

Abstract
Although co-ordinate interaction between different signal transduction pathways is essential for developmental decisions, interpathway connections are often obscured and difficult to identify due to cross-talk. Here signals from the fission yeast stress-activated MAPK Spc1 are shown to regulate Cgs2, a negative regulator of the cAMP-dependent protein kinase (protein kinase A) pathway. Pathway integration is achieved via Spc1-dependent binding of Atf1-Pcr1 heterodimer to an M26 DNA site in the cgs2+ promoter, which remodels chromatin to regulate expression of cgs2+ and targets downstream of protein kinase A. This direct interpathway connection co-ordinates signals of nitrogen and carbon source depletion to affect a G0 cell-cycle checkpoint and sexual differentiation. The Atf1-Pcr1-M26 complex-dependent chromatin remodeling provides a molecular mechanism whereby Atf1-Pcr1 heterodimer can function differentially as either a transcriptional activator, or as a transcriptional repressor, or as an inducer of meiotic recombination. We also show that the Atf1-Pcr1-M26 complex functions as both an inducer and repressor of chromatin remodeling, which provides a way for various chromatin remodeling-dependent effector functions to be regulated.
In the presence of sufficient nutrients haploid cells of the fission yeast Schizosaccharomyces pombe proliferate by mitotic division. In response to depleted nitrogen and carbon source, cells of opposite mating types conjugate, undergo karyogamy, enter meiosis, and produce haploid spores that remain metabolically dormant until nutritional conditions improve. Ste11 (a TR-box transcription factor) and Mei2 regulate the induction of a set of meiosis-specific genes; overexpression of mei2+ or ste11+ is sufficient to drive sexual differentiation ectopically, even under conditions where nutrients are not limiting (1-5).
At least three signal transduction pathways are required for sexual differentiation in fission yeast (Fig. 1). Signals about mating type heterozygosity are transduced by the mating pheromone signaling pathway; inactivation of Pat1 (Ran1) kinase de-represses Mei2 and Ste11 (6 - 8). High energy (glucose) levels repress sexual differentiation via the cAMP-dependent protein kinase PKA1; attenuation of the PKA pathway de-represses downstream activators (9-11). Nitrogen starvation triggers a stress-activated MAPK pathway; MAPK Spc1 (Sty1/Phh1) and one of its targets for phosphorylation, Atf1 protein (Mts1/Gad7), are positive effecters of sexual differentiation (12-16). Components of the PKA and stress-activated MAPK pathways are structurally and functionally conserved from fission yeast to humans. The MAPK pathway is required for expression of some genes under control of the PKA pathway (17), but the molecular basis for this connection is unknown.
Fig.1. Signal transduction pathways of nutritional stress response and sexual differentiation in S. pombe.

The central components of three pathways are shown (arrows, activation; bars, repression). Signals from the stress-activated MAPK Spc1, the cAMP-dependent kinase PKA, and the mating pheromone-responsive kinase Pat1 converge upon Ste11 and Mei2, which are key positive effecters of sexual differentiation. This study demonstrates that Atf1-Pcr1-M26 complex transduces signals from the stress-activated MAPK pathway to help regulate cgs2+ and components downstream of the PKA pathway. Additional Spc1-dependent signals are transduced by an Atf1-Pcr1-independent mechanism (“?” and dotted lines). We also hypothesize that PKA represses Atf1-Pcr1 to coordinate signals from two sensing pathways and to activate a feedback amplification loop that ensures commitment to sexual differentiation (“*” and dotted lines).
Atf1 protein forms heterodimers with Pcr1 (Mts2) protein (12, 16), but each of these proteins can also form homodimers that bind to DNA (12). Atf1 and Pcr1 harbor basic, leucine zipper (bZIP) motifs characteristic of dimeric, DNA-binding transcription factors of the CREB/ATF family. The pcr1 mutants also exhibit defects in sexual development (13, 16, 18), suggesting that Atf1 and Pcr1 may function together in sexual differentiation. However, some stress responses require only Atf1 or Pcr1 (not both) (13, 19). This indicates that Atf1, Pcr1, and other bZIP proteins can form different combinations of homodimers and heterodimers, each of which regulates the expression of a specific set of genes in response to a particular stimulus.
Atf1-Pcr1 heterodimer binds with high affinity and specificity to a DNA site at the ade6-M26 meiotic recombination hotspot; single base pair mutations at the “M26” DNA site coordinately affect protein-DNA complex formation and hotspot activation (12). By extension, if a complex containing Atf1 and Pcr1 directly regulates the transcription of any particular gene in response to a given stress, then one would expect that gene to contain an Atf1-Pcr1 binding site whose mutation (at the endogenous locus) recapitulates the effect of loss of Atf1-Pcr1 heterodimer itself. Here we report that the Atf1-Pcr1 heterodimer directly links the stress-activated MAPK and cAMP-dependent kinase pathways by regulating cgs2+.
EXPERIMENTAL PROCEDURES
S. pombe Culture
The genotypes of S. pombe strains used are listed in Table I. Culture media and genetic methods were as described previously (20). Cells were grown with good aeration at 32 °C in rich YEL medium (0.5% yeast extract, 3% glucose) or defined PM medium (containing nitrogen and essential growth factors) to a density of 5 × 106 cells/ml. For nitrogen starvation, cells grown in complete PM were transferred to PM medium lacking nitrogen, cultured with good aeration at 32 °C, and harvested at the time points indicated. Cells were processed immediately for analysis of chromatin structure, for preparation of protein extracts, or for fixation. Aliquots of cells were stored at −20 °C (as cell pellets) for subsequent preparation of mRNA.
Table I.
Genotypes of S. pombe strains used in this study
| Strain | Genotype |
|---|---|
| WSP 571 | h+ ade6-M26 his3-D1 ura4-D18 leu1–32 |
| WSP 644 | h+ ade6-M26 mts1-D15::ura4 his3-D1 ura4-D18 leu1–32 |
| WSP 666 | h+ ade6-M26 mts2-D1::ura4 his3-D1 ura4-D18 leu1–32 |
| WSP 675 | h− ade6-M26 mts1-D15::ura4 mts2-D1::his3 his3-D1 ura4-D18 leu1–32 |
| WSP 1040 | h+ ade6-M26 spc1::ura4 his3-D1 ura4-D18 leu1–32 |
| WSP 1098 | h− ade6-M26 mts2-D1::his3 spc1::ura4 his3-D1 ura4-D18 leu1–32 |
| WSP 1974 | h+ ade6-M26 cgs2-EcoR1 his3-D1 ura4-D18 leu1–32 |
| WSP 2097 | h− ade6-M26 cgs2-EcoR1 mts1-D15::ura4 his3-D1 ura4-D18 leu1–32 |
| WSP 2209 | h+ ade6-M26 cgs2-EcoR1 mts2-D1::his3 his3-D1 ura4-D18 leu1–32 |
| WSP 2105 | h+ ade6-M26 cgs2-EcoR1 spc1::ura4 his3-D1 ura4-D18 leu1–32 |
| WSP 2195 | h+ hade6-M26 cgs2-EcoR1 mts1-D15::ura4 mts2-D1::his3 his3-D1 ura4-D18 leu1–32 |
Gene Replacement
Mutation of the M26 DNA site at the endogenous cgs2+ locus was achieved by transformation and a pop-in, pop-out approach (21). Two-step, PCR-based mutagenesis (22) was used to simultaneously introduce a 2-bp mutation within the M26 DNA site and to create an EcoRI site for diagnostic purposes (5′-TATGACGTC-3′ → 5′-TATGAatTC-3′, with the M26 site underlined and the EcoRI site italicized). A mutated PCR product of 858 bp in length was cloned into pURA4, and clones were subject to DNA sequencing to confirm the presence of the desired mutations and to eliminate clones with spurious mutations. pURA4-cgs2+-EcoRI was linearized by digestion with HpaI. Transformation, forward selection for Uracil prototrophy, and reverse selection for FOA resistance were as described (21). Candidates were screened with a combination of PCR analysis, restriction mapping, and DNA sequencing to identify those with successful allele replacement.
Protein Preparation and Gel Mobility Shift Assays
Cell culture, preparation of whole cell extracts, and protein fraction III were as described (12). Gel mobility shift assays were as described (12, 23) using the DNA fragments indicated in Fig. 2. One microliter of polyclonal α-Atf1 or α-Pcr1 antisera (13) was added to the indicated binding reactions 15 min prior to electrophoresis.
Fig.2. Spc1 MAPK-dependent binding of Atf1-Pcr1 complex to M26 DNA site of cgs2+.
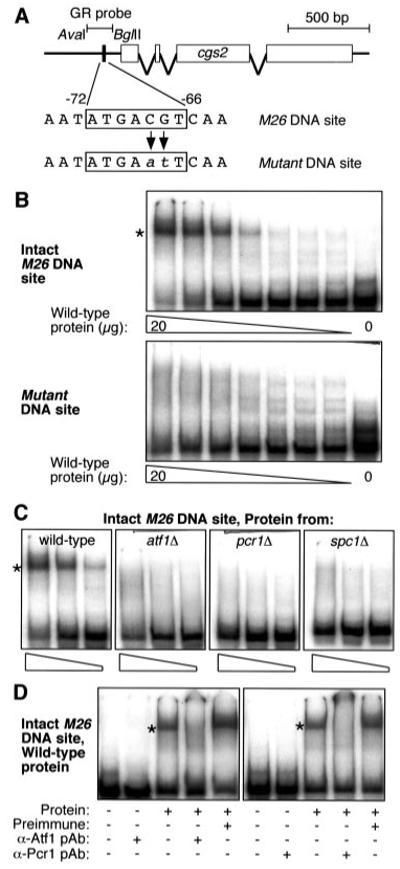
A, diagram of cgs2+ locus showing locations of M26 DNA site and radiolabeled DNA fragments used for gel mobility shift assay (GR probe). Gene targeting was used to replace the endogenous M26 DNA site with a mutant DNA site. Gel mobility shift assays were conducted with partially fractionated whole-cell extracts prepared from cells at early log phase. B, an intact M26 DNA site is required for efficient protein-DNA binding (asterisk). Assays differed only in the probe used and were run on the same gel; the autoradiogram was divided for the sake of presentation. C, dependence of M26-specific protein-DNA complex (asterisk) upon Atf1, Pcr1, and Spc1 kinase. Binding reactions contained 20, 5, and 1 μg of protein prepared from atf1Δ, pcr1Δ, and spc1Δ deletion mutants. D, Atf1 and Pcr1 are components of protein-DNA complex. Pre-immune serum, α-Atf1, and α-Pcr1 polyclonal antibodies were added to the indicated binding reactions 15 min prior to electrophoresis.
Real-time Quantitative PCR
RNA was prepared (13) and subject to real-time, quantitative, reverse-transcription PCR using TaqMan technology and an Applied Biosystems Inc. Prism 7700 fluorometric thermal cycler using established methods (24). Primers and probes were designed using the Primer Express software (version 1.5; Applied Biosystems). The gene names, forward and reverse primers, and TaqMan probe sequences are provided in Table II. Three independent cultures were assayed for each strain/experimental condition. For each reaction, expression levels were normalized to those of an internal control, cam1+, a housekeeping gene. To facilitate comparison, expression levels in cells cultured under inducing conditions are presented relative to the expression levels in wild-type cells under non-inducing conditions.
Table II.
TaqMan primer and probe sequences
| Gene | Forward primer | Reverse primer | TaqMan probe |
|---|---|---|---|
| cgs2 + | 5′-CCACCTATCGGCGTTGGTT-3′ | 5′-ACCAAGAATCGACGGACAGG-3′ | 5′-6FAM-AGTTGATCCAGTCAAAAC-TAMRA-3′ |
| ste11 + | 5′-CGAACCCGAAAAGCCTGTT-3′ | 5′-AGCGGGAGCTAATAAAGCTGC-3′ | 5′-6FAM-ATGAGGAGGAAACTTT-TAMRA-3′ |
| cam1 + | 5′-CGATGGTAATGGCACAATTGAT-3′ | 5′-TTCGTTGTCGGTATCCTTCATTT-3′ | 5′-6FAM-TACCGAATTTTTGACTATGATGGCCCGA-TAMRA-3′ |
Chromatin Mapping
Methods for analysis of chromatin structure were described in detail elsewhere (25). In brief, intact chromatin within isolated nuclei was treated with a range of concentrations of micrococcal nuclease (MNase: 0, 10, 20, and 30 units/ml). Nuclei were extracted, and DNA was deproteinized, digested to completion with PstI and ClaI, fractionated by gel electrophoresis, and subject to Southern blotting using a probe that hybridizes to the 3′-end of the PstI-ClaI restriction fragment of cgs2+. Chromatin treated without MNase (0 units/ml) served as a control to ensure that DNA cleavage was catalyzed by the MNase, rather than by endogenous nucleases. Naked DNA treated with MNase served as a control to ensure that preferential DNA cleavage was dependent upon chromatin proteins, rather than being directed by DNA sequence alone.
Fluorescence-activated Cell Sorting Analysis of DNA Content
Cells were transferred from defined minimal medium containing nitrogen to minimal medium lacking nitrogen and further cultured for the indicated times. One-ml samples of each culture were harvested by centrifugation, the cells were fixed for at least 15 min at −20 °C in 70% ethanol, washed with H2O, and stained with 1 μm Sytox Green (Molecular Probes). Cell suspensions were sonicated for 15 s at power level 2 with a microtip Branson Sonifier prior to analysis using a FACSCalibur machine (BD Biosciences). Data analysis was conducted using ModFit v3.0 software (Verity Software House, Inc.) with FL1-H channel maximum set at 125.
RESULTS
A candidate gene approach for targets of Atf1-Pcr1 was used. We searched S. pombe genomic DNA sequence for genes that met two criteria: First, the genes should contain at least one copy of the M26 DNA site in the promoter region. Second, there should be some indication (experimental or hypothetical) that the genes have a role in sexual differentiation in response to nutritional stress conditions.
The cgs2+ (pde1+) gene was selected as a good candidate for further study: It was identified in a screen for mutants unable to undergo normal meiosis (26) and independently in a screen for S. pombe cDNA clones that could suppress the heat-shock-sensitive phenotypes of iraΔ mutants of Saccharomyces cerevisiae (27, 28). Cgs2 protein is a phosphodiesterase that has a major role in regulating the single cAMP-dependent protein kinase (PKA) pathway of fission yeast (9, 26, 28, 29). Cgs2 cleaves an essential cofactor (cyclic AMP) to negatively regulate PKA, which is itself a negative regulator of mating and meiosis. As a consequence, cgs2Δ null mutants are partially sterile and are unable to undergo meiosis. They also continue to grow in stationary phase under stress conditions in which wild-type cells undergo a G0 cell-cycle arrest (26). The presence of an M26 DNA site located at positions −66 to −72 of the cgs2+ promoter region (Fig. 2A) suggested to us that Atf1-Pcr1 heterodimer (and the nitrogen stress-activated MAPK cascade) helps to regulate cgs2+ (and the energy-sensing PKA cascade) in response to nutritional deprivation.
Spc1-dependent Binding of Atf1-Pcr1 to M26 DNA Site of cgs2+
The core DNA sequence of the M26 site in the cgs2+ promoter is flanked by different base pairs (5′-AATATGACGTCAA-3′) than the core of the M26 site at ade6+ (5′-TGGATGACGTGAG-3′). Because base pairs flanking the core region may be essential for binding of Atf1-Pcr1 heterodimer (30), we determined whether Atf1-Pcr1 complex could bind to the M26 site at cgs2+ in the context of the different flanking sequences. A gel mobility shift assay using partially fractionated protein extracts and a DNA fragment from the cgs2+ promoter (Fig. 2A) revealed one predominant protein-DNA complex and a number of less abundant protein-DNA complexes (Fig. 2B). Mutation of two base pairs within the M26 DNA site abolished formation of the predominant protein-DNA complex, demonstrating that one or more proteins in the extract bind directly to the M26 DNA site located in the promoter of the cgs2+ gene. The less abundant complexes were insensitive to mutations within M26 (Fig. 2B), suggesting that the proteins responsible for those complexes bind DNA sites distant from M26.
Protein extracts obtained from atf1Δ, pcr1Δ, and spc1Δ null mutants were unable to produce M26-specific protein-DNA complexes under conditions in which wild-type extracts exhibited robust binding (Fig. 2C). Thus, the Atf1, Pcr1, and Spc1 proteins are essential for formation of a protein-DNA complex at the M26 DNA site. The diagnostic, M26-dependent protein-DNA complex in wild-type extracts was supershifted entirely by the addition of α-Atf1 or α-Pcr1 antibodies, but not by the addition of corresponding preimmune serum (Fig. 2D). We conclude that Atf1-Pcr1 heterodimer binds directly to the M26 DNA site of cgs2+ and that the stress-activated MAPK Spc1 is required for formation of the Atf1-Pcr1-M26 complex.
Atf1-Pcr1-M26 Complex Regulates Expression of cgs2+
The Atf1-Pcr1-M26 complex forms avidly at the cgs2+ promoter (Fig. 2). To test whether this complex regulates expression of cgs2+ in vivo, we introduced a mutated M26 DNA site (cgs2-EcoRI) into the endogenous cgs2+ locus (Fig. 2A). Cells were subject to nitrogen starvation, and expression of cgs2+ mRNA was analyzed by reverse transcription, real-time, quantitative PCR (24). In wild-type cells a rapid induction of cgs2+ expression was observed (Fig. 3A). Disruption of the Atf1-Pcr1-M26 complex, either by ablation of the M26 site or by loss of either subunit of the Atf1-Pcr1 heterodimer, abolished the induction of cgs2+ mRNA in response to nitrogen starvation (Fig. 3A). We conclude that Atf1-Pcr1 heterodimer is a transcription factor that directly regulates a known component of the PKA path-way, cgs2+.
Fig.3. Spc1 MAPK-regulated expression of cgs2+ and PKA pathway target ste11+ require Atf1-Pcr1-M26 complex.

Cells were cultured in minimal medium lacking nitrogen for the indicated times. mRNA was prepared and real-time PCR was used to determine expression levels relative to an internal control, cam1+; data are mean ± S.D. from three independent experiments and are normalized to t = 0 values from wild-type cells. A, dependence of cgs2+ expression upon components of Atf1-Pcr1-M26 complex and the MAPK Spc1. B, regulation of ste11+, which is downstream of and regulated by the cAMP-dependent kinase pathway (see Fig. 1).
Atf1-Pcr1-M26 Complex Mediates Chromatin Remodeling at cgs2+
The finding that Atf1-Pcr1 heterodimer is a transcription factor that induces expression of cgs2+ is intriguing, because Atf1-Pcr1 activates the ade6-M26 meiotic recombination hotspot without substantially affecting transcription at ade6 (19). It has been suggested that Atf1-Pcr1-dependent modification of chromatin structure could mediate such disparate out-comes (13, 31). We therefore used micrococcal nuclease (MNase) mapping (25) to determine the chromatin configuration of the cgs2+ promoter region in response to nitrogen starvation in the presence or absence of the Atf1-Pcr1-M26 complex.
Treatment of chromatin with MNase revealed five prominent MNase cleavage sites mapping to a ~150-bp region of the cgs2+ promoter centered upon the M26 DNA site (Fig. 4). The prominent cleavage sites were not present in naked (deproteinized) DNA, demonstrating that they required chromatin. In wild-type cells DNA cleavage was enhanced at four of the MNase sites after nitrogen starvation, demonstrating stress-induced increases in the accessibility of DNA within chromatin. Intact Atf1-Pcr1 complex was required for the stress-induced chromatin remodeling at each of those four MNase cleavage sites; this chromatin remodeling correlated positively with complex-dependent, stress-induced transcription of cgs2+ (Fig. 3). Interestingly, loss of Atf1-Pcr1 heterodimer led to a constitutive increase (not inducible by nitrogen starvation) in DNA accessibility at MNase sites 1 and 2 (Fig. 4, B and C). Atf1-Pcr1-M26 complex either directly suppresses chromatin accessibility at those sites or loss of Atf1-Pcr1 heterodimer allows access for additional chromatin remodeling factors that are otherwise excluded by Atf1-Pcr1. We conclude that Atf1-Pcr1-M26 complex has dual specificity as both an inducer and repressor of chromatin remodeling.
Fig.4. Nitrogen stress promotes Atf1-dependent chromatin remodeling at cgs2+ promoter.
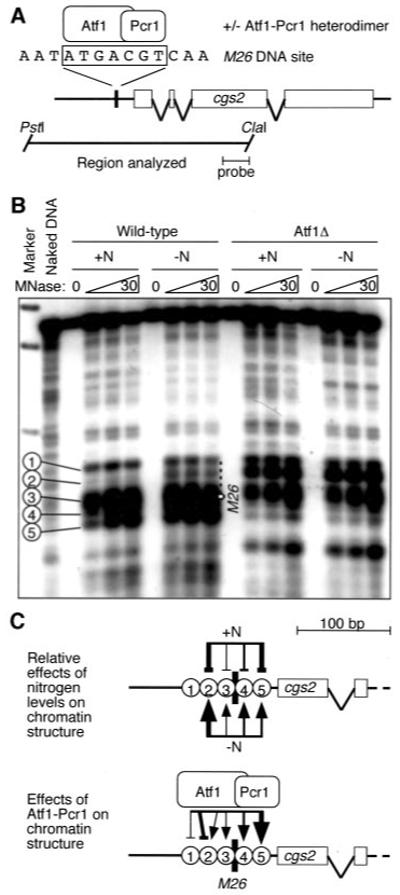
A, diagram of cgs2+ locus showing locations of M26 DNA site and DNA fragments analyzed. B, chromatin structure of cgs2+ revealed by micrococcal nuclease digestion and indirect end labeling. Balloons indicate five prominent hypersensitive sites of the promoter region that are affected by nitrogen starvation and/or Atf1-Pcr1 heterodimer. C, summary of stress-induced, Atf1-Pcr1-dependent chromatin remodeling: positive (arrows) and negative (bars) effects are indicated, and the line width indicates magnitude of the effect.
The preceding data demonstrate that Atf1-Pcr1-M26 complex functions as a mediator of chromatin remodeling and transcription activation at a nexus of the stress-activated MAPK and PKA pathways. To build upon this conclusion, we determined whether the Atf1-Pcr1-M26 complex transduces signals from the MAPK Spc1 to regulate cgs2+ and, if so, whether those signals were subsequently required for regulation of PKA pathway targets.
Stress-activated Kinase Spc1 Regulates cgs2+ via Atf1-Pcr1-M26 Complex
The Spc1 kinase binds to and phosphorylates Atf1 in vitro (32, 33) and is required for binding of Atf1-Pcr1 to the M26 DNA site of cgs2+ (Fig. 2). We therefore used quantitative PCR analyses to determine whether Atf1-Pcr1-M26-dependent transcription of cgs2+ also required the Spc1 kinase. Cells lacking Spc1 alone or in combination with deficiencies in the other factors were incapable of inducing cgs2+ gene expression in response to nitrogen starvation (Fig. 3A). These data confirm a link between the stress-activated kinase pathway and the Atf1-Pcr1-M26 complex at cgs2+. The single and triple mutant phenotypes were indistinguishable, demonstrating that the Spc1-dependent induction of cgs2+ transcription is mediated predominantly, if not exclusively, by Atf1-Pcr1-M26 complex.
Regulation of Targets Downstream of the cAMP-dependent Kinase Pathway
Cgs2 is a phosphodiesterase that cleaves cAMP, which is an essential cofactor for the cAMP-dependent protein kinase PKA. If Atf1-Pcr1-M26 complex-dependent regulation of cgs2+ feeds into the cAMP-dependent protein kinase pathway, then attenuation of cgs2+ should affect the regulation of factors known to be downstream of PKA. We therefore used quantitative PCR to determine whether expression of the ste11+ gene, which encodes an inducer of sexual differentiation known to be under the control of the PKA pathway, required components of the Atf1-Pcr1-M26 complex and the MAPK Spc1.
Nitrogen starvation induced a 20-fold increase in ste11+ mRNA levels, and this induction was strictly dependent upon the simultaneous presence of Atf1, Pcr1, and the Spc1 kinase (Fig. 3B). The induction kinetics of ste11+ were substantially slower than those of cgs2+, suggesting that regulation of ste11+ is an indirect, secondary consequence of Atf1-Pcr1 heterodimer functioning elsewhere. In support of this inference, there are no obvious binding sites for Atf1-Pcr1 heterodimer in the ste11+ promoter region and gel shift assays with ste11+ promoter fragments failed to reveal any Atf1-Pcr1-dependent protein-DNA interactions (data not shown). Notably, Spc1-dependent, Atf1-Pcr1-dependent induction of ste11+ transcription was strictly dependent upon the M26 DNA site of cgs2+ (Fig. 3B). We conclude that there is a direct connection between the MAPK and PKA signal transduction pathways and that this connection is mediated by the action of Atf1-Pcr1-M26 complex in the promoter of the cgs2+ gene (Fig. 1).
The Interpathway Connection Regulates a G0 Cell Cycle Arrest
Stress-activated MAPK Spc1 and Atf1-Pcr1 heterodimer likely have multiple targets, so it was necessary to determine the biological relevance of the M26 DNA site at cgs2+. In fission yeast sexual differentiation is entered from the G1 phase of the cell cycle. In response to nutritional starvation diploid cells heterozygous at mat1 (the mating type locus) enter meiosis, whereas haploids or diploids homozygous at mat1 enter a G0 (G1) cell cycle arrest (5). In the presence of partners of opposite mating type the haploids in G0 conjugate and enter meiosis; otherwise, they remain at G0 arrest and can maintain viability for extended periods of time. The cgs2 null mutants (and other mutants that cannot repress the PKA pathway) fail to arrest in G0, and this likely contributes to their partial sterility (26). Similarly, the atf1Δ and pcr1Δ mutants are partially sterile and lose viability under stress, perhaps as a result of inability to arrest in G0 (16, 18, 34).
To gain insight into the biological consequences of Atf1-Pcr1-M26 complex acting on the cgs2+ promoter, we examined cell cycle regulation in response to nitrogen starvation. The majority of wild-type cells entered G0 arrest by 16 h (Fig. 5A). Mutation of the cis-acting M26 site in the cgs2+ promoter reduced significantly the fraction of cells entering G0 arrest (Fig. 5B), demonstrating that M26 DNA site-dependent regulation of cgs2+ transcription is biologically meaningful. Loss of Atf1 or Pcr1 (or both) had similar consequences and there was no additivity of phenotypes in double mutants lacking M26 and Atf1 or Pcr1, again demonstrating a linear, dependent pathway relationship. Loss of Spc1 kinase eliminated the G0 arrest at 16 h (Fig. 5B), so it was not possible to make conclusions about pathway dependence from cell cycle data of double mutants harboring the spc1Δ allele. It is clear from the data, however, that signals from Spc1 kinase that are required for G0 arrest are not transduced exclusively via Atf1-Pcr1-M26 complex acting on the cgs2+ promoter. This is not surprising, because Spc1 kinase affects many downstream targets, of which only a subset would likely be dependent upon Atf1 and/or Pcr1.
Fig.5. Spc1-dependent regulation of a G0 cell cycle arrest by Atf1-Pcr1-M26 complex at cgs2+.

Cells were subject to nitrogen stress for the indicated times, and DNA content was monitored using fluorescence-activated cell sorting. A, kinetics of G0 cell cycle arrest in wild-type cells. Actively dividing S. pombe cells are predominantly in G2 phase of the cell cycle and S phase occurs at the time of cytokenesis, so cells in log-phase cultures exhibit almost exclusively a DNA content of 2C. Cells arrested in G0 exhibit a 1C DNA content. B, requirement for Spc1-regulated Atf1-Pcr1-M26 complex in G0 cell cycle arrest. Histograms indicate the percentage of cells in G0 arrest after 16 h of nitrogen starvation; data are mean ± S.D. from three experiments.
DISCUSSION
Atf1-Pcr1-M26 Complex at cgs2+ Links Directly the Stress-activated MAPK and cAMP-dependent Protein Kinase Pathways
The Atf1-Pcr1 (Mts1-Mts2) heterodimer was first identified and purified as a protein complex that binds with high affinity to a DNA site (M26) that activates a meiosis-specific recombination hotspot in the ade6 gene (12, 19). Subsequently, the atf1+ and pcr1+ genes were identified independently in screens for multicopy suppressors of a partial mating defect of spc1 mutants and of a sporulation defect in spo5 mutants, respectively (18, 32, 34). The Atf1 and Pcr1 proteins each harbor basic, leucine zipper (bZIP) motifs characteristic of dimeric, DNA-binding transcription factors of the CREB/ATF family. bZIP proteins can form combinatorial homodimers and heterodimers (35), each of which presumably binds preferentially to its “own” DNA site in vivo to regulate a specific subset of genes in response to one or more stimuli. It is therefore important to determine which bZIP protomers associate into dimers, to identify which genes are regulated by specific bZIP dimers, to elucidate whether such regulation is due to direct DNA binding or due to indirect (downstream) action, and to reveal molecular mechanisms by which each dimer species elicits a biological response.
The atf1+ gene is required for a variety of stress responses, including those caused by nutritional starvation, oxidative stress, osmotic stress, cold stress, UV damage, and nucleotide pool depletion (13, 16, 18, 32-34, 36-40). For most of these stress responses, it is not known whether Atf1 functions as a homodimer or as a heterodimer with some other bZIP protein. Although Atf1-Pcr1 heterodimer is strictly required to activate the ade6-M26 meiotic recombination hotspot (19), some Atf1-dependent stress responses do not require Pcr1 (13). Furthermore, the individual homodimers (Atf1-Atf1 and Pcr1-Pcr1) are capable of binding DNA sites in vitro that are related to, but distinct from, the M26 DNA site (12, 18, 34).
Atf1 can be phosphorylated by stress-activated (MAPK) Spc1 (13, 15, 16); this study reveals molecular mechanisms by which Atf1 transduces signals from the stress-activated kinase pathway to downstream targets (Fig. 1). We report that Spc1 kinase is essential for the induction of cgs2+ transcription in response to nitrogen starvation (Fig. 3A). One can conclude that this Spc1-dependent, transcriptional regulation of cgs2+ is mediated directly and predominantly by an Atf1-Pcr1-M26 complex in the promoter region of cgs2+ for the following reasons: First, formation of the M26 DNA site-specific protein-DNA complex requires the simultaneous presence of the Spc1, Atf1, and Pcr1 proteins (Fig. 2, A–C). (Although other proteins may bind at or near to M26, no M26-specific protein-DNA complexes are present in mutants lacking Spc1, or Atf1, or Pcr1.) Second, 100% of those protein-DNA complexes contain both Atf1 and Pcr1 (Fig. 2D). Third, mutants lacking Spc1, or Atf1, or Pcr1, or the M26 DNA site in the cgs2+ promoter fail to properly induce cgs2+ transcription in response to nitrogen starvation (Fig. 3A). Fourth, there is no additivity to the cgs2+ transcriptional phenotypes of triple mutants, relative to mutants lacking individual components (Fig. 3A).
The Spc1-regulated, Atf1-Pcr1-M26 complex induces transcription of cgs2+ in response to nitrogen starvation, but the magnitude of that regulation is relatively modest (4-fold) (Fig. 3B). Nevertheless, this nominal change in expression of cgs2+ has clear and profound biological effects. This is best illustrated by comparing the situation in wild-type cells to what happens when two base pairs within the M26 DNA site of cgs2+ are mutated (Fig. 2A). Under these circumstances, any observed differential effects must be attributed specifically to the cis-acting DNA site in the cgs2+ gene, rather than to indirect action of the various trans-acting factors.
Cgs2 is a repressor of PKA, which is in turn a repressor of meiotic development (Fig. 1). A repressor of a repressor serves as an activator, so induction of cgs2+ leads to induction of factors normally repressed by PKA. In wild-type cells, nitrogen starvation triggers a 20-fold induction in the expression of ste11+ (Fig. 3B). In mutants lacking the M26 DNA site of cgs2+, there is no induction of ste11+ transcription (Fig. 3B). This proves that regulation of ste11+ is a secondary consequence of M26-dependent activation of cgs2+. In accord with this conclusion, the ste11+ promoter region lacks binding sites for Atf1-Pcr1 heterodimer and induction of ste11+ occurs significantly later than induction of cgs2+ (Fig. 3, A and B). So, for the nitrogen stress-activated transcriptional cascade, a relatively modest direct regulation of cgs2+ (by Atf1-Pcr1 heterodimer) undergoes signal amplification when regulating downstream, indirect targets such as ste11+. Other downstream targets are similarly dependent upon function of the Atf1-Pcr1-M26 complex at cgs2+.2 The M26 DNA site-dependent regulation of cgs2+ also has a clear and pronounced cellular phenotype. In wild-type cells, nitrogen starvation triggers a G0 cell-cycle arrest (Fig. 5). In mutants lacking the M26 DNA site, the G0 cell-cycle arrest is compromised (Fig. 5). It is therefore apparent that the M26 DNA site of cgs2+ is required for secondary regulation of downstream targets and for the normal G0 cell-cycle arrest, which is in turn required for subsequent sexual differentiation and production of ascospores.
In response to nitrogen starvation, signals from the stress-activated MAPK Spc1 are transduced by the Atf1-Pcr1-M26 complex to cgs2+ (Figs. 2 and 3). Induction of cgs2+ transcription then feeds into the PKA pathway to help induce a G0 cell-cycle arrest (Figs. 3 and 5). However, the cell-cycle arrest phenotype of mutants lacking Spc1 is more severe than that of mutants lacking the Atf1-Pcr1-M26 complex (Fig. 5). We conclude that signals from Spc1 kinase that are required for G0 arrest are not transduced exclusively via Atf1-Pcr1-M26 complex acting on the cgs2+ promoter. This finding is consistent with, and provides an explanation for, previous findings: First, some Spc1-dependent stress responses do not require Atf1 and Pcr1 (13, 36, 41-45). Second, recent transcriptional profiling revealed that only about one quarter of the Spc1-dependent genes are dependent upon Atf1 (dependence upon Pcr1 was not tested) (46). Third, overexpression of pyp1+ (which encodes a protein phosphatase that negatively regulates Spc1) inhibits some phenotypes of cells lacking the cAMP-dependent protein kinase (41). It is thus clear that the stress-activated kinase Spc1 regulates components in (and downstream of) the PKA pathway by at least two distinct mechanisms, one that uses the direct action of Atf1-Pcr1-M26 complex at cgs2+, and one that uses some factor(s) other than Atf1-Pcr1 heterodimer (Fig. 1).
Atf1-Pcr1-M26 Complex Mediates Chromatin Remodeling, a Mechanistic Solution to the Paradox of Disparate Effector Functions
Upon nitrogen starvation, the Atf1-Pcr1-M26 complex induces transcription of the cgs2+ gene (Figs. 2 and 3), so in that context it functions as an activator of transcription. However, Atf1 and Pcr1 seem to function (presumably as a heterodimer, although this has not been proven) as repressors of transcription for a reporter gene inserted near the mat loci (47). Unphosphorylated Atf1 (perhaps in concert with Pcr1) may repress expression of endogenous ctt1+ (36). Furthermore, the Atf1-Pcr1-M26 complex activates a meiosis-specific recombination hotspot in the ade6 gene (12, 19). This raises an interesting paradox: how can one protein-DNA complex affect such seemingly disparate functions?
Our analysis of the chromatin structure of the cgs2+ locus, and our recent work on the ade6-M26 meiotic recombination hotspot (31), suggests that one common, underlying molecular mechanism can explain these seemingly paradoxical functions of Atf1-Pcr1-M26 complex. In response to nitrogen deprivation, the complex induces hypersensitivity to MNase at discrete sites in the DNA of chromatin at cgs2+ promoter region (Fig. 4). The hypersensitive sites are absent in naked (deproteinized) DNA, demonstrating that they require chromatin. In the presence of nitrogen, Atf1-Pcr1-M26 complex represses chromatin remodeling at some sites (i.e. removal of the complex induces chromatin remodeling) (Fig. 4). Thus, Atf1-Pcr1-M26 complex exhibits dual specificity as both an inducer and repressor of chromatin remodeling at cgs2+. This dual specificity may be conserved: The bZIP protein Sko1 of budding yeast, which shares some localized homology with the Atf1 subunit of Atf1-Pcr1 heterodimer, can also mediate differential chromatin remodeling (48). Our finding that Atf1-Pcr1 can both repress and induce chromatin remodeling (Fig. 4) provides a mechanism by which Atf1-Pcr1-dependent effector functions can be regulated directly and has implications for additional interpathway connections (see below).
The chromatin remodeling activity of Atf1-Pcr1-M26 complex provides a solution to the paradox of how the complex can mediate disparate functions at different locations. We propose that Atf1-Pcr1-M26 complex-dependent chromatin remodeling facilitates the assembly of components of the transcription and meiotic recombination apparatus. The net outcome could be either positive or negative regulation of function, depending upon the local constellation of cis-acting DNA sites (e.g. enhancers or repressors) and trans-acting factors (e.g. transcription factors or recombination enzymes) whose access is promoted by the Atf1-Pcr1-M26 complex itself and by the complex-dependent chromatin remodeling.
Additional Interpathway Connections, Co-ordination of Signals, and Amplification Loops
Although the focus of this study is on the transmission of activating signals from the stress-activated MAPK pathway into the PKA pathway, we note that components of the PKA pathway can repress some Atf1-dependent functions (38, 50). We suggest that such negative regulation may be exerted directly through Atf1-Pcr1 heterodimer by virtue of its ability to repress chromatin remodeling (Fig. 4). Repression of Atf1-Pcr1 by PKA would have two implications (Fig. 1). First, Atf1-Pcr1 at cgs2+ could serve as a binary “switch” that measures the relative signal intensity from two different pathways (stress-activated protein kinase/nitrogen and PKA/glucose). In that case, Atf1-Pcr1 would be switched into the chromatin-remodeling, transcription-activating state in response to relative, graded levels of both carbon source and nitrogen source, rather than to absolute levels of either factor alone (Fig. 1). Second, once the binary switch has been activated, the cgs2+-mediated attenuation of the PKA pathway would attenuate the PKA-dependent repression of Atf1-Pcr1. This would trigger an amplification loop, via Atf1-Pcr1, to ensure full activation of downstream factors once the cells have made the developmental decision to enter the pathway of sexual differentiation (Fig. 1).
Summary
An Atf1-Pcr1-M26 complex acting at cgs2+ directly links the stress-activated MAPK and cAMP-dependent kinase pathways to regulate a G0 cell cycle arrest. The molecular mechanism by which the complex functions at cgs2+ (via chromatin remodeling) reveals how Atf1-Pcr1 heterodimer can mediate such disparate outcomes as induction or repression of transcription or activation of meiotic recombination. The discovery of a direct interpathway connection in fission yeast may be relevant to signal transduction pathways in multicellular eukaryotes, because components of the stress-activated and cAMP-dependent kinase pathways are conserved in organisms ranging from yeast to humans. Now that the fission yeast genome sequence has been determined (49), one could conduct whole genome DNA array studies of factor binding sites and gene expression profiles, with the goal of identifying additional inter-pathway connections that are refractory to identification by other means.
Acknowledgments
We thank Charla Wiley and Audrey Stone for laboratory support and the Flow Cytometry Core and Microarray Core Facilities at University of Arkansas for Medical Sciences for use of their resources.
Footnotes
The abbreviations used are: PKA, protein kinase A; MAPK, mito-gen-activated protein kinase; bZIP, basic, leucine zipper; MNase, micrococcal nuclease.
This study was supported by Grants GM62244 and GM62801 from the NIGMS, National Institutes of Health. Additional support was provided by the Bioarchitect Research Program of The Institute of Physical and Chemical Research and the Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation, and the Ministry of Education, Science, Culture & Sports, Japan.
REFERENCES
- 1.Sugimoto A, Iino Y, Maeda T, Watanabe Y, Yamamoto M. Genes Dev. 1991;5:1990–1999. doi: 10.1101/gad.5.11.1990. [DOI] [PubMed] [Google Scholar]
- 2.Kitamura K, Katayama S, Dhut S, Sato M, Watanabe Y, Yamamoto M, Toda T. Dev. Cell. 2001;1:389–399. doi: 10.1016/s1534-5807(01)00037-5. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe Y, Yamamoto M. Cell. 1994;78:487–498. doi: 10.1016/0092-8674(94)90426-x. [DOI] [PubMed] [Google Scholar]
- 4.Sato M, Watanabe Y, Akiyoshi Y, Yamamoto M. Curr. Biol. 2002;12:141–145. doi: 10.1016/s0960-9822(01)00654-6. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto M. Cell Struct. Function. 1996;21:431–436. doi: 10.1247/csf.21.431. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe Y, Lino Y, Furuhata K, Shimoda C, Yamamoto M. EMBO J. 1988;7:761–767. doi: 10.1002/j.1460-2075.1988.tb02873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLeod M, Beach D. EMBO J. 1986;5:3665–3671. doi: 10.1002/j.1460-2075.1986.tb04697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng Z, Wang W, Schettino A, Leung B, McLeod M. Curr. Genet. 2003;43:178–185. doi: 10.1007/s00294-003-0384-5. [DOI] [PubMed] [Google Scholar]
- 9.Maeda T, Watanabe Y, Kunitomo H, Yamamoto M. J. Biol. Chem. 1994;269:9632–9637. [PubMed] [Google Scholar]
- 10.Yu G, Li J, Young D. Gene (Amst.) 1994;151:215–220. doi: 10.1016/0378-1119(94)90659-9. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi T, Watanabe Y, Yamamoto M. Mol. Cell. Biol. 2002;22:1–11. doi: 10.1128/MCB.22.1.1-11.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wahls WP, Smith GR. Genes Dev. 1994;8:1693–1702. doi: 10.1101/gad.8.14.1693. [DOI] [PubMed] [Google Scholar]
- 13.Kon N, Schroeder SC, Krawchuk MD, Wahls WP. Mol. Cell. Biol. 1998;18:7575–7583. doi: 10.1128/mcb.18.12.7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato T, Jr., Okazaki K, Murakami H, Stettler S, Fantes PA, Okayama H. FEBS Lett. 1996;378:207–212. doi: 10.1016/0014-5793(95)01442-x. [DOI] [PubMed] [Google Scholar]
- 15.Degols G, Shiozaki K, Russell P. Mol. Cell. Biol. 1996;16:2870–2877. doi: 10.1128/mcb.16.6.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanoh J, Watanabe Y, Ohsugi M, Iino Y, Yamamoto M. Genes Cells. 1996;1:391–408. doi: 10.1046/j.1365-2443.1996.d01-247.x. [DOI] [PubMed] [Google Scholar]
- 17.Stettler S, Warbrick E, Prochnik S, Mackie S, Fantes P. J. Cell Sci. 1996;109:1927–1935. doi: 10.1242/jcs.109.7.1927. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe Y, Yamamoto M. Mol. Cell. Biol. 1996;16:704–711. doi: 10.1128/mcb.16.2.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kon N, Krawchuk MD, Warren BG, Smith GR, Wahls WP. Proc. Natl. Acad. Sci. U. S. A. 1997;94:13765–13770. doi: 10.1073/pnas.94.25.13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krawchuk MD, DeVeaux LC, Wahls WP. Genetics. 1999;153:57–68. doi: 10.1093/genetics/153.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharif WD, Glick GG, Davidson MK, Wahls WP. Cell Chromosome. 2002;1:1. doi: 10.1186/1475-9268-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higuchi R. In: PCR Protocols: A Guide to Methods and Applications. Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. Academic Press; San Diego: 1990. pp. 177–183. [Google Scholar]
- 23.Wahls WP, Swenson G, Moore PD. Nucleic Acids Res. 1991;19:3269–3274. doi: 10.1093/nar/19.12.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- 25.Mizuno K, Emura Y, Baur M, Kohli J, Ohta K, Shibata T. Genes Dev. 1997;11:876–886. doi: 10.1101/gad.11.7.876. [DOI] [PubMed] [Google Scholar]
- 26.DeVoti J, Seydoux G, Beach D, McLeod M. EMBO J. 1991;10:3759–3768. doi: 10.1002/j.1460-2075.1991.tb04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mochizuki N, Yamamoto M. Mol. Gen. Genet. 1992;233:17–24. doi: 10.1007/BF00587556. [DOI] [PubMed] [Google Scholar]
- 28.Matviw H, Li J, Young D. Biochem. Biophys. Res. Commun. 1993;194:79–82. doi: 10.1006/bbrc.1993.1787. [DOI] [PubMed] [Google Scholar]
- 29.Wu SY, McLeod M. Mol. Cell. Biol. 1995;15:1479–1488. doi: 10.1128/mcb.15.3.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fox ME, Yamada T, Ohta K, Smith GR. Genetics. 2000;156:59–68. doi: 10.1093/genetics/156.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada T, Mizuno KI, Hirota K, Kon N, Wahls WP, Hartsuiker E, Murofushi H, Shibata T, Ohta K. EMBO J. 2004;23:1792–1803. doi: 10.1038/sj.emboj.7600138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiozaki K, Russell P. Genes Dev. 1996;10:2276–2288. doi: 10.1101/gad.10.18.2276. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson MG, Samuels M, Takeda T, Toone WM, Shieh J-C, Toda T, Millar JBA, Jones N. Genes Dev. 1996;10:2289–2301. doi: 10.1101/gad.10.18.2289. [DOI] [PubMed] [Google Scholar]
- 34.Takeda T, Toda T, Kominami K, Kohnosu A, Yanagida M, Jones N. EMBO J. 1995;14:6193–6208. doi: 10.1002/j.1460-2075.1995.tb00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Struhl K. Nucleic Acids Res. 1995;23:2531–2537. doi: 10.1093/nar/23.13.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degols G, Russell P. Mol. Cell. Biol. 1997;17:3356–3363. doi: 10.1128/mcb.17.6.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinn J, Findlay VJ, Dawson K, Millar JB, Jones N, Morgan BA, Toone WM. Mol. Biol. Cell. 2002;13:805–816. doi: 10.1091/mbc.01-06-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirota K, Hasemi T, Yamada T, Mizuno KI, Hoffman CS, Shibata T, Ohta K. Nucleic Acids Res. 2004;32:855–862. doi: 10.1093/nar/gkh251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soto T, Beltran FF, Paredes V, Madrid M, Millar JB, Vicente-Soler J, Cansado J, Gacto M. Eur. J. Biochem. 2002;269:5056–5065. doi: 10.1046/j.1432-1033.2002.03214.x. [DOI] [PubMed] [Google Scholar]
- 40.Madrid M, Soto T, Franco A, Paredes V, Vicente J, Hidalgo E, Gacto M, Cansado J. J. Biol. Chem. 2004;279:41594–41602. doi: 10.1074/jbc.M405509200. [DOI] [PubMed] [Google Scholar]
- 41.Dal Santo P, Blanchard B, Hoffman CS. J. Cell Sci. 1996;109:1919–1925. doi: 10.1242/jcs.109.7.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaits F, Shiozaki K, Russell P. J. Biol. Chem. 1997;272:17873–17879. doi: 10.1074/jbc.272.28.17873. [DOI] [PubMed] [Google Scholar]
- 43.Nakagawa CW, Yamada K, Mutoh N. J. Biochem. (Tokyo) 1998;123:1048–1054. doi: 10.1093/oxfordjournals.jbchem.a022042. [DOI] [PubMed] [Google Scholar]
- 44.Toone WM, Kuge S, Samuels M, Morgan BA, Toda T, Jones N. Genes Dev. 1998;12:1453–1463. doi: 10.1101/gad.12.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen AN, Lee A, Place W, Shiozaki K. Mol. Biol. Cell. 2000;11:1169–1181. doi: 10.1091/mbc.11.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen D, Toone WM, Mata J, Lyne R, Burns G, Kivinen K, Brazma A, Jones N, Bahler J. Mol. Biol. Cell. 2003;14:214–229. doi: 10.1091/mbc.E02-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia S, Noma K, Grewal SI. Science. 2004;304:1971–1976. doi: 10.1126/science.1099035. [DOI] [PubMed] [Google Scholar]
- 48.Proft M, Struhl K. Mol. Cell. 2002;9:1307–1317. doi: 10.1016/s1097-2765(02)00557-9. [DOI] [PubMed] [Google Scholar]
- 49.Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, Basham D, Bowman S, Brooks K, Brown D, Brown S, Chillingworth T, Churcher C, Collins M, Connor R, Cronin A, Davis P, Feltwell T, Fraser A, Gentles S, Goble A, Hamlin N, Harris D, Hidalgo J, Hodgson G, Holroyd S, Hornsby T, Howarth S, Huckle EJ, Hunt S, Jagels K, James K, Jones L, Jones M, Leather S, McDonald S, McLean J, Mooney P, Moule S, Mungall K, Murphy L, Niblett D, Odell C, Oliver K, O’Neil S, Pearson D, Quail MA, Rabbinowitsch E, Rutherford K, Rutter S, Saunders D, Seeger K, Sharp S, Skelton J, Simmonds M, Squares R, Squares S, Stevens K, Taylor K, Taylor RG, Tivey A, Walsh S, Warren T, Whitehead S, Woodward J, Volckaert G, Aert R, Robben J, Grymonprez B, Weltjens I, Vanstreels E, Rieger M, Schafer M, Muller-Auer S, Gabel C, Fuchs M, Fritzc C, Holzer E, Moestl D, Hilbert H, Borzym K, Langer I, Beck A, Lehrach H, Reinhardt R, Pohl TM, Eger P, Zimmermann W, Wedler H, Wambutt R, Purnelle B, Goffeau A, Cadieu E, Dreano S, Gloux S, Lelaure V, Mottier S, Galibert F, Aves SJ, Xiang Z, Hunt C, Moore K, Hurst SM, Lucas M, Rochet M, Gaillardin C, Tallada VA, Garzon A, Thode G, Daga RR, Cruzado L, Jimenez J, Sanchez M, del Rey F, Benito J, Dominguez A, Revuelta JL, Moreno S, Armstrong J, Forsburg SL, Cerrutti L, Lowe T, McCombie WR, Paulsen I, Potashkin J, Shpakovski GV, Ussery D, Barrell BG, Nurse P. Nature. 2002;415:871–880. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- 50.Mizuno K, Hasemi T, Ubukata T, Yamada T, Lehmann E, Kohli J, Watanabe Y, Iino Y, Yamamoto M, Fox ME, Smith GR, Murofushi H, Shibata T, Ohta K. Genetics. 2001;159:1467–1478. doi: 10.1093/genetics/159.4.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]