming growth factor-beta and tumour necrosis factor-alpha is mediated by protein kinase A-dependent RelA acetylation. Biochem J. 2009;417:583–591. doi: 10.1042/BJ20080781. [DOI] [PubMed] [Google Scholar]
- Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Bernitsas DG, Ichikawa H, Takada Y, Myers JN, Lin XL, Darnay BG, et al. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene. 2007;26:1385–1397. doi: 10.1038/sj.onc.1209945. [DOI] [PubMed] [Google Scholar]
- Jamaluddin M, Tian B, Boldogh I, Garofalo RP, Brasier AR. Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J Virol. 2009;83:10605–10615. doi: 10.1128/JVI.01090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SJ, Radonovich M, Brady JN, Pise-Masison CA. HTLV-I Tax induces a novel interaction between p65/RelA and p53 that results in inhibition of p53 transcriptional activity. Blood. 2004;104:1490–1497. doi: 10.1182/blood-2003-12-4174. [DOI] [PubMed] [Google Scholar]
- Jiang R, Xia Y, Li J, Deng L, Zhao L, Shi J, et al. High expression levels of IKKalpha and IKKbeta are necessary for the malignant properties of liver cancer. Int J Cancer. 2010;126:1263–1274. doi: 10.1002/ijc.24854. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M, Yasuda S, Imai S, Toyota M, Kanoh H, Sakane F. Diacylglycerol kinase alpha enhances protein kinase Czeta-dependent phosphorylation at Ser311 of p65/RelA subunit of nuclear factor-kappaB. FEBS Lett. 2009;583:3265–3268. doi: 10.1016/j.febslet.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kato T, Jr, Delhase M, Hoffmann A, Karin M. CK2 is a C-terminal IkappaB kinase responsible for NF-kappaB activation during the UV response. Mol Cell. 2003;12:829–839. doi: 10.1016/s1097-2765(03)00358-7. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SP, Park JW, Lee SH, Lim JH, Jang BC, Jang IH, et al. Homeodomain protein CDX2 regulates COX-2 expression in colorectal cancer. Biochem Biophys Res Commun. 2004;315:93–99. doi: 10.1016/j.bbrc.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Kleinberg L, Dong HP, Holth A, Risberg B, Trope CG, Nesland JM, et al. Cleaved caspase-3 and nuclear factor-kappaB p65 are prognostic factors in metastatic serous ovarian carcinoma. Hum Pathol. 2009;40:795–806. doi: 10.1016/j.humpath.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Kobori M, Yang Z, Gong D, Heissmeyer V, Zhu H, Jung YK, et al. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 2004;11:123–130. doi: 10.1038/sj.cdd.4401325. [DOI] [PubMed] [Google Scholar]
- Koumakpayi IH, Le Page C, Mes-Masson AM, Saad F. Hierarchical clustering of immunohistochemical analysis of the activated ErbB/PI3K/Akt/NF-kappaB signalling pathway and prognostic significance in prostate cancer. Br J Cancer. 2010;102:1163–1173. doi: 10.1038/sj.bjc.6605571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- Kumar A, Lin Z, SenBanerjee S, Jain MK. Tumor necrosis factor alpha-mediated reduction of KLF2 is due to inhibition of MEF2 by NF-kappaB and histone deacetylases. Mol Cell Biol. 2005;25:5893–5903. doi: 10.1128/MCB.25.14.5893-5903.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- Le Page C, Koumakpayi IH, Lessard L, Mes-Masson AM, Saad F. EGFR and Her-2 regulate the constitutive activation of NF-kappaB in PC-3 prostate cancer cells. Prostate. 2005;65:130–140. doi: 10.1002/pros.20234. [DOI] [PubMed] [Google Scholar]
- Ledford H. Big science: The cancer genome challenge. Nature. 2010;464:972–974. doi: 10.1038/464972a. [DOI] [PubMed] [Google Scholar]
- Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- Lee SK, Kim JH, Lee YC, Cheong J, Lee JW. Silencing mediator of retinoic acid and thyroid hormone receptors, as a novel transcriptional corepressor molecule of activating protein-1, nuclear factor-kappaB, and serum response factor. J Biol Chem. 2000;275:12470–12474. doi: 10.1074/jbc.275.17.12470. [DOI] [PubMed] [Google Scholar]
- Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23:16–43. doi: 10.1634/stemcells.2004-0117. [DOI] [PubMed] [Google Scholar]
- Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR, et al. Proteasome inhibitor PS-341 (bortezomib) induces calpain-dependent IkappaB(alpha) degradation. J Biol Chem. 2010;285:16096–16104. doi: 10.1074/jbc.M109.072694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–635. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- Linnekin D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int J Biochem Cell Biol. 1999;31:1053–1074. doi: 10.1016/s1357-2725(99)00078-3. [DOI] [PubMed] [Google Scholar]
- Liptay S, Weber CK, Ludwig L, Wagner M, Adler G, Schmid RM. Mitogenic and antiapoptotic role of constitutive NF-kappaB/Rel activity in pancreatic cancer. Int J Cancer. 2003;105:735–746. doi: 10.1002/ijc.11081. [DOI] [PubMed] [Google Scholar]
- Liu B, Yang R, Wong KA, Getman C, Stein N, Teitell MA, et al. Negative regulation of NF-kappaB signaling by PIAS1. Mol Cell Biol. 2005;25:1113–1123. doi: 10.1128/MCB.25.3.1113-1123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Guerra M, Colomer D. NF-kappaB as a therapeutic target in chronic lymphocytic leukemia. Expert Opin Ther Targets. 2010;14:275–288. doi: 10.1517/14728221003598930. [DOI] [PubMed] [Google Scholar]
- Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, et al. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci USA. 2010;107:46–51. doi: 10.1073/pnas.0912493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Maeda G, Chiba T, Kawashiri S, Satoh T, Imai K. Epigenetic inactivation of IkappaB Kinase-alpha in oral carcinomas and tumor progression. Clin Cancer Res. 2007;13:5041–5047. doi: 10.1158/1078-0432.CCR-07-0463. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Maine GN, Mao X, Komarck CM, Burstein E. COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J. 2007;26:436–447. doi: 10.1038/sj.emboj.7601489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW, et al. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kappaB/RelA. Genes Dev. 2009;23:849–861. doi: 10.1101/gad.1748409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Subero JI, Ibbotson R, Klapper W, Michaux L, Callet-Bauchu E, Berger F, et al. A comprehensive genetic and histopathologic analysis identifies two subgroups of B-cell malignancies carrying a t(14;19)(q32;q13) or variant BCL3-translocation. Leukemia. 2007;21:1532–1544. doi: 10.1038/sj.leu.2404695. [DOI] [PubMed] [Google Scholar]
- Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Chiao PJ, Verma IM. Enhanced I kappa B alpha degradation is responsible for constitutive NF-kappa B activity in mature murine B-cell lines. Mol Cell Biol. 1994;14:3276–3282. doi: 10.1128/mcb.14.5.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–334. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008;118:868–878. doi: 10.1172/JCI33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosialos G, Gilmore TD. v-Rel and c-Rel are differentially affected by mutations at a consensus protein kinase recognition sequence. Oncogene. 1993;8:721–730. [PubMed] [Google Scholar]
- Mukhopadhyay A, Manna SK, Aggarwal BB. Pervanadate-induced nuclear factor-kappaB activation requires tyrosine phosphorylation and degradation of IkappaBalpha. Comparison with tumor necrosis factor-alpha. J Biol Chem. 2000;275:8549–8555. doi: 10.1074/jbc.275.12.8549. [DOI] [PubMed] [Google Scholar]
- Na SY, Kang BY, Chung SW, Han SJ, Ma X, Trinchieri G, et al. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFkappaB. J Biol Chem. 1999;274:7674–7680. doi: 10.1074/jbc.274.12.7674. [DOI] [PubMed] [Google Scholar]
- Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res. 2002;62:7001–7003. [PubMed] [Google Scholar]
- Nawata R, Yujiri T, Nakamura Y, Ariyoshi K, Takahashi T, Sato Y, et al. MEK kinase 1 mediates the antiapoptotic effect of the Bcr-Abl oncogene through NF-kappaB activation. Oncogene. 2003;22:7774–7780. doi: 10.1038/sj.onc.1206901. [DOI] [PubMed] [Google Scholar]
- Neil JR, Schiemann WP. Altered TAB1: I kappaB kinase interaction promotes transforming growth factor beta-mediated nuclear factor-kappaB activation during breast cancer progression. Cancer Res. 2008;68:1462–1470. doi: 10.1158/0008-5472.CAN-07-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmark JR, Hardy DO, Tonb DC, Carter BS, Epstein JI, Isaacs WB, et al. Androgen receptor gene mutations in human prostate cancer. Proc Natl Acad Sci USA. 1992;89:6319–6323. doi: 10.1073/pnas.89.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Giancotti FG. Integrin beta4 signaling promotes tumor angiogenesis. Cancer Cell. 2004;6:471–483. doi: 10.1016/j.ccr.2004.09.029. [DOI] [PubMed] [Google Scholar]
- O’Dea EL, Kearns JD, Hoffmann A. UV as an amplifier rather than inducer of NF-kappaB activity. Mol Cell. 2008;30:632–641. doi: 10.1016/j.molcel.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Palvimo JJ, Reinikainen P, Ikonen T, Kallio PJ, Moilanen A, Janne OA. Mutual transcriptional interference between RelA and androgen receptor. J Biol Chem. 1996;271:24151–24156. doi: 10.1074/jbc.271.39.24151. [DOI] [PubMed] [Google Scholar]
- Pandey MK, Sandur SK, Sung B, Sethi G, Kunnumakkara AB, Aggarwal BB. Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue. J Biol Chem. 2007;282:17340–17350. doi: 10.1074/jbc.M700890200. [DOI] [PubMed] [Google Scholar]
- Pandey MK, Sung B, Kunnumakkara AB, Sethi G, Chaturvedi MM, Aggarwal BB. Berberine modifies cysteine 179 of IkappaBalpha kinase, suppresses nuclear factor-kappaB-regulated anti-apoptotic gene products, and potentiates apoptosis. Cancer Res. 2008;68:5370–5379. doi: 10.1158/0008-5472.CAN-08-0511. [DOI] [PubMed] [Google Scholar]
- Park J, Lee JH, La M, Jang MJ, Chae GW, Kim SB, et al. Inhibition of NF-kappaB acetylation and its transcriptional activity by Daxx. J Mol Biol. 2007;368:388–397. doi: 10.1016/j.jmb.2007.02.047. [DOI] [PubMed] [Google Scholar]
- Park KJ, Krishnan V, O’Malley BW, Yamamoto Y, Gaynor RB. Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell. 2005;18:71–82. doi: 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Rosenfeldt HM, Lyons R, Servitja JM, Bustelo XR, Siroff M, et al. Persistent activation of Rac1 in squamous carcinomas of the head and neck: evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis. 2007;28:1145–1152. doi: 10.1093/carcin/bgm008. [DOI] [PubMed] [Google Scholar]
- Pepper C, Hewamana S, Brennan P, Fegan C. NF-kappaB as a prognostic marker and therapeutic target in chronic lymphocytic leukemia. Future Oncol. 2009;5:1027–1037. doi: 10.2217/fon.09.72. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993;12:3551–3558. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pianetti S, Arsura M, Romieu-Mourez R, Coffey RJ, Sonenshein GE. Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene. 2001;20:1287–1299. doi: 10.1038/sj.onc.1204257. [DOI] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Pileri SA, Zinzani PL, Gaidano G, Falini B, Gaulard P, Zucca E, et al. Pathobiology of primary mediastinal B-cell lymphoma. Leuk Lymphoma. 2003;44(Suppl 3):S21–S26. doi: 10.1080/10428190310001623810. [DOI] [PubMed] [Google Scholar]
- Planavila A, Rodriguez-Calvo R, Jove M, Michalik L, Wahli W, Laguna JC, et al. Peroxisome proliferator-activated receptor beta/delta activation inhibits hypertrophy in neonatal rat cardio-myocytes. Cardiovasc Res. 2005;65:832–841. doi: 10.1016/j.cardiores.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010a;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010b;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi C, Del Turco D, Sie JM, Golinski PA, Tegeder I, Deller T, et al. Accumulation of phosphorylated I kappaB alpha and activated IKK in nodes of Ranvier. Neuropathol Appl Neurobiol. 2008;34:357–365. doi: 10.1111/j.1365-2990.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336:25–37. doi: 10.1007/s11010-009-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey CS, Yeung F, Stoddard PB, Li D, Creutz CE, Mayo MW. Copine-I represses NF-kappaB transcription by endoproteolysis of p65. Oncogene. 2008;27:3516–3526. doi: 10.1038/sj.onc.1211030. [DOI] [PubMed] [Google Scholar]
- Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reber L, Vermeulen L, Haegeman G, Frossard N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS One. 2009;4:e4393. doi: 10.1371/journal.pone.0004393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner F, Moreno R, Schmitz ML. SUMOylation-dependent localization of IKKepsilon in PML nuclear bodies is essential for protection against DNA-damage-triggered cell death. Mol Cell. 2010;37:503–515. doi: 10.1016/j.molcel.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Reuther JY, Reuther GW, Cortez D, Pendergast AM, Baldwin AS., Jr A requirement for NF-kappaB activation in Bcr-Abl-mediated transformation. Genes Dev. 1998;12:968–981. doi: 10.1101/gad.12.7.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukos DH. Novel clinico-genome network modeling for revolutionizing genotype-phenotype-based personalized cancer care. Expert Rev Mol Diagn. 2010;10:33–48. doi: 10.1586/erm.09.69. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, et al. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–36923. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- Salminen A, Paimela T, Suuronen T, Kaarniranta K. Innate immunity meets with cellular stress at the IKK complex: regulation of the IKK complex by HSP70 and HSP90. Immunol Lett. 2008;117:9–15. doi: 10.1016/j.imlet.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- Schmitz ML, Stelzer G, Altmann H, Meisterernst M, Baeuerle PA. Interaction of the COOH-terminal transactivation domain of p65 NF-kappa B with TATA-binding protein, transcription factor IIB, and coactivators. J Biol Chem. 1995;270:7219–7226. doi: 10.1074/jbc.270.13.7219. [DOI] [PubMed] [Google Scholar]
- Scortegagna M, Cataisson C, Martin RJ, Hicklin DJ, Schreiber RD, Yuspa SH, et al. HIF-1alpha regulates epithelial inflammation by cell autonomous NFkappaB activation and paracrine stromal remodeling. Blood. 2008;111:3343–3354. doi: 10.1182/blood-2007-10-115758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Sethi G, Ahn KS, Aggarwal BB. Targeting nuclear factor-kappa B activation pathway by thymoquinone: role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol Cancer Res. 2008;6:1059–1070. doi: 10.1158/1541-7786.MCR-07-2088. [DOI] [PubMed] [Google Scholar]
- Sethi G, Ahn KS, Chaturvedi MM, Aggarwal BB. Epidermal growth factor (EGF) activates nuclear factor-kappaB through IkappaBalpha kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IkappaBalpha. Oncogene. 2007;26:7324–7332. doi: 10.1038/sj.onc.1210544. [DOI] [PubMed] [Google Scholar]
- Sgarbanti M, Remoli AL, Marsili G, Ridolfi B, Borsetti A, Perrotti E, et al. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J Virol. 2008;82:3632–3641. doi: 10.1128/JVI.00599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher T, Dy GK, Adjei AA. Small cell lung cancer. Mayo Clin Proc. 2008;83:355–367. doi: 10.4065/83.3.355. [DOI] [PubMed] [Google Scholar]
- Shishodia S, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates activation of cigarette smoke-induced nuclear factor (NF)-kappaB by suppressing activation of IkappaBalpha kinase in human non-small cell lung carcinoma: correlation with suppression of cyclin D1, COX-2, and matrix metalloproteinase-9. Cancer Res. 2004;64:5004–5012. doi: 10.1158/0008-5472.CAN-04-0206. [DOI] [PubMed] [Google Scholar]
- Singh S, Aggarwal BB. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected] J Biol Chem. 1995;270:24995–25000. doi: 10.1074/jbc.270.42.24995. [DOI] [PubMed] [Google Scholar]
- Singh S, Darnay BG, Aggarwal BB. Site-specific tyrosine phosphorylation of IkappaBalpha negatively regulates its inducible phosphorylation and degradation. J Biol Chem. 1996;271:31049–31054. doi: 10.1074/jbc.271.49.31049. [DOI] [PubMed] [Google Scholar]
- Smith D, Shimamura T, Barbera S, Bejcek BE. NF-kappaB controls growth of glioblastomas/astrocytomas. Mol Cell Biochem. 2008;307:141–147. doi: 10.1007/s11010-007-9593-4. [DOI] [PubMed] [Google Scholar]
- Son PS, Park SA, Na HK, Jue DM, Surh YJ. Piceatannol, a catechol-type polyphenol, inhibits phorb. Carcinogenesis. 2010;31:1442–1449. doi: 10.1093/carcin/bgq099. [DOI] [PubMed] [Google Scholar]
- Sriskantharajah S, Ley SC. Cell biology. Turning off inflammation signaling. Science. 2010;327:1093–1094. doi: 10.1126/science.1187271. [DOI] [PubMed] [Google Scholar]
- Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein B, Baldwin AS, Jr, Ballard DW, Greene WC, Angel P, Herrlich P. Cross-coupling of the NF-kappa B p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 1993;12:3879–3891. doi: 10.1002/j.1460-2075.1993.tb06066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrecher KA, Wilson W, III, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol. 2005;25:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–526. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650–665. doi: 10.1038/nrc1169. [DOI] [PubMed] [Google Scholar]
- Sun HZ, Yang TW, Zang WJ, Wu SF. Dehydroepiandroster-one-induced proliferation of prostatic epithelial cell is mediated by NFKB via PI3K/AKT signaling pathway. J Endocrinol. 2010;204:311–318. doi: 10.1677/JOE-09-0270. [DOI] [PubMed] [Google Scholar]
- Sung B, Pandey MK, Ahn KS, Yi T, Chaturvedi MM, Liu M, et al. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood. 2008;111:4880–4891. doi: 10.1182/blood-2007-10-117994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tago K, Funakoshi-Tago M, Sakinawa M, Mizuno N, Itoh H. {kappa}B-Ras is a nuclear-cytoplasmic small GTPase that inhibits the NF-{kappa}B activation through the suppression of transcriptional activation of p65/RelA. J Biol Chem. 2010;285:30622–30633. doi: 10.1074/jbc.M110.117028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Harigae H, Ishii KK, Inomata M, Fujiwara T, Yokoyama H, et al. Over-expression of Flt3 induces NF-kappaB pathway and increases the expression of IL-6. Leuk Res. 2005;29:893–899. doi: 10.1016/j.leukres.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Tang X, Liu D, Shishodia S, Ozburn N, Behrens C, Lee JJ, et al. Nuclear factor-kappaB (NF-kappaB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer. 2006;107:2637–2646. doi: 10.1002/cncr.22315. [DOI] [PubMed] [Google Scholar]
- Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino ML, et al. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66:11831–11839. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- Toualbi-Abed K, Daniel F, Guller MC, Legrand A, Mauriz JL, Mauviel A, et al. Jun D cooperates with p65 to activate the proximal kappaB site of the cyclin D1 promoter: role of PI3K/PDK-1. Carcinogenesis. 2008;29:536–543. doi: 10.1093/carcin/bgm293. [DOI] [PubMed] [Google Scholar]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- Vainer GW, Pikarsky E, Ben-Neriah Y. Contradictory functions of NF-kappaB in liver physiology and cancer. Cancer Lett. 2008;267:182–188. doi: 10.1016/j.canlet.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Van Waes C, Yu M, Nottingham L, Karin M. Inhibitor-kappaB kinase in tumor promotion and suppression during progression of squamous cell carcinoma. Clin Cancer Res. 2007;13:4956–4959. doi: 10.1158/1078-0432.CCR-07-1287. [DOI] [PubMed] [Google Scholar]
- Vatsyayan J, Qing G, Xiao G, Hu J. SUMO1 modification of NF-kappaB2/p100 is essential for stimuli-induced p100 phosphorylation and processing. EMBO Rep. 2008;9:885–890. doi: 10.1038/embor.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30:383–391. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004;64:8804–8807. doi: 10.1158/0008-5472.CAN-04-2623. [DOI] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- Wang D, Westerheide SD, Hanson JL, Baldwin AS., Jr Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275:32592–32597. doi: 10.1074/jbc.M001358200. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941–951. doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- Warren JL, Mariotto AB, Meekins A, Topor M, Brown ML. Current and future utilization of services from medical oncologists. J Clin Oncol. 2008;26:3242–3247. doi: 10.1200/JCO.2007.14.6357. [DOI] [PubMed] [Google Scholar]
- Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- Wieland GD, Nehmann N, Muller D, Eibel H, Siebenlist U, Suhnel J, et al. Early growth response proteins EGR-4 and EGR-3 interact with immune inflammatory mediators NF-kappaB p50 and p65. J Cell Sci. 2005;118:3203–3212. doi: 10.1242/jcs.02445. [DOI] [PubMed] [Google Scholar]
- Wilson W, III, Baldwin AS. Maintenance of constitutive IkappaB kinase activity by glycogen synthase kinase-3alpha/beta in pancreatic cancer. Cancer Res. 2008;68:8156–8163. doi: 10.1158/0008-5472.CAN-08-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittwer T, Schmitz ML. NIK and Cot cooperate to trigger NF-kappaB p65 phosphorylation. Biochem Biophys Res Commun. 2008;371:294–297. doi: 10.1016/j.bbrc.2008.04.069. [DOI] [PubMed] [Google Scholar]
- Wolf JS, Chen Z, Dong G, Sunwoo JB, Bancroft CC, Capo DE, et al. IL (interleukin)-1alpha promotes nuclear factor-kappaB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin Cancer Res. 2001;7:1812–1820. [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Wright CJ, Zhuang T, La P, Yang G, Dennery PA. Hyperoxia-induced NF-kappaB activation occurs via a maturationally sensitive atypical pathway. Am J Physiol Lung Cell Mol Physiol. 2009;296:L296–L306. doi: 10.1152/ajplung.90499.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZH, Miyamoto S. Many faces of NF-kappaB signaling induced by genotoxic stress. J Mol Med. 2007;85:1187–1202. doi: 10.1007/s00109-007-0227-9. [DOI] [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- Yang J, Splittgerber R, Yull FE, Kantrow S, Ayers GD, Karin M, et al. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J Clin Invest. 2010a;120:2563–2574. doi: 10.1172/JCI42358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–L576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- Yang XD, Tajkhorshid E, Chen LF. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol Cell Biol. 2010b;30:2170–2180. doi: 10.1128/MCB.01343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Kumar A, Koyama Y, Peng H, Arman A, Boch JA, et al. Interleukin 1 activates STAT3/nuclear factor-kappaB cross-talk via a unique TRAF6- and p65-dependent mechanism. J Biol Chem. 2004;279:1768–1776. doi: 10.1074/jbc.M311498200. [DOI] [PubMed] [Google Scholar]
- Yu Z, Kone BC. The STAT3 DNA-binding domain mediates interaction with NF-kappaB p65 and iuducible nitric oxide synthase transrepression in mesangial cells. J Am Soc Nephrol. 2004;15:585–591. doi: 10.1097/01.asn.0000114556.19556.f9. [DOI] [PubMed] [Google Scholar]
- Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J, et al. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- Zhou W, Cao Q, Peng Y, Zhang QJ, Castrillon DH, DePinho RA, et al. FoxO4 inhibits NF-kappaB and protects mice against colonic injury and inflammation. Gastroenterology. 2009;137:1403–1414. doi: 10.1053/j.gastro.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Park E, Liu B, Xia X, Fischer SM, Hu Y. Critical role of IkappaB kinase alpha in embryonic skin development and skin carcinogenesis. Histol Histopathol. 2009;24:265–271. doi: 10.14670/HH-24.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Xia X, Liu B, Shen J, Hu Y, Person M. IKKalpha shields 14-3-3sigma, a G(2)/M cell cycle checkpoint gene, from hypermethylation, preventing its silencing. Mol Cell. 2007;27:214–227. doi: 10.1016/j.molcel.2007.05.042. [DOI] [PubMed] [Google Scholar]
- Zwergal A, Quirling M, Saugel B, Huth KC, Sydlik C, Poli V, et al. C/EBP beta blocks p65 phosphorylation and thereby NF-kappa B-mediated transcription in TNF-tolerant cells. J Immunol. 2006;177:665–672. doi: 10.4049/jimmunol.177.1.665. [DOI] [PubMed] [Google Scholar]