

Abstract
The spectrum of chronic dysimmune neuropathies has widened well beyond chronic demyelinating polyradiculoneuropathy (CIDP). Pure motor (multifocal motor neuropathy), sensorimotor with asymmetrical involvement (multifocal acquired demylinating sensory and motor neuropathy), exclusively distal sensory (distal acquired demyelinating sensory neuropathy) and very proximal sensory (chronic immune sensory polyradiculopathy) constitute the variants of CIDP. Correct diagnosis of these entities is of importance in terms of initiation of appropriate therapy as well as prognostication of these patients. The rates of detection of immune-mediated neuropathies with monoclonal cell proliferation (monoclonal gammopathy of unknown significance, multiple myeloma, etc.) have been facilitated as better diagnostic tools such as serum immunofixation electrophoresis are being used more often. Immune neuropathies associated with malignancies and systemic vasculitic disorders are being defined further and treated early with better understanding of the disease processes. As this field of dysimmune neuropathies will evolve in the future, some of the curious aspects of the clinical presentations and response patterns to different immunosuppressants or immunomodulators will be further elucidated. This review also discusses representative case studies.
Keywords: Multifocal motor neuropathy, multifocal acquired demyelinating sensory and motor, chronic inflammatory demyelinating neuropathy, distal acquired demyelinating predominately sensory
Introduction
Dysimmune neuropathies are etiologically heterogeneous disorders affecting the peripheral nervous system, having diverse clinical presentations [Table 1]. The underlying causes encompass a variety of benign and neoplastic syndromes. Early recognition of the immunologic disturbance or malignancy with appropriate diagnostic testing is necessary to initiate potentially effective therapies.
Table 1.
Chronic immune-mediated neuropathies
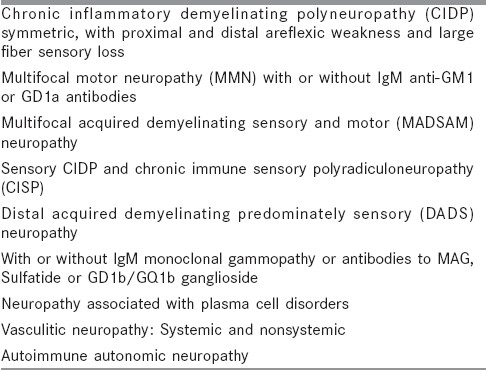
Chronic inflammatory demyelinating neuropathy (CIDP) is a chronic progressive or relapsing, clinically symmetric, sensorimotor disorder with proximal and distal involvement. It is considered to be the chronic equivalent of acute inflammatory demyelinating neuropathy (AIDP). It is characterized by albuminocytological dissociation in the cerebrospinal fluid analysis and demyelinating neuropathy with partial conduction blocks on electrophysiological evaluation. Treatment options include corticosteroids, IV Ig (short term), plasmapheresis or other immunosupressants. Apart from this idiopathic CIDP which is the prototype of dysimmune neuropathies, there are several polyneuropathies [Table 1] which share some of the features of CIDP but with distinctive clinical, electrophysiological and immune attributes and different responses to various treatment options.
This review discusses evaluation, clinical aspects, differential diagnosis, pathophysiology and treatment of disease states with abnormal immunoglobulin production that are associated with peripheral neuropathy, beyond the prototype chronic inflammatory demyelinating neuropathy (CIDP). Representative clinical case studies are provided.
Multifocal Motor Neuropathy with Conduction Blocks
Multifocal motor neuropathy (MMN) is an acquired immune-mediated neuropathy characterized by chronic or stepwise progressive asymmetrical limb weakness without sensory deficits.
Early clinical descriptions of patients having chronic asymmetric, distal motor neuropathy without sensory loss and proximal multifocal persistent conduction blocks were given by Roth[1] and Chad.[2] The term “multifocal motor neuropathy” was coined in 1988 by Pestronk et al.,[3] who first recognized the association of MMN with anti-GM1-IgM antibodies and the responsiveness to immune-modulating therapies.
Clinical Features and Disease Course MMN is a rare disease, with an estimated prevalence of 1–2/100,000 individuals. It is more frequent in men than in women, with an approximate ratio of 3:1. The mean age at disease onset is 40 years. Almost 80% of the patients develop first symptoms between 20 and 50 years of age.[4–6] Clinically, MMN is characterized by slowly progressive or stepwise progressive, asymmetric and distal involvement related to individual peripheral nerves. The upper limbs are usually affected earlier, and this is more severe than the lower limbs. Initial involvement tends to be distal, and the most common presenting symptom is wrist drop and impaired grip strength. Only 5–10% of all cases of MMN manifests with proximal muscle weakness.[7] Muscle atrophy is often mild in the early stages, but tends to become prominent during the course of the disease.[8] Other symptoms comprise of fasciculations and muscle cramps seen in about 50% of the patients, while myokymia has only been reported occasionally.
Another feature of MMN is the absence of sensory symptoms. Only a few patients complain of discrete paraesthesiae or numbness during the course of the disease, and a minor loss of vibration sense has been documented in 20% of the subjects.[5] Tendon reflexes from the paretic muscles are usually reduced but may be normal or, rarely, brisk. In the latter case, differentiation from amyotrophic lateral sclerosis or lower motor-neuron disease can be difficult. Cranial nerve involvement is uncommon, except for the hypoglossal nerve in few cases.[9]
Most patients develop a slowly progressive disease course, and the degree of disability correlates with the overall duration of the disease.[10] Uncommonly, other evolutions of MMN showing acute deterioration, stepwise progression as well as spontaneous remissions have been described.[11]
Pathophysiology of multifocal motor neuropathy
The electrophysiological hallmarks of MMN are conduction blocks (CB). These are supposed to be causally related to muscle weakness. However, patients exist who present with clinical symptoms typical for MMN but in whom CBs cannot be detected by routine nerve conduction studies. In such cases, very proximal or distal CBs inaccessible to standard neurography might be present. Interestingly, the majority of nerve-conduction studies in MMN demonstrate significant improvement of CBs after treatment with IVIgs, although muscle strength in these patients rarely recovers to normal.
Beyond focal demyelination, generalized axonal dysfunction might be present in MMN.[12] Pathological and electrophysiological findings have highlighted the functional role of axonal disintegration and impaired axon–myelin interaction. Anti-GM1 antibodies are found in 20–80% of the patients suffering from MMN, but their causal relationship is speculative.[13]
Electrophysiological findings
The most prominent electrophysiological features in MMN are multifocal, persistent, partial CBs present in the motor but not in the sensory nerve fibers and located outside the common entrapment sites. However, consensus on the required magnitude of amplitude or area reduction that unambiguously defines partial CB has not yet been reached. This is mainly due to the fact that besides CB, several other mechanisms can lead to significant Compound Muscle Action Potential (CMAP) reduction (pseudo-CB). Presently, the commonly applied cut-off level of 50% CMAP decline (amplitude or area) is the most validated electrophysiological criterion of partial CB.[14] However, the strict appliance of 50% limit has its pitfalls if CMAP amplitudes are below 20% of the normal value. In that case, potentials are often too polyphasic to allow accurate quantification. The relatively restrictive American and European electrophysiological criteria aim to avoid confusion between real CB and temporal dispersion (TD).This approach, however, may underdiagnose MMN, which represents a potentially treatable neuropathy[15] Moreover, CB is a dynamic phenomenon in time frame.[16] Hence, repeated evaluations increase the yield. Subtle focal CB can be unearthed using the so-called “inching technique,” wherein several nerve sites, with an inter stimulation distance of 10–15 mm, are stimulated sequentially.[17]
Detection of proximal CB is technically challenging and can be helped by the application of transcutaneous magnetic coils or high-voltage stimulators.[18,19] F-waves provide information on the integrity of a peripheral nerve over its entire course, but the F-wave persistence depends on several other factors such as axonal integrity, and its reduction does not necessarily indicate proximal CB.[20] Newer tests like “Magnetic fatigue test”[21] and triple stimulation technique[22] are expected to help in the analysis of the proximal segment.
Although CB clearly is an important hallmark of MMN, whether its presence is mandatory for the diagnosis of MMN is still under debate.[23] About 30 cases of MMN with typical clinical presentation and a good response to IVIg but without CB have been reported so far.[24] In a recent retrospective analysis, patients with and without CB showed similar clinical features and a comparable response to IVIg treatment after a long median follow-up of 7 years.[25] Final appraisal of the existence of CB-negative MMN is hampered, in that it is not clear whether these subjects really never had CB or whether CB merely disappeared over time due to secondary axonal degeneration with reduction of the distal CMAP amplitudes.[26]
Case study: Efficacy of IVIg in the Absence of Conduction Blocks
SR, a 62-year-old medical practitioner, developed progressive weakness of the right upper limb over a period of 4 months. It began initially in the thumb and the fore finger and progressed to weaken all the fingers and forearm muscles, with mild wasting. At this stage, the left hand also got weaker in a similar pattern. Electrophysiology detected denervation and reinnervation of the distal upper limb muscles with milder forearm involvement. The paraspinal muscles were normal. No conduction blocks were identified on repeated detailed examinations. Anti-GM1 antibody was not detected. A provisional diagnosis of motor neuron disease was made and he underwent physiotherapy. At the end of 1 year and 4 months, he had not developed any bulbar involvement or upper motor neuron signs, and the daily activities were worsening. A trial of IVIg was given with the presumption of MMN without CB, with remarkable improvement in the weakness over 6–8 weeks. He took further courses of maintenance IVIg and remained well for the next 2 years, at which stage he succumbed to a myocardial infarction.
The case highlights the efficacy of IVIg in the absence of CB and anti-GM1 positivity.
Laboratory findings
The most common laboratory findings in MMN are IgM serum antibodies against the ganglioside GM1, which can be detected at high titers in 30–80% of the patients. The reported variations in the incidence of GM1 antibodies are probably related to the different enzyme-linked immunosorbent assays used in the different studies as well as heterogeneous control populations.[27] Besides GM1 antibodies, immunoreactivity against other axon or myelin components such as the glycolipids GD1a or GM2 can be infrequently found.[28] Similar to CB, anti-GM1 antibodies are not specific for MMN. They are also present in 5–10% of the patients with motor neuron disease (MND), other immune-mediated neuropathies (Guillain Barre Syndrome (GBS), Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)) and even in healthy individuals, although GM1 titers are usually lower under these conditions compared with MMN.[29] Interestingly, anti-IgG GM1-antibodies are frequently found at high concentrations in patients with multifocal acquired demyelinating sensory and motor (MADSAM) neuropathy,[30] GBS and MND. Taken together, the detection of anti-GM1-IgM antibodies supports the diagnosis of MMN, while a negative finding does not exclude the disease.
Most routine laboratory parameters in MMN are normal. Muscle creatine kinase may show mild elevation in approximately two-thirds of the patients. Analysis of the cerebrospinal fluid in most cases shows a discrete increase in overall protein concentration (up to 80 mg/dl) with normal cell counts.[31] At times, serum protein electrophoresis reveals elevated polyclonal antibody formation, while monoclonal peaks typical for IgM gammopathy are generally absent.[32]
About 40–50% of the patients with MMN show hyperintense signals on T2-weighted magnetic resonance imaging (MRI) or contrast-enhanced T1 sequences of the brachial plexus.[33] The pattern of signal alterations closely correlates with the distribution of muscle weakness, and might co-localize with CB. MRI may help differentiate MMN from CIDP and MND.[34]
Nerve biopsy
Biopsies taken from the sensory nerves (e.g., sural nerve) are naturally not helpful for the diagnosis of MMN and should only be performed if significant sensory deficits are present and when differential diagnosis of CIDP, Lewis-Sumner syndrome or vasculitis has to be considered. Hence, only a few reports on sensory nerve biopsies in MMN exist, and those describe either normal findings or nonspecific signs of mild axonal degeneration, demyelination or both. Tissue samples taken from the motor nerves of MMN patients are likewise rare. Auer et al. reported thinly myelinated axons and the formation of onion bulbs at the site of the suspected CB, which typically indicate simultaneous de- and remyelination.[35] In another study, axonal degeneration outweighed myelin pathology, and onion bulb formation as well as para- and internodal demyelination were absent. In contrast to CIDP, inflammatory cells invading the nerve are only sporadically found in MMN, underlining that different disease mechanisms are operational.[35]
Therapy of multifocal motor neuropathy
As MMN is believed to be an immune-mediated disease, various immunomodulatory treatment strategies have been applied to date in MMN patients. In contrast to CIDP and Lewis-Sumner syndrome, studies have demonstrated that glucocorticoids and plasma exchange are ineffective in MMN. In fact, these modalities worsen the symptoms in up to 20% of the MMN patients, underlining the differences in pathophysiological mechanisms.[36,18]
Presently, IVIgs are regarded as first-line therapy, and their efficacy in MMN has been proven in four large double-blind, placebo-controlled trials. In addition, four retrospective trials also confirmed that IVIg is initially effective in 70–86% of the patients, and that most individuals require periodic maintenance therapy for several years.[37] Whether the subcutaneous route of IVIg administration is advantageous compared with the regular intravenous infusions with respect to steady IVIg plasma concentrations, patients′ quality of life or cost-effectiveness needs further evaluation.[38]
The precise mechanism of action of IVIg in MMN is unclear at present.[38] It is also not known whether patients with high titers of anti-GM1 antibodies respond better to IVIg compared with those with lower titers. The clinical effect of IVIg is usually impressive, and muscle strength improves substantially within the first week of treatment. Chronic paresis and muscle atrophy may not recover satisfactorily after IVIg application in most of the cases. While anti-GM1 antibody titers are not affected by IVIg and, thus, are not suitable as therapeutic markers, disappearance of partial CB often parallels clinical improvement.[39] The common IVIg dose at the beginning of the disease is 2 g/kg body weight given on 2–5 consecutive days. However, the treatment effect declines after several weeks. Therefore, it is important to find an applicable maintenance regime with individualized IVIg doses (e.g.. 0.4 g/kg IVIg once weekly or 1–2 g/kg IVIg in monthly intervals) in order to optimize the cost-to-benefit ratio.[40] The efficacy of IVIg decreases after several years of treatment in most patients, necessitating higher dosage or shortened infusion intervals to stabilize the symptoms.[40,41] The recent observation that higher doses of IVIg might be superior at the initial stage[42] and be able to prevent secondary axonal degeneration or promote remyelination[42] awaits confirmation.
Soon after the initial description of MMN, cyclophosphamide was tested for this indication in several small uncontrolled trials. Taken together, high doses of cyclophosphamide seem to have a moderate effect, especially when given intravenously, while lower oral doses do not influence disease progression.[43] Given its unfavorable risk-to-benefit ratio, cyclophosphamide is currently recommended only if IVIg is not sufficiently effective Immunosuppressive agents such as Azathioprine, Methotrexate, Cyclosporine A, Mycophenolate Mofetil and Β-Interferon have occasionally been used in MMN, but controlled trials on these therapies are not available.[44] Data concerning the efficacy of the monoclonal antibody Rituximab, which targets the CD20 molecule on B cells and therefore reduces pathological autoantibody levels in MMN, are inconclusive at present.[45]
Multifocal acquired demyelinating sensory and motor neuropathy (Lewis-Sumner syndrome)
Originally described in 1982 as a mononeuritis multiplex, MADSAM or Lewis-Sumner syndrome is characterized by its striking multifocal presentation.[46] MADSAM is a slowly progressive demyelinating neuropathy affecting the upper limbs more than the lower limbs, and is associated with proximal as well as distal weakness. It is most often seen in females, generally manifesting in the 4th decade onwards. Many patients report of painful sensory symptoms in the extremities.
Similar to CIDP, the cerebro spinal fluid (CSF) protein content is increased in 60–70% of the patients with MADSAM.[47] This increase in CSF protein is more pronounced as compared with MMN, probably due to the involvement of the proximal nerve roots. IgM antibodies against GM1 are rarely detected in these patients. Although MADSAM has similar laboratory and electrophysiological characteristics, it is differentiated from CIDP due to its conspicuous asymmetrical involvement of multiple nerves and from MMN by involvement of the sensory nerves [Table 2]. There are anecdotal reports of patients who present initially with pure motor syndrome like MMN but then develop sensory symptoms years later. Occasional patients with MMN show very subtle sensory abnormalities, making it difficult to decide whether or not these changes are sufficient for the diagnosis of MADAM. Sensory involvement is more evident in electrophysiological studies with proximal stimulation. Most patients tend to show some decrease in the sensory amplitudes. Sural nerve biopsies show demyelination in a remarkably high number of patients with MADSAM.
Table 2.
Clinical and laboratory differences in neuropathies and motor neuron disease
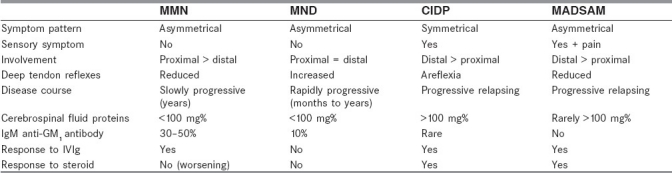
About 60–70% of the patients show improvement after the administration of IVIg after several months. Unlike in MMN where steroids are remarkably ineffective or sometimes deleterious, 50–70% of the patients with MADSAM show improvement with corticosteroids.[48,49] Hence, it is imperative to diagnose this distinct clinical entity for better management of the patients.
Case Study: Multifocal Acquired Demyelinating Sensory and Motor Neuropathy with Hypoglossal Nerve Involvement
A 35-year-old female presented with weakness of her ring and little fingers of the right hand in 2001. Ten days later, she started noticing slipping of footwear. After 1 month, her grip with right hand and then of the left hand became weak. Weakness gradually progressed over a period of the next 8 months, when she could not walk without support. She also developed mild wasting in the hands, forearms and calves. She had tingling numbness in all her extremities and in the perioral region and difficulty walking in the dark. Examination confirmed distal weakness associated with mild wasting. She had tremors in both hands. She had total areflexia, and sensory examination revealed asymmetrical proprioceptive loss in all four limbs. The tongue showed severe weakness and atrophy with resultant dysarthria.
She had normal blood counts with ESR of 5 mm at 1 h. The CSF examination was normal. Ultrasonography abdomen and X-ray chest were normal. Electrophysiological evaluation showed widespread, motor more than sensory, multifocal demyelinating polyneuropathy with secondary axonal changes affecting all four limbs, with evidence of persistent motor conduction blocks. There was hypoglossal nerve involvement, with the tongue showing chronic partial dennervation.
This case highlights MADSAM presentation with cranial nerve affection. The affectation of the hypoglossal nerve is noteworthy, emphasizing the clinical overlap, as it is documented more with MMN than with MADSAM. Trial of oral prednisolone showed marginal benefit while subsequent plasmapheresis gave her partial relief to. She was put on immunomodulation and remained stable for the available follow-up of 1 year.
Distal acquired demyelinating sensory neuropathy
This variant of CIDP is characterized by symmetric, exclusively distal sensory and motor deficits in the hands and feet. Such patients complain of numbness or tingling in the hands and feet and sometimes notice weakness in these regions. On examination, the physician finds distal sensory loss with either no evidence of weakness or weakness that is exclusively distal. There is no weakness in the facial, neck or proximal arm or leg muscles. Some patients in this group also have gait unsteadiness and tremor.
Motor nerve conduction studies in this group of patients reveal demyelination, and they often have markedly prolonged distal motor latencies. The most striking laboratory finding in this group of patients is positivity of an IgM kappa monoclonal protein in the serum and myelin-associated glycoprotein (MAG) in 50% of the patients. This group of patients has been referred to as a subgroup of CIDP with IgM kappa/MAG antibodies.[50]
It is important to identify this DADS neuropathy subset of patients because they have a different response to therapy compared with typical CIDP and the other chronic acquired demyelinating variants. DADS neuropathy patients are very resistant to therapy with prednisone, IVIg, plasmapheresis or chemotherapy. Some patients may show mild improvement over many years of therapy, but it is often difficult to detect. This is in contrast to most MADSAM and MMN patients, in whom beneficial response to therapy can usually be seen within a month or two after initiating treatment. Therefore, while attempting a course of immunosuppressive therapy in patients with DADS neuropathy, patients should be counseled at the onset that they may not improve, or improvement may take many months, and the benefit may be modest.
Chronic immune sensory polyradiculoneuropathy
Involvement of nerve roots is common in varieties of both acute (AIDP) and chronic (CIDP) inflammatory demyelinating polyradiculoneuropathies, but generally the distal nerve affection is also present in these diseases. CISP is a syndrome likely due to immune-mediated demyelination predominantly affecting the dorsal roots proximal to the dorsal root ganglion. Supporting evidence for this entity is the presence of large fiber sensory loss, which is confirmed pathologically and documented on quantitative sensory testing. These patients present with predominant sensory ataxia and loss of reflexes. Nerve conduction study is normal while abnormalities are detected on somatosensory-evoked potentials (SSEP).[51] CSF examination shows elevated protein levels. MRI of the spine showing thickened lumbosacral rootlets is a characteristic finding. Inflammatory demyelinating changes on the lumbar rootlet biopsies and favorable response to immunomodulating treatment also suggest immune-mediated pathology in this syndrome.
Acute or subacute progressive sensory ataxia is also seen in Sjögren's syndrome, paraneoplastic sensory ganglionopathy, patients with high titers of anti-MAG antibodies and some patients with IgM-monoclonal gammopathy of unknown significance (MGUS). In a majority of patients, establishing the etiology is not possible, and this group of patients may represent idiopathic immune-mediated sensory ganglionopathy, which generally responds to some modality of immunosuppression.
Case Study: Treatable Sensory Ataxia with Normal Nerve Conduction Study
TM, a 40-year-old male, presented with tingling numbness of 6 years duration that initially started in the left lower limb, followed by right lower limb, over a period of 6 months. The patient then started having occasional slipping of foot wear and the sensory complaints started ascending up to the thighs. The patient was investigated for the same and was found to have marginally low serum B12 levels, which were corrected with injectable vitamin B12, without any significant improvement in the symptoms. Since the last 1 year, the patient's symptoms progressed rapidly to an extent that he started having severe difficulty in maintaining balance while standing, which used to get aggravated in the dark. He had no complaints in the upper limbs.
On examination, he was found to have loss of joint position sense and vibration up to the anterior superior iliac spine in the lower limbs while decrease in pain and temperature sensation up to both ankles. The patient had areflexia in both the lower limbs, while only the right triceps reflex was present in the upper limb. He had only minimal weakness in the lower limbs. He had no cerebellar signs. He had gait ataxia with severely positive Romberg sign. His nerve conduction study was within normal limits while SSEP in both upper and lower limbs was abnormal, suggestive of posterior column involvement. CSF examination showed proteins of 123 mg% with 10 cells. All blood investigations, including serum B12, anti-ro, anti-la, ESR, ANA, dsDNA, serum protein electrophoresis and routine biochemical parameters, were normal. His MRI spine on a 3 Tesla system showed characteristic lumbosacral root enlargement with contrast enhancement [Figure 1]. The patient has been recently started on immunomodulation.
Figure 1.
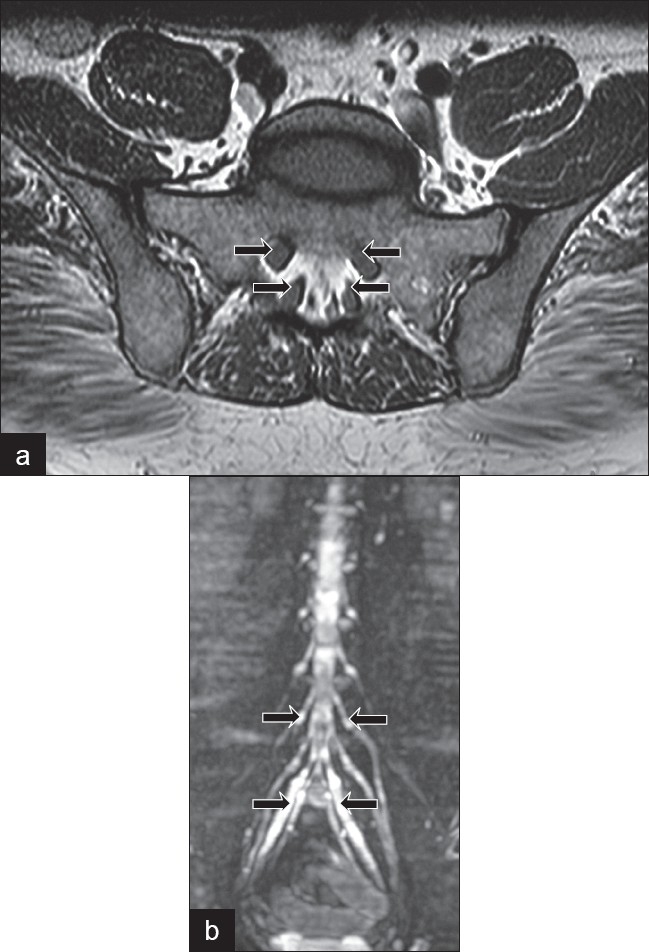
(a) Magnetic resonance imaging axial scan at L5-S1 (arrows show the extreme thickening of the sacral nerve roots) (b) Whole body substraction magnetic resonance image showing tubershaped lumbosacral root enlargement from the L1 to the S1 levels (most prominent in S1 root as shown by the arrow)
This case demonstrates selectively proximal demyelinating neuropathy resulting in sensory ataxia.
Monoclonal gammopathy of unknown significance
Patients undergoing evaluation for chronic peripheral neuropathy need to be screened for the presence of a monoclonal protein.[52] Monoclonal protein is produced by a single clone of plasma cells and is usually composed of four polypeptides: two identical heavy chains and two light chains.
The pathophysiological relationship between the M protein and the neuropathy is often obscure, but some M protein may have antibody-like properties directed against components of myelin or axolemma. Finding of M protein among patients with neuropathy lead to the discovery of underlying primary disorders such as primary amyloidosis, multiple or osteosclerotic myeloma, plasma cell dyscrasias, macrogloulinemia, Castleman's disease, cryoglobulinemia and lymphoma. In two-thirds of the patients, no detectable underlying disease is found and they are described as having MGUS.[53] The risk of progression of MGUS to a malignant plasma cell proliferative disorder is about 1% per year.[54] Hence, these patients require regular clinical and laboratory follow-up.
Approximately 5% of the patients with MUGS have an associated polyneuropathy. Symptoms begin later in life, with median age of onset in the sixth decade, appear insidiously and progresses slowly over months to years. Males are predominantly affected. The most common presentation is a distal symmetrical sensorimotor polyneuropathy. Cranial nerves and autonomic functions are generally preserved. The lower limbs are involved earlier and to a greater extent than the upper limbs. In approximately half of the patients, a polyradiculoneuropathy occurs, which shares clinical and laboratory features of CIDP. Gait ataxia and upper limb postural tremor can be prominent in patients with IgM MGUS.
Electrophysiological studies show evidence of demyelination, particularly in IgM MGUS, in patients who have a predilection for distal demyelination. Sensory Nerve Action Potentials (SNAPs) are reduced in amplitude or are unobtainable. A small number of patients, particularly with IgG MGUS, has electrophysiological evidence of pure axonal neuropathy.
Routine serum electrophoresis frequently lacks the sensitivity required to detect small M proteins. Immunoelectrophoresis or immunofixation is required to detect small amounts of protein and to confirm the monoclonal nature and to characterize light and heavy chains. If monoclonal spike exceeds 1.5 g/dl, a bone marrow aspirate and biopsy should be performed to differentiate malignant plasma cell dyscrasias from MGUS.
When the higher frequency of MGUS in older patients is considered, the causal relationship between MGUS and this type of neuropathy becomes less clear.[55] Elevation of CSF protein is common, sometimes in excess of 100 mg/dl. In at least 50% of the patients with IgM MGUS neuropathy, the IgM monoclonal protein demonstrate reactivity against MAG.[55] Ultrastructurally, the myelin lamellae show a widened periodicity (myelin splitting), which is considered the pathological hallmark of anti-MAG antibodies.[56]
Approximately 15% of the patients with IgM MGUS neuropathy have autoantibodies against GD1b and GQ1b. These patients predominantly have sensory ataxic neuropathies.[57] Patients who have disialosyl ganglioside IgM antibodies and cold agglutinins present with a chronic sensory ataxic neuropathy, areflexia and fixed or relapsing–remitting ophthalmoplegia.
Treatment
The optimal treatment of MGUS neuropathy has not been established. Immunomodulatory treatments have resulted in serious adverse effects in half of the patients. However, in general, the more closely the neuropathy fulfills the criteria for CIDP, the more likely patients will respond to immunomodulatory therapies. Patients with a CIDP-like picture and IgG gammopathy should be treated like patients with CIDP without gammopathy.
However, in IgM-associated neuropathies, the role of these therapies is less-clear and may require aggressive immune interventions like pulse cyclophosphamide.[58] The purine anologue, Cladirabine, resulted in prolonged remission in one patient with IgM neuropathy.[59] Rituximab has been reported to be effective in some cases of anti-MAG neuropathy.[60]
Case Study: Monoclonal Gammopathy of Unknown Significance with Asymmetrical Neuropathy
RA, a 64-year-old man, initially developed weakness and wasting of the left upper limb of 2 years and 5 months duration and inability to walk on toes and heels evolved over 6 months. Examination showed an asymmetric sensorimotor neuropathy with distal involvement. The electrophysiology showed widespread multifocal motor more than sensory, upper limbs more than lower limbs demyelinating polyneuropathy. Serum protein electrophoresis showed M band (0.9) consistent with the diagnosis of MGUS. No skeletal lesions or organomegaly was observed. The above case highlights the occurrence of asymmetric neuropathy in a case of MGUS.
Plasma Cell Proliferative Disorders
Waldenstrom's macrogloulinemia
It is characterized by proliferation of malignant lymphoplasmacytic cells in the bone marrow and lymph nodes that secrete IgM monoclonal spike of more than 3 g/dl. It affects elderly males and manifests with fatigue, anemia, bleeding and hyperviscosity syndrome. One-third of the patients have chronic symmetrical, predominantly sensory, polyneuropathy.
Paresthesias in the distal leg mark the onset, with mild motor weakness following later. Anti-MAG antibodies are present in 50% of the patients who have neuropathy.[60] Prolongation of P100 latency on visual-evoked response in these patients is suggestive of subclinical central involvement.[61]
Nerve biopsy findings are indistinguishable from those seen in IgM MGUS neuropathies. Response to treatment (plasmapheresis and Chlorambucil) appears less consistent than in IgM MGUS-related neuropathy. Treatment with autologous stem cell transplantation has been rarely shown to have a positive response even in patients resistant to other therapies.
Multiple myeloma
Polyneuropathy occurs in approximately 5% of the patients with multiple myeloma. Most patients present with mild distal sensorimotor polyneuropathy. Painful dysesthesias, preferential involvement of small fiber sensory nerves, autonomic dysfunction and carpal tunnel syndrome are suggestive of amyloid neuropathy.
Nerve conduction studies and sural nerve biopsies are consistent with primary axonal process causing secondary loss of myelinated fibers. Treatment of underlying myeloma does not improve neuropathy, and polyneuropathy also may be caused by medications that are part of the therapeutic regimen for multiple myeloma, such as Thalidomide or Bortezomib]
Osteosclerotic myeloma
In this disorder, plasma cell proliferation occurs as a single or multiple plasmacytomas that manifest as sclerotic bone lesions. It occurs in only 3% of the patients with myeloma; however, 85% of these patients present with peripheral neuropathy. Neuropathy in osteosclerotic myeloma is different from that associated with multiple myeloma in various aspects. It occurs at an earlier age and mostly in men; it is demyelinating, predominantly motor, with a striking resemblance to CIDP. Patients with multiple bone lesions are treated with radiation therapy along with steroids and Melphalan. Substantial improvements in both neurological and systemic features occur in some patients, but the response may take a long time.
Polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes syndrome
Polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes syndrome (POEMS syndrome) is an uncommon cause of demyelinating and axonal sensorimotor polyneuropathy. The acronym enumerates the main components of the syndrome. One or more of these could dominate the clinical features. Neuropathy tends to be symmetrical and sensorimotor, with proximal and distal areflexic involvement. Electrophysiology confirms the primary demyelinating nature of the process with secondary axonal involvement. Prolongation of distal motor latencies and conduction blocks are less common than in typical CIDP. These patients have a favorable prognosis, with median survival up to 13.8 years in patients who do not receive peripheral blood stem cell transplant.[62]
Case Study: Chronic Demyelinating Polyradiculoneuropathy Heralding Recurrence of Osteosclerotic Myeloma
PK, a 35-year-old male patient, presented with complaints of severe back ache in 2005, which lasted for 8–10 months. Investigations detected solitary plasmacytoma at the D4 level. It was operated and a metallic loop was fixed. Serum protein electrophoresis showed M band up to level of 4.5. The patient received six cycles of radiotherapy and chemotherapy till 2008, after which the M band came to under 2 and the patient was asymptomatic. Since November 2010, he started having pain in the lower limbs with tingling and numbness, with progressive difficulty in walking. Electrophysiological evaluation showed the presence of sensorimotor demyelinating neuropathy characteristic of CIDP. He was started on corticosteroids, but no response was seen even after 2 weeks of therapy. His repeat MRI study showed multiple hypointense lesions in the cervical, dorsal and lumbar vertebrae, suggestive of relapse of myeloma. This was later confirmed with whole-body Positron Emission Tomography (PET) scan. The M band was up to 2.5. Bone marrow biopsy showed presence of 15% of plasma cells. Bence jones proteins were negative. The patient was restarted on chemotherapy with improvement in neuropathy (Courtesy Dr. R.K. Singh).
Primary (light chain) amyloidosis
Amyloidosis is not a single pathologic entity but describes several disorders characterized by extracellular deposition of misfolded proteins that aggregate as insoluble fibrils in various soft tissues.[63]
Unlike multiple myeloma in which the most common light chain is kappa, lambda light chains predominate in primary amyloidosis, with the ratio of kappa to lambda light chain of 1:3.[64] Sensorimotor axonal neuropathy with prominent autonomic nervous system involvement may be observed in up to 17% of the patients. Predominant proximal motor involvement is not uncommon. The median duration of survival ranges from 2 to 3.8 years. Melphalan and steroid combinations are shown to prolong survival.
Vasculitic neuropathy (systemic and nonsystemic)
Peripheral neuropathy is an important and, at times, presenting, clinical feature of the vasculidities. The following table [Table 3] charts the various vasculitic disorders and associated neuropathic presentations in decreasing order of frequency.
Table 3.
Vasculitic neuropathies
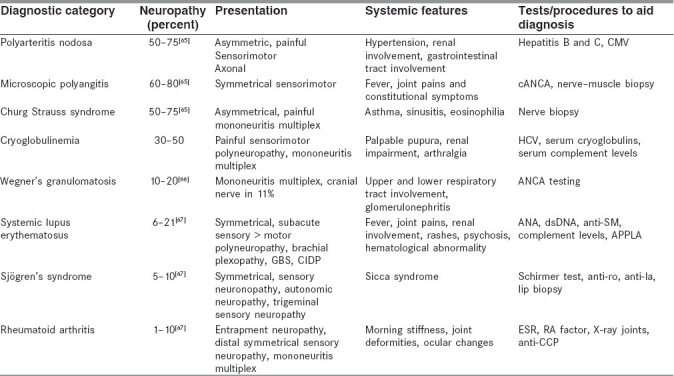
Analysis of data of patients with systemic vasculitic diseases at a tertiary medical research center in Mumbai concurred that neuropathy can be the presenting feature or an accompaniment of an established syndrome.
Neuropathy was very common in microscopic polyangitis and in hepatitis B virus-induced polyarteritis nodosa (70–80%) It was less frequent in diseases such as sarcoidosis, systemic lupus erythematosus and rheumatoid arthritis (up to 10%). Symmetrical sensorimotor axonal neuropathy was most common, except for Churg Strauss syndrome, where it tended to be asymmetrical. In sarcoidosis, systemic lupus erythematosus and rheumatoid arthritis, peripheral nervous system involvement was seen at the later stages (personal communication: Dr. S.N. Amin). In another study of 20 patients from Mumbai, with biopsy-proven vasculitic neuropathies, syndromic diagnosis could be achieved in only about half of the patients at the time of presentation.[68]
Nonsystemic vasculitis
These patients are often seen first by neurologists and have definite nerve biopsy-proven vasculitis without any evidence of underlying disease, central nervous system vasculitis or evidence of vasculitis affecting any other part of the body. Most common presentation is a mononeuritis multiplex. This syndrome has an indolent course and relatively good prognosis as compared with systemic vasculitis. A study of 16 patients with vasculitic neuropathy from a tertiary research center in south India showed the presence of nonsystemic vasculitis in nine patients, of which seven showed complete recovery while two were left with minimal disability.[69]
Treatment
Oral or intravenous pulse cyclophosphamide along with corticosteroids generally improves the sensory symptoms over a few weeks, and motor symptoms take longer time to recover. Earlier diagnosis and treatment definitely influences prognosis favorably. The residual deficits in these patients range from 20 to 80%.
Chronic demyelinating polyradiculoneuropathy associated with malignancy
The association of CIDP with malignant disease is rare, but has been described with several malignancies. Paraneoplastic polyneuropathy is usually associated with small cell carcinoma of the lung[70] and neoplasm of the ovary or uterus.[71] Malignancies associated with CIDP include Hodgkin's lymphoma,[72] carcinomas of the colon, pancreas and cholangiocarcinomas,[72] carcinomas of the larynx, lung, hepatocellular carcinoma[71] and malignant melanoma.
Case Study: Hodgkin Lymphoma with Demyelinating Neuropathy
SP, a 33-year-old male patient, presented with tingling numbness of palms and soles of 15 days duration. One week later, he started having calf pains and fatigability followed by difficulty in climbing stairs and getting up from the sitting position. His handgrip also became weak. The patient was a chronic smoker. On examination, he had mild right-sided lower motor neuron facial palsy with proptosis and moderate proximal muscle weakness in both the girdles. Sensory examination revealed reduced pinprick, temperature and vibration in both palms and sole with generalized areflexia. General examination showed presence of swelling in the right inguinal region. The patient's routine blood counts and biochemical tests were within normal limits. Electrophysiological study showed the presence of sensorimotor demyelinating neuropathy. CSF examination showed proteins of 140 mg%, cells 6 with elevated IgG levels. Serum protein electrophoresis did not show any M band. Excision biopsy of the inguinal swelling was performed and histopathological examination showed the presence of Reed Sternberg cells suggestive of Hodgkin lymphoma of the nodular sclerosis type [Figure 2]. The patient showed a very good response to IVIg (courtesy Dr. J.A. Lalkaka).
Figure 2.
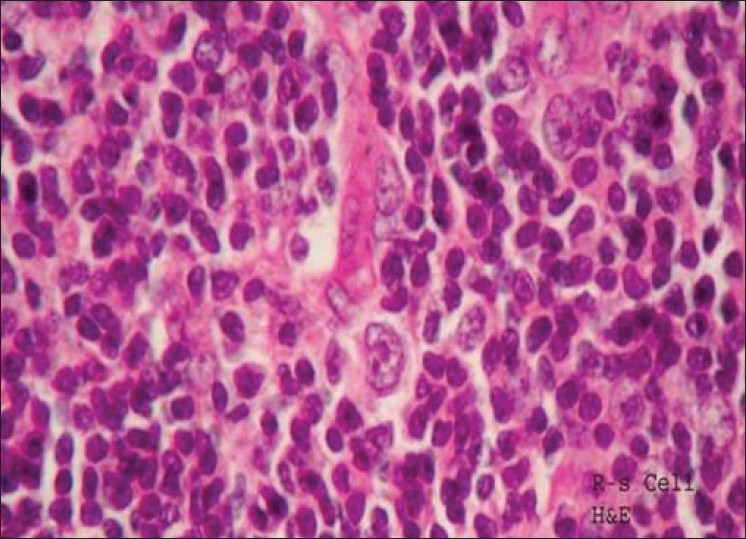
Loss of architecture, vascular proliferation, infiltrate of small and large lymphocytes with prominent nucleoli, mitotic figures and Reed Sternberg cells Hodgkin's disease – nodular sclerosis ×40 Haematoxylin and eosin
This case highlights the importance of searching for secondary causes of immune-mediated neuropathies with atypical course and clinical features.
Concluding Remarks
This review has focused on the immune-mediated neuropathies beyond CIDP. This group of disorders shares immune pathogenesis but has differentiating clinical and electrophysiological features and, in some cases, biochemical markers. The differentiation is important as the therapeutic responses of various entities differ significantly. The optimal choice of immunomodulation or immunosuppression is the key to successful alteration of the clinical course of these disorders. As our understanding of these conditions improve, more specific and more effective modalities of management will emerge in the future.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Roth G, Rohr J, Magistris MR, Ochsner F. Motor neuropathy with proximal multifocal persistent conduction block, fasciculations and myokymia. Evolution to tetraplegia. Eur Neurol. 1986;25:416–23. doi: 10.1159/000116045. [DOI] [PubMed] [Google Scholar]
- 2.Chad DA, Hammer K, Sargent J. Slow resolution of multifocal weakness and fasciculation: A reversible motor neuron syndrome. Neurology. 1986;36:1260–3. doi: 10.1212/wnl.36.9.1260. [DOI] [PubMed] [Google Scholar]
- 3.Pestronk A, Cornblath DR, Ilyas AA, Baba H, Quarles RH, Griffin JW, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol. 1988;24:73–8. doi: 10.1002/ana.410240113. [DOI] [PubMed] [Google Scholar]
- 4.Van Asseldonk JT, Franssen H, Van den Berg-Vos RM, Wokke JH, Van den Berg LH. Multifocal motor neuropathy. Lancet Neurol. 2005;4:309–19. doi: 10.1016/S1474-4422(05)70074-0. [DOI] [PubMed] [Google Scholar]
- 5.Nobile-Orazio E. What's new in multifocal motor neuropathy in 2007–2008? J Peripher Nerv Syst. 2008;13:261–3. doi: 10.1111/j.1529-8027.2008.00190.x. [DOI] [PubMed] [Google Scholar]
- 6.Leger JM, Behin A. Multifocal motor neuropathy. Curr Opin Neurol. 2005;18:567–73. doi: 10.1097/01.wco.0000175937.31569.15. [DOI] [PubMed] [Google Scholar]
- 7.Nobile-Orazio E, Cappellari A, Priori A. Multifocal motor neuropathy: Current concepts and controversies. Muscle Nerve. 2005;31:663–80. doi: 10.1002/mus.20296. [DOI] [PubMed] [Google Scholar]
- 8.Kieseier BC, Kiefer R, Gold R, Hemmer B, Willison HJ, Hartung HP. Advances in understanding and treatment of immune-mediated disorders of the peripheral nervous system. Muscle Nerve. 2004;30:131–56. doi: 10.1002/mus.20076. [DOI] [PubMed] [Google Scholar]
- 9.Kaji R, Shibasaki H, Kimura J. Multifocal demyelinating motor neuropathy: Cranial nerve involvement and immunoglobulin therapy. Neurology. 1992;42:506–9. doi: 10.1212/wnl.42.3.506. [DOI] [PubMed] [Google Scholar]
- 10.Taylor BV, Wright RA, Harper CM, Dyck PJ. Natural history of 46 patients with multifocal motor neuropathy with conduction block. Muscle Nerve. 2000;23:900–8. doi: 10.1002/(sici)1097-4598(200006)23:6<900::aid-mus9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 11.Van den Berg-Vos RM, Franssen H, Wokke JH, Van Es HW, Van den Berg LH. Multifocal motor neuropathy: Diagnostic criteria that predict the response to immunoglobulin treatment. Ann Neurol. 2000;48:919–26. [PubMed] [Google Scholar]
- 12.Priori A, Cinnante C, Pesenti A, Carpo M, Cappellari A, Nobile-Orazio E, et al. Distinctive abnormalities of motor axonal strength-duration properties in multifocal motor neuropathy and in motor neuron disease. Brain. 2002;125:2481–90. doi: 10.1093/brain/awf255. [DOI] [PubMed] [Google Scholar]
- 13.Thomas FP. Anti-Gm1 antibodies in motor system diseases and neuropathies (in German) Nervenarzt. 1990;61:704–10. [PubMed] [Google Scholar]
- 14.Schulte-Mattler WJ, Müller T, Georgiadis D, Kornhuber ME, Zierz S. Length dependence of variables associated with temporal dispersion in human motor nerves. Muscle Nerve. 2001;24:527–33. doi: 10.1002/mus.1036. [DOI] [PubMed] [Google Scholar]
- 15.Oh SJ, Kim DE, Kuruoglu HR. What is the best diagnostic index of conduction block and temporal dispersion? Muscle Nerve. 1994;17:489–93. doi: 10.1002/mus.880170504. [DOI] [PubMed] [Google Scholar]
- 16.Lange DJ, Trojaborg W, McDonald TD, Blake DM. Persistent and transient ‘conduction block’ in motor neuron diseases. Muscle Nerve. 1993;16:896–903. doi: 10.1002/mus.880160903. [DOI] [PubMed] [Google Scholar]
- 17.Brown WF, Feasby TE. Conduction block and denervation in Guillain-Barré polyneuropathy. Brain. 1984;107:219–39. doi: 10.1093/brain/107.1.219. [DOI] [PubMed] [Google Scholar]
- 18.Carpo M, Cappellari A, Mora G, Pedotti R, Barbieri S, Scarlato G, et al. Deterioration of multifocal motor neuropathy after plasma exchange. Neurology. 1998;50:1480–2. doi: 10.1212/wnl.50.5.1480. [DOI] [PubMed] [Google Scholar]
- 19.Olney RK. Guidelines in electrodiagnostic medicine.Consensus criteria for the diagnosis of partial conduction block. Muscle Nerve Suppl. 1999;8:S225–9. [PubMed] [Google Scholar]
- 20.Arunachalam R, Osei-Lah A, Mills KR. Transcutaneous cervical root stimulation in the diagnosis of multifocal motor neuropathy with conduction block. J Neurol Neurosurg Psychiatry. 2003;74:1329–31. doi: 10.1136/jnnp.74.9.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nodera H, Bostock H, Izumi Y, Nakamura K, Urushihara R, Sakamoto T, et al. Activity-dependent conduction block in multifocal motor neuropathy: Magnetic fatigue test. Neurology. 2006;67:280–7. doi: 10.1212/01.wnl.0000225048.20239.e4. [DOI] [PubMed] [Google Scholar]
- 22.Deroide N, Uzenot D, Verschueren A, Azulay JP, Pouget J, Attarian S. Triple-stimulation technique in multifocal neuropathy with conduction block. Muscle Nerve. 2007;35:632–6. doi: 10.1002/mus.20742. [DOI] [PubMed] [Google Scholar]
- 23.Chaudhry V, Swash M. Multifocal motor neuropathy: Is conduction block essential? Neurology. 2006;67:558–9. doi: 10.1212/01.wnl.0000233832.13749.ac. [DOI] [PubMed] [Google Scholar]
- 24.Pakiam AS, Parry GJ. Multifocal motor neuropathy without overt conduction block. Muscle Nerve. 1998;21:243–5. doi: 10.1002/(sici)1097-4598(199802)21:2<243::aid-mus14>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Delmont E, Azulay JP, Giorgi R, Attarian S, Verschueren A, Uzenot D, et al. Multifocal motor neuropathy with and without conduction block: A single entity? Neurology. 2006;67:592–6. doi: 10.1212/01.wnl.0000234063.51897.20. [DOI] [PubMed] [Google Scholar]
- 26.Terenghi F, Cappellari A, Bersano A, Carpo M, Barbieri S, Nobile-Orazio E. How long is IVIg effective in multifocal motor neuropathy? Neurology. 2004;62:666–8. doi: 10.1212/01.wnl.0000110185.23464.a1. [DOI] [PubMed] [Google Scholar]
- 27.Pestronk A, Choksi R. Multifocal motor neuropathy.Serum IgM anti-GM1 ganglioside antibodies in most patients detected using covalent linkage of GM1 to ELISA plates. Neurology. 1997;49:1289–92. doi: 10.1212/wnl.49.5.1289. [DOI] [PubMed] [Google Scholar]
- 28.Cavanna B, Carpo M, Pedotti R, Scarpini E, Meucci N, Allaria S, et al. Anti-GM2 IgM antibodies: Clinical correlates and reactivity with a human neuroblastoma cell line. J Neuroimmunol. 1999;94:157–64. doi: 10.1016/s0165-5728(98)00245-8. [DOI] [PubMed] [Google Scholar]
- 29.Latov N, Hays AP, Donofrio PD, Liao J, Ito H, McGinnis S, et al. Monoclonal IgM with unique specificity to gangliosides GM1 and GD1b and to lacto-N-tetraose associated with human motor neuron disease. Neurology. 1988;38:763–8. doi: 10.1212/wnl.38.5.763. [DOI] [PubMed] [Google Scholar]
- 30.Alaedini A, Sander HW, Hays AP, Latov N. Antiganglioside antibodies in multifocal acquired sensory and motor neuropathy. Arch Neurol. 2003;60:42–6. doi: 10.1001/archneur.60.1.42. [DOI] [PubMed] [Google Scholar]
- 31.Taylor BV, Gross L, Windebank AJ. The sensitivity and specificity of anti-GM1 antibody testing. Neurology. 1996;47:951–5. doi: 10.1212/wnl.47.4.951. [DOI] [PubMed] [Google Scholar]
- 32.Freddo L, Yu RK, Latov N, Donofrio PD, Hays AP, Greenberg HS, et al. Gangliosides GM1 and GD1b are antigens for IgM M-protein in a patient with motor neuron disease. Neurology. 1986;36:454–8. doi: 10.1212/wnl.36.4.454. [DOI] [PubMed] [Google Scholar]
- 33.Taylor BV, Dyck PJ, Engelstad J, Gruener G, Grant I, Dyck PJ. Multifocal motor neuropathy: Pathologic alterations at the site of conduction block. J Neuropathol Exp Neurol. 2004;63:129–37. doi: 10.1093/jnen/63.2.129. [DOI] [PubMed] [Google Scholar]
- 34.Van Es HW, Van den Berg LH, Franssen H, Witkamp TD, Ramos LM, Notermans NC, et al. Magnetic resonance imaging of the brachial plexus in patients with multifocal motor neuropathy. Neurology. 1997;48:1218–24. doi: 10.1212/wnl.48.5.1218. [DOI] [PubMed] [Google Scholar]
- 35.Auer RN, Bell RB, Lee MA. Neuropathy with onion bulb formations and pure motor manifestations. Can J Neurol Sci. 1989;16:194–7. doi: 10.1017/s0317167100028894. [DOI] [PubMed] [Google Scholar]
- 36.Viala K, Renié L, Maisonobe T, Béhin A, Neil J, Léger JM, et al. Follow-up study and response to treatment in 23 patients with Lewis-Sumner syndrome. Brain. 2004;127:2010–7. doi: 10.1093/brain/awh222. [DOI] [PubMed] [Google Scholar]
- 37.Van den Berg LH, Lokhorst H, Wokke JH. Pulsed high-dose dexamethasone is not effective in patients with multifocal motor neuropathy. Neurology. 1997;48:1135. doi: 10.1212/wnl.48.4.1135. [DOI] [PubMed] [Google Scholar]
- 38.Federico P, Zochodne DW, Hahn AF, Brown WF, Feasby TE. Multifocal motor neuropathy improved by IVIg: Randomized, double-blind, placebo-controlled study. Neurology. 2000;55:1256–62. doi: 10.1212/wnl.55.9.1256. [DOI] [PubMed] [Google Scholar]
- 39.Comi G, Amadio S, Galardi G, Fazio R, Nemni R. Clinical and neurophysiological assessment of immunoglobulin therapy in five patients with multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 1994;57(Suppl):35–7. doi: 10.1136/jnnp.57.suppl.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van den Berg LH, Franssen H, Wokke JH. The long-term effect of intravenous immunoglobulin treatment in multifocal motor neuropathy. Brain. 1998;121:421–42. doi: 10.1093/brain/121.3.421. [DOI] [PubMed] [Google Scholar]
- 41.Van den Berg-Vos RM, Franssen H, Wokke JH, Van den Berg LH. Multifocal motor neuropathy: Long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment. Brain. 2002;125:1875–86. doi: 10.1093/brain/awf193. [DOI] [PubMed] [Google Scholar]
- 42.Baumann A, Hess CW, Sturzenegger M. IVIg dose increase in multifocal motor neuropathy: A prospective six month follow-up. J Neurol. 2009;256:608–14. doi: 10.1007/s00415-009-0130-0. [DOI] [PubMed] [Google Scholar]
- 43.Pringle CE, Belden J, Veitch JE, Brown WF. Multifocal motor neuropathy presenting as ophthalmoplegia. Muscle Nerve. 1997;20:347–51. doi: 10.1002/(SICI)1097-4598(199703)20:3<347::AID-MUS12>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 44.Piepers S, Van den Berg-Vos R, Van der Pol WL, Franssen H, Wokke J, Van den Berg L. Mycophenolate mofetil as adjunctive therapy for MMN patients: A randomized, controlled trial. Brain. 2007;130:2004–10. doi: 10.1093/brain/awm144. [DOI] [PubMed] [Google Scholar]
- 45.Rojas-García R, Gallardo E, de Andrés I, de Luna N, Juarez C, Sánchez P, et al. Chronic neuropathy with IgM anti-ganglioside antibodies: Lack of long term response to rituximab. Neurology. 2003;61:1814–6. doi: 10.1212/01.wnl.0000098996.02934.86. [DOI] [PubMed] [Google Scholar]
- 46.Parry GJ. Are multifocal motor neuropathy and lewis summer syndrome distinct nosologic entities? Muscle nerve. 1999;22:557–9. doi: 10.1002/(sici)1097-4598(199905)22:5<557::aid-mus1>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 47.Gorson KC, Ropper AH, Weinberg DH. Upper limb predominant multifocal chronic demyelinating polyneuropathy. Muscle Nerve. 1999;22:758–65. doi: 10.1002/(sici)1097-4598(199906)22:6<758::aid-mus13>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 48.Oh SJ, Claussen GC, Kim DS. Motor and sensory demyelinating mononeuropathy multiplex: A separate entity or variant of CIDP? J Peripher Nerv Syst. 1997;2:362–9. [PubMed] [Google Scholar]
- 49.Saperstein DS, Amato AA, Wolfe GI, Katz JS, Nations SP, Jackson CE, et al. Multifocal acquired demyelinating sensory and motor neuropathy. Muscle Nerve. 1999;22:560–6. doi: 10.1002/(sici)1097-4598(199905)22:5<560::aid-mus2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 50.Katz JS, Saperstein DS, Gronseth G, Amato AA, Barohn RJ. Distal acquired demyelinating neuropathy. Neurology. 2000;54:615–20. doi: 10.1212/wnl.54.3.615. [DOI] [PubMed] [Google Scholar]
- 51.Sinnreich M, Klein CJ, Daube JR, Engelstad J, Spinner RJ, Dyck PJ. Chronic immune sensory polyradiculopathy: A possibly treatable sensory ataxia. Neurology. 2004;63:1662–9. doi: 10.1212/01.wnl.0000142507.12763.58. [DOI] [PubMed] [Google Scholar]
- 52.Rajkumar SV, Dispenzieri A, Kyle RA. Monoclonal gammopathy of undetermined significance, Waldenström macroglobulinemia, AL amyloidosis, and related plasma cell disorders: Diagnosis and treatment. Mayo Clin Proc. 2006;81:693–703. doi: 10.4065/81.5.693. [DOI] [PubMed] [Google Scholar]
- 53.Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346:564–9. doi: 10.1056/NEJMoa01133202. [DOI] [PubMed] [Google Scholar]
- 54.Steck AJ, Stalder AK, Renaud S. Anti-myelin-associated glycoprotein neuropathy. Curr Opin Neurol. 2006;19:458–63. doi: 10.1097/01.wco.0000245368.36576.0d. [DOI] [PubMed] [Google Scholar]
- 55.Kwan JY. Paraproteinemic neuropathy. Neurol Clin. 2007;25:47–69. doi: 10.1016/j.ncl.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 56.Eurelings M, Ang CW, Notermans NC, Van Doorn PA, Jacobs BC, Van den Berg LH. Antiganglioside antibodies in polyneuropathy associated with monoclonal gammopathy. Neurology. 2001;57:1909–12. doi: 10.1212/wnl.57.10.1909. [DOI] [PubMed] [Google Scholar]
- 57.Nobile-Orazio E, Cappellari A, Priori A. Multifocal motor neuropathy: Current concepts and controversies. Muscle Nerve. 2005;31:663–80. doi: 10.1002/mus.20296. [DOI] [PubMed] [Google Scholar]
- 58.Notermans NC, Lokhorst HM, Franssen H, Van der Graaf Y, Teunissen LL, Jennekens FG, et al. Intermittent cyclophosphamide and prednisone treatment of polyneuropathy associated with monoclonal gammopathy of undetermined significance. Neurology. 1996;47:2227–33. doi: 10.1212/wnl.47.5.1227. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh A, Littlewood T, Donaghy M. Cladirabine in the treatment of IgM paraproteinemic polyneuropathy. Neurology. 2002;59:1290–1. doi: 10.1212/wnl.59.8.1290. [DOI] [PubMed] [Google Scholar]
- 60.Dalakas MC, Rakocevic G, Salajegheh M, Dambrosia JM, Hahn AF, Raju R, McElroy B. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann Neurol. 2009;65:286–93. doi: 10.1002/ana.21577. [DOI] [PubMed] [Google Scholar]
- 61.Barbieri S, Nobile-Orazio E, Baldini L, Fayoumi Z, Manfredini E, Scarlato G. Visual evoked potentials in patients with neuropathy and macroglobulinemia. Ann Neurol. 1987;22:663–6. doi: 10.1002/ana.410220520. [DOI] [PubMed] [Google Scholar]
- 62.Dispenzieri A, Kyle RA, Lacy MQ, Rajkumar SV, Therneau TM, Larson DR, et al. POEMS syndrome: Definition and long term outcome. Blood. 2003;101:2496–506. doi: 10.1182/blood-2002-07-2299. [DOI] [PubMed] [Google Scholar]
- 63.Nomenclature Sub Committee. WHO-IUIS. 1993;71:105–12. [PMC free article] [PubMed] [Google Scholar]
- 64.Falk RH, Comenzo RL, Skinner M. The systemic amyloidosis. N Engl J Med. 1997;337:898–909. doi: 10.1056/NEJM199709253371306. [DOI] [PubMed] [Google Scholar]
- 65.Guillevin L, Lhote F, Gherardi R. Polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: Clinical aspects, neurologic manifestations, and treatment. Neurol Clin. 1997;15:865–86. doi: 10.1016/s0733-8619(05)70352-2. [DOI] [PubMed] [Google Scholar]
- 66.Nadeau SE. Diagnostic approach to central and peripheral nervous system vasculitis. Neurol Clin. 1997;15:759–77. doi: 10.1016/s0733-8619(05)70346-7. [DOI] [PubMed] [Google Scholar]
- 67.Rosenbaum R. Neuromuscular complications of connective tissue disease. Muscle Nerve. 2001;24:154–9. doi: 10.1002/1097-4598(200102)24:2<154::aid-mus20>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 68.Singhal BS, Khadilkar SV, Gursahani RD, Surya N. Vasculitic neuropathy: Profile of twenty patients. J Assoc Physicians India. 1995;43:459–61. [PubMed] [Google Scholar]
- 69.Murthy JMK, Sundram C, Meena AK. Vasculitic neuropathy: Clinical electrophysiological and histopathological characteristics. Neurol India. 1998;46:18–22. [PubMed] [Google Scholar]
- 70.Graus F, Cordon-Cardo C, Posner JB. Neuronal antinuclear antibodies in sensory neuronopathy from lung cancer. Neurology. 1985;35:538–43. doi: 10.1212/wnl.35.4.538. [DOI] [PubMed] [Google Scholar]
- 71.Sugai F, Abe K, Fujimoto T, Nagano S, Fujimura H, Kayanoki Y, et al. Chronic inflammatory demyelinating polyneuropathy accompanied by hepatocellular carcinoma. Intern Med. 1997;36:53–5. doi: 10.2169/internalmedicine.36.53. [DOI] [PubMed] [Google Scholar]
- 72.Barohn RJ, Kissel JT, Warmolts JR, Mendell JR. Chronic inflammatory demyelinating polyradiculoneuropathy: Clinical characteristics, course, and recommendations for diagnostic criteria. Arch Neurol. 1989;46:878–84. doi: 10.1001/archneur.1989.00520440064022. [DOI] [PubMed] [Google Scholar]