

Abstract
Background:
Neurological affection in Sjogren's syndrome (SS) can occur in the central and peripheral nervous system. Literature describing the neurological involvement in SS among Indian patients is lacking.
Materials and Methods:
Six patients of SS fulfilling the histological or serological criteria of the American European Consensus Group for SS were studied prospectively. The patients underwent clinical examination and laboratory investigations. Their clinical and investigation features are described.
Results:
The age of the patients ranged from 26 to 48 years, with a male to female ratio of 2:4. In our series, peripheral sensori-motor neuropathy and sensory ataxic neuropathy was seen in 3/6, mononeuritis multiplex in 2/6, cranial neuropathy in 2/6, autonomic neuropathy in 1/6, myelopathy in 4/6, optic neuropathy in 2/6, with presence of classical sicca features in 5/6 patients. Positive lip biopsy was seen in three, altitudinal field defect in one and positive Schirmer's test in five patients. Nerve conduction study abnormalities were seen in three and evidence of vasculitis was seen in nerve biopsy of one patient and chronic nonuniform axonopathy was seen in another. Antibody to Ro (SSA) or La (SSB) was positive in five patients.
Conclusions:
SS involves different parts of the nervous system with varied presentations. Clinical suspicion and adequate laboratory testing helps to diagnose and manage this disorder that is relatively rare in Indian patients.
Keywords: Myelitis, polyneuropathies, Sjogren's syndrome, trigeminal neuropathy
Introduction
Primary Sjogren's syndrome (SS) is a multisystem autoimmune connective tissue disorder with xerophthalmia and xerostomia as its defining features, resulting from lymphocytic infiltration of the lacrimal, salivary and other exocrine glands. Secondary SS combines these features and a defined rheumatic disease.[1] Diagnostic criteria in SS have been a subject of controversy in view of the variability of clinical features, especially at disease onset, occasional absence of key diagnostic features during the disease course, and lack of a single distinguishing feature for diagnosis. Several criteria such as the Copenhagen Criteria, Las Vegas Criteria and The European Study Group Classification Criteria have attempted to address this by using multiple clinical and laboratory parameters but with partial success. The European Criteria have been criticized for their lack of emphasis on serological and histological features. This has been rectified in the current criteria proposed by the American-European consensus group, which in addition to objective evidence of the sicca complex makes serological or histological evidence mandatory for diagnosis and also lists the exclusion criteria for the first time.[1] The European criteria for primary SS have a lesser specificity (75%), but a higher sensitivity (65.7%) compared to the current criteria.[2] Although more than half of the patients develop extraglandular manifestations, neurological complications are uncommon.[3]
Indian scenario
A study from a tertiary care center from North India described 26 patients of SS seen over a 10-year period which comprised 0.5% of the total patients with rheumatic diseases.[4] Patients in this series had presented primarily with sicca syndrome, and the common extraglandular manifestations were purpura, glomerulonephritis and renal tubular acidosis. One patient in this series had neuropathy.[4] The prevalence of SS varies from 0.5 to 3% depending on the criteria employed (American-European, San Diego or San Francisco criteria). SS was reported to be uncommon in a large series of rheumatic diseases from a dedicated rheumatology center in India.[5]
Materials and Methods
Patients attending the neurology services at a tertiary care center and teaching hospital in South India and fulfilling the histological or serological criteria for SS as per the American-European consensus group were studied prospectively from December 2008 to December 2009. Six patients were evaluated with history, including sicca symptoms, detailed examination including visual field charting, Schirmer's test, complete hemogram and erythrocyte sedimentation rate (ESR), biochemical profile, urine analysis, cerebrospinal fluid (CSF) analysis for cells, proteins, sugar and oligoclonal bands (OCB), rheumatoid factor (RA), antinuclear antibody (ANA), other markers for connective tissue diseases, magnetic resonance imaging (MRI) of brain and spine, visual evoked potentials (VEP), brainstem auditory evoked potentials (BAER) and somatosensory evoked potentials (SSEP), wherever indicated, nerve biopsy when indicated, lip biopsy for minor salivary gland studies and serology for human immunodeficiency virus (HIV) and hepatitis C virus (HCV). Ocular involvement was documented by Schirmer's test (abnormal if less than 5 mm of the filter paper was moistened in 5 minutes). Xerostomia could not be confirmed by abnormal salivary gland scintigraphy due to nonavailability of the test. Biopsy samples of the minor salivary glands were considered suggestive of SS if the lymphocytic focus score was equal to or greater than an aggregate of 50 mononuclear cells in 4 mm2 of glandular tissue.[6] Screening for autoantibodies to Ro/SSA, La/SSB and other connective tissue diseases was systematically performed. Exclusion criteria were the following: Past head and neck radiation treatment, hepatitis C infection, acquired immunodeficiency disease, pre-existing lymphoma, sarcoidosis, graft versus host disease, use of anticholinergic drugs or other drugs inducing sicca symptoms.
Results
Patient 1
A 26-year-old lady (P1) presented with a history of acute myelopathy with paraparesis, sensory loss and sphincter disturbance 3 years back with full recovery over 2 weeks with treatment. Her current hospitalization was for acute impairment of vision in her left eye followed by the right eye. She had significant loss of weight and appetite, and dryness of mouth since several years. Examination showed impaired vision in her right eye with a relative afferent pupillary defect, a superior altitudinal field defect and mild optic atrophy. Schirmer's test was positive in both eyes. Investigations showed anemia, high ESR (93 mm at 1 st hour), prolonged VEP in both eyes, abnormal MRI brain which showed a single T1 hypointense and T2 and T2 FLAIR hyperintense, non-enhancing lesion in the left parietal white matter and raised CSF protein (75 mg/dl). MRI of the spine was normal.
Patient 2
A 37-year-old man (P2) presented with progressive asymmetric paresthesias in the lower limbs for 12 months, followed by similar symptoms in the right median and left ulnar nerve distribution. One month after the onset, he developed progressive ataxia and limb incoordination which worsened in the dark, distal wasting and weakness of left upper and both the lower limbs. He had dryness of mouth and Schirmer's test was positive. He had significant loss of appetite and weight since the onset of symptoms. Examination showed generalized hypotonia and areflexia; fine touch was impaired on the right foot and left ulnar two fingers, while pain was impaired on the medial aspect of left foot. He had impaired vibration and proprioception in all limbs with severe sensory ataxia. Investigations showed ESR of 45 mm at 1st hour, abnormal nerve conduction studies suggestive of axonal type of mononeuritis multiplex, involving bilateral median and ulnar, right radial, common peroneal and sural nerves and left superficial peroneal nerve. Peroneus tertius muscle and superficial peroneal nerve biopsy showed features of vasculitic neuropathy [Figure 1]. Evoked potential studies (visual and somatosensory) were normal.
Figure 1.
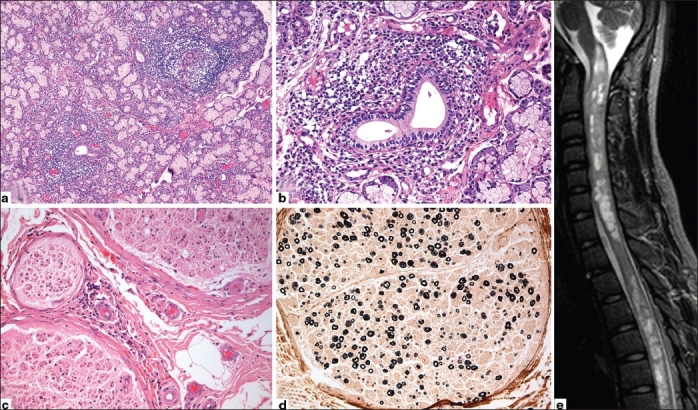
(a) Lip salivary gland biopsy showing two large lymphocytic foci, >1 per 4 mm2 of glandular tissue, thus giving focus score of >1. (b) Higher magnifi cation shows single focus composed of mature lymphocytes centered around duct. (c) Sural nerve biopsy with vasculitis involving the epineurial small arterioles. (d) Myelin stain reveals nonuniform, multifocal myelinated fi ber loss of moderate severity. (e) MRI spine showing T2W intramedullary hyperintensities of gray and white matter from cervical to lumbar level associated with cord expansion in cervical region in patient with Sjogren's myelopathy (P3)
Patient 3
A 14-year-old girl (P3) presented with three episodes of myelopathy over a period of 10 months. The first episode was characterized by acute onset of asymmetric quadriplegia with sensory and sphincter impairment. She improved completely over 15 days with steroids. The second episode developed 7 months later when she developed acute left lower limb weakness with intermittent diplopia. There was also loss of weight and appetite. She had spasticity, asymmetric paraparesis, extensor plantar response and normal sensory examination. Her vision was normal. She was diagnosed to have long segment transverse myelitis and improved almost completely with steroids. Despite being on immunomodulation, she developed the third episode 1 month later, characterized by right lower limb weakness, urinary urgency and frequency. She never had sicca symptoms, but Schirmer's test was positive. Her ESR was 17 mm at 1st hour, and she had normal VEP and abnormal SSEP. Her CSF showed 116 cells/mm3 with 95% lymphocytes and 5% neutrophils, protein level of 75 mg/dl and normal glucose. MRI spine showed extensive zone of T2 intramedullary hyperintensities from cervicomedullary junction to just above the conus, associated with cord expansion in the cervical region and patchy contrast enhancement. MRI brain showed T2 hyperintensities in the dorsal aspect of medulla.
Patient 4
This 43-year-old man (P4) presented with facial sensory impairment, impaired sensations in the limbs, distal weakness and wasting of right upper limb, and later left upper limb, all of 9 months duration. Examination revealed impairment of touch and pain in the ophthalmic and maxillary divisions of left trigeminal nerve, below the level of shoulders, particularly over the medial aspect of both hands and forearms, lateral aspects of legs and dorsum of both feet. He had claw hand deformity and absent ankle jerks and plantar response. He developed right-sided lower motor neuron type of facial weakness with taste involvement prior to admission. He reported significant loss of weight and appetite. He had a similar constellation of symptoms 11 years ago which improved completely with steroids. He had poorly controlled diabetes and history of recurrent crops of erythematous papular skin eruptions over the body. He did not have sicca symptoms, and Schirmer's test was negative. ESR was 14 mm at 1st hour, SSEP were not recordable, and MRI of brain and spine was normal. CSF analysis showed two cells, protein of 61 mg/dl, and normal glucose levels. Nerve conduction studies showed asymmetrical demyelinating sensorimotor polyneuropathy, but sural nerve biopsy was normal. Lip biopsy features were compatible with SS.
Patient 5
A 39-year-old lady (P5) presented with sensory ataxia of 10 years duration, along with incoordination, proximal and distal limb weakness. This was followed by dryness of mouth, dental caries, tremulousness of the head and painful lower limb paresthesias since 6 years. She also reported multiple asymmetrical large joint pains since 6 years without any joint swelling or early morning stiffness. She developed two episodes of painless blurring of vision in both eyes, 12 years apart, with impaired color vision, which was subacute in onset, progressed over 3 days, and recovered completely over the next 3 months. She also developed hearing impairment and dysphagia since 2 months, with occasional cough during feeds. She had pallor, dry mouth and eyes with positive Schirmer's test, numerous dental caries, left corneal opacity and sluggishly reactive pupils. Examination showed postural drop in systolic blood pressure of 24 mm Hg, hypotonia in all limbs, truncal and lower limb ataxia, sensory ataxia, generalized areflexia and a normal plantar response. She had symmetrical impairment of touch, vibration and proprioception all over the body and face, especially below the knees in both lower limbs and below the elbows in both upper limbs. She had anemia, ESR was 20 mm at 1st hour, VEP and SEP were absent, and nerve conduction studies showed evidence of sensory polyneuropathy, and cardiac autonomic dysfunction was noted.
Patient 6
This 48-year-old lady (P6) presented with slowly progressive paresthesias and sensory impairment starting distally and extending up to thighs and shoulders over 8 years, associated with distal weakness and wasting of hands and feet. She had allodynia and sphincter disturbances. For 4 months, she had dryness of mouth and pain in small and large joints of limbs. Examination revealed hypertension, multinodular goiter, contractures in both upper limbs with painful restriction of movements at the fingers, wrist, elbow and shoulders. Schirmer's test was positive. She had sluggishly reactive pupils with mild asymmetry, spasticity in lower limbs, weakness of neck and trunk muscles, mild weakness of all four limbs and generalized areflexia. Proprioceptive loss was up to the knees and elbows, while vibration was impaired up to the clavicles. Fine touch was impaired up to the posterior scalp, whereas pain was impaired only in both hands and feet. Plantar response was extensor. There was limb incoordination and severe sensory ataxia. Investigations showed anemia, ESR of 60 mm at 1st hour, absent sensory evoked potentials and normal CSF. MRI spine showed hyperintense signal changes involving the posterior cord at lower cervical level on T2W sequences. A sensory polyneuropathy was detected on nerve conduction studies, and left sural nerve biopsy showed marked, nonuniform loss of myelinated fibers and occasional regenerating clusters consistent with asymmetric chronic axonopathy. In the absence of inflammation, the findings were consistent with possible vasculitis. The patient was diagnosed as secondary SS.
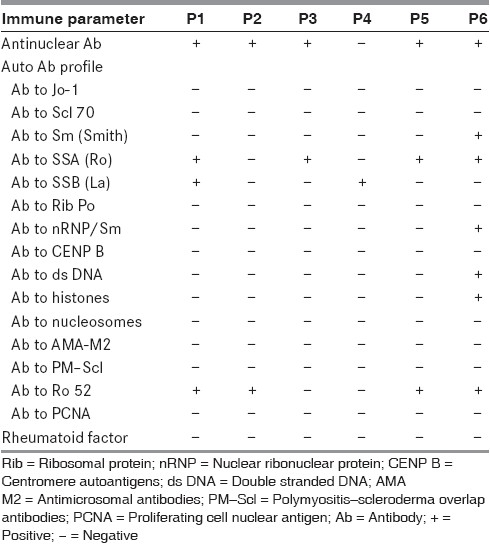
The diagnostic features and immune profile of individual patients are given in Tables 1 and 2. Rheumatoid factor was negative in all the patients.
Table 1.
Revised international classifi cation (American-European consensus group) criteria for Sjogren's syndrome (SS)
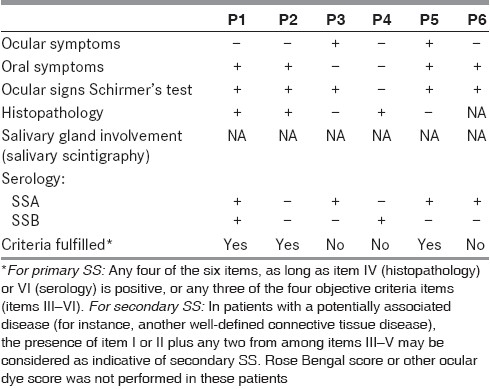
Table 2.
The description of the antibody profile of the patients
Our series of Neuro-Sjogrens syndrome highlights the neurological manifestations like peripheral sensorimotor neuropathy and sensory ataxic neuropathy (3/6: P2, P5 and P6), mononeuritis multiplex (2/6: P2 and P4), cranial neuropathy (2/6: P1 and P5), autonomic neuropathy, central nervous system (CNS) involvement (4/6: P1, P3, P5 and P6), myelopathy (3/6: P1, P2 and P6) and optic neuropathy (1/6: P1) with variable presence of classical sicca features (5/6: P1, P2, P3, P5 and P6).
Response to treatment
P3 made significant recovery in myelopathy after steroids and was completely ambulant within 2 weeks of treatment. The patients with sensory ataxia and painful sensory symptoms showed only partial improvement with steroids but responded to symptomatic treatment for neuropathic pain (P2, P4, P5 and P6). There was significant benefit in sicca symptoms in P1, P2, P3, P5 and P6. Patient P2 was lost to follow-up. Patient P2 also received Cyclophosphamide pulse therapy in addition to the symptomatic management.
Discussion and Brief Review of Neuro-Sjogren's Syndrome
In 1933, Henrik Sjögren, a Swedish ophthalmologist, described the neurological manifestations in SS for the first time in his doctoral thesis. Absence of sicca symptoms does not exclude the diagnosis of SS as sicca symptoms occur only in 38% patients before the onset of neurologic features; 15% develop both at the same time, while in 47% of patients neurologic features precede the sicca symptoms, often by up to 12 years.[7]
Peripheral nervous system involvement
In a large series of 82 patients of SS with neurological manifestations from the USA, cranial neuropathies were reported in 19.5% and peripheral neuropathy (PN) was reported in 44.5% of the patients.[8] In the same study, 68% patients had CNS involvement, 62% patients had peripheral nervous system involvement, 24% patients had spinal cord involvement, and 11% patients had visual loss secondary to optic neuritis.[7]
SS associated neuropathy has a broad clinical spectrum, including sensory ataxic neuropathy, painful sensory neuropathy without sensory ataxia, multiple mononeuropathy, multiple cranial neuropathy, trigeminal neuropathy, autonomic neuropathy, ganglionopathy and radiculoneuropathy.[8] The most common conditions are symmetric sensorimotor PN and symmetric pure-sensory PN.[8]
Appropriate serologic tests (especially SSA and SSB) have to be done in all patients with polyneuropathy of unknown cause or with multiple mononeuropathy or cranial neuropathy. A clue to the diagnosis of SS is the prominence of sensory symptoms and signs. Some patients with painful sensory neuropathy eventually develop sensory ataxia during long-term follow-up. In contrast to sensory neuropathies, multiple mononeuropathy and multiple cranial neuropathies often include motor nerve involvement with predominantly acute and subacute onset. These forms are not seen in the sensory ataxic and painful sensory forms of neuropathy, suggesting that these neuropathies are distinct from the sensory neuropathies. A study from Norway examined 62 patients with primary SS and 27% of them were found to have neuropathy.[3]
Cranial neuropathies
Isolated cranial nerve lesions affecting the third,[9] fourth,[10] fifth,[11] seventh,[12] ninth and tenth[11] nerves have been reported previously. In a study from Japan involving 92 patients of primary SS, most of these patients (93%) were diagnosed as SS after neuropathic symptoms had appeared. About 12% (n = 11) of them had multiple mononeuropathy, 5% (n = 5) had multiple cranial neuropathy and 16% (n = 15) had trigeminal neuropathy.[8]
CNS involvement
The manifestations of CNS involvement may be localized (optic neuropathy, hemiparesis, myelitis, dystonia) or diffuse (encephalopathy, dementia).[9] CNS involvement is usually multifocal, additive, and progressive, with a clinical course of fixed and cumulative deficits or a relapsing-remitting course that may mimic multiple sclerosis.[13] The occurrence of myelopathy in primary SS is rare, with a clear preponderance in women; however, men are notably younger at diagnosis.[14] Acute transverse myelitis is the most common form of spinal cord involvement resulting from thoracic or cervico-thoracic lesions.[14]
The antibodies to Ro and La have been recognized as one of the important steps for the diagnosis of SS. The Ro/La system is a heterogeneous antigenic complex formed by three different proteins (52 kDa Ro, 60 kDa Ro and La) and four small RNAs particles. Anti-Ro/SSA are the most prevalent among many autoimmune diseases such as systemic lupus erythematosus (SLE), SS/SLE overlap syndrome, subacute cutaneous LE (SCLE), neonatal lupus and primary biliary cirrhosis, but anti-La/SSB is more associated with SS.[15]
Vasculitis and subsequent axonopathy is the likely etiology in neuropathic disorders. Lymphocytic (T-cell) infiltration of the dorsal ganglia, cryoglobulin-mediated and necrotizing vasculitis, ischemic mechanisms, anti-neuronal antibodies and a direct role of anti-Ro antibodies have all been suggested as possible causes of the nervous system involvement in SS.[9]
Treatment of neuro-Sjogren's syndrome
For the therapy of neuro-Sjogren's syndrome, corticosteroids, other immunosuppressants, plasmapheresis, D-penicillamine, Infliximab and immunoglobulin administration have been reported anecdotally and suggest a favorable therapeutic response.[10] Corticosteroid therapy is preferred for multiple mononeuropathy and multiple cranial neuropathies; favorable improvement of polyradiculoneuropathy and dysesthesias in the painful sensory neuropathy has been observed with intravenous immunoglobulin IVIG therapy.[8] In our series, the improvement with immunosuppressants was significant in 5/6 patients.
The drawbacks of our study were the small sample size, nonavailability of salivary gland scintigraphy which is an important diagnostic tool, and absence of long-term follow-up to ascertain how many of them eventually developed all the features of SS. Despite this, our series raises an important issue about the difficulties in diagnosing SS in Indian patients who present with complex neurological features and positive serology but without fulfilling all the criteria of SS, especially the sicca complex.
Conclusions
Neuro-Sjogren's syndrome with its varied clinical and radiological manifestations can mimic more common disorders and can make diagnosis challenging in the absence of sicca symptoms. However, neurological manifestations often precede sicca symptoms, which themselves may be mild. This warrants a high index of suspicion and investigation with lip biopsy and autoantibody profile for the diagnosis. This study highlights the need to revise the overemphasis of sicca symptoms in various current diagnostic criteria in order to improve early recognition and institution of treatment.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjögren's syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. 2002;61:554–8. doi: 10.1136/ard.61.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gálvez J, Sáiz E, López P, Pina MF, Carrillo A, Nieto A, et al. Diagnostic evaluation and classification criteria in Sjögren's Syndrome. Joint Bone Spine. 2009;76:44–9. doi: 10.1016/j.jbspin.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 3.Gøransson LG, Herigstad A, Tjensvoll AB, Harboe E, Mellgren SI, Omdal R. Peripheral neuropathy in primary sjogren syndrome: A population-based study. Arch Neurol. 2006;63:1612–5. doi: 10.1001/archneur.63.11.1612. [DOI] [PubMed] [Google Scholar]
- 4.Misra R, Hissaria P, Tandon V, Aggarwal A, Krishnani N, Dabadghao S. Primary Sjogren's syndrome: rarity in India. J Assoc Physicians India. 2003;51:859–62. [PubMed] [Google Scholar]
- 5.Malaviya AN, Singh RR, Kapoor SK, Sharma A, Kumar A, Singh YN. Prevalence of rheumatic diseases in India. Results of a population survey. J Indian Rheum Assoc. 1994;2:13–7. [Google Scholar]
- 6.Morbini P, Manzo A, Caporali R, Epis O, Villa C, Tinelli C, et al. Multilevel examination of minor salivary gland biopsy for Sjogren's syndrome significantly improves diagnostic performance of AECG classification criteria. Arthritis Res Ther. 2005;7:R343–8. doi: 10.1186/ar1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delalande S, de Seze J, Fauchais AL, Hachulla E, Stojkovic T, Ferriby D, et al. Neurologic manifestations in primary Sjögren syndrome: A study of 82 patients. Medicine (Baltimore) 2004;83:280–91. doi: 10.1097/01.md.0000141099.53742.16. [DOI] [PubMed] [Google Scholar]
- 8.Mori K, Iijima M, Koike H, Hattori N, Tanaka F, Watanabe H, et al. The wide spectrum of clinical manifestations in Sjögren's syndrome-associated neuropathy. Brain. 2005;128:2518–34. doi: 10.1093/brain/awh605. [DOI] [PubMed] [Google Scholar]
- 9.Galbussera A, Tremolizzo L, Tagliabue E, Ceresa C, Cilia R, Ruffmann C, et al. Third cranial nerve palsy? Look for a sicca syndrome. J Neurol Sci. 2007;253:88–9. doi: 10.1016/j.jns.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 10.Chu K, Kang DW, Song YW, Yoon BW. Trochlear nerve palsy in Sjögren's syndrome. J Neurol Sci. 2000;177:157–9. doi: 10.1016/s0022-510x(00)00344-0. [DOI] [PubMed] [Google Scholar]
- 11.Urban PP, Keilmann A, Teichmann EM, Hopf HC. Sensory neuropathy of the trigeminal, glossopharyngeal, and vagal nerves in Sjögren's syndrome. J Neurol Sci. 2001;186:59–63. doi: 10.1016/s0022-510x(01)00501-9. [DOI] [PubMed] [Google Scholar]
- 12.Hadithi M, Stam F, Donker AJ, Dijkmans BA. Sjögren's syndrome: An unusual cause of Bell's palsy. Ann Rheum Dis. 2001;60:724–5. doi: 10.1136/ard.60.7.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander E. Central nervous system disease in Sjogren's syndrome: New insights into immunopathogenesis. Rheum Dis Clin North Am. 1992;18:637–72. [PubMed] [Google Scholar]
- 14.Williams CS, Butler E, Roman GC. Treatment of myelopathy in Sjogren syndrome with a combination of Prednisone and Cyclophosphamide. Arch Neurol. 2001;58:815–9. doi: 10.1001/archneur.58.5.815. [DOI] [PubMed] [Google Scholar]
- 15.Franceschini F, Cavazzana I. Anti-Ro/SSA and La/SSB antibodies. Autoimmunity. 2005;38:55–63. doi: 10.1080/08916930400022954. [DOI] [PubMed] [Google Scholar]