

Abstract
We demonstrate a technique for detecting magnetically labeled Listeria monocytogenes and for measuring the binding rate between antibody-linked magnetic particles and bacteria. This sensitive assay quantifies specific bacteria in a sample without the need to immobilize them or wash away unbound magnetic particles. In the measurement, we add 50-nm-diameter superparamagnetic magnetite particles, coated with antibodies, to an aqueous sample containing L. monocytogenes. We apply a pulsed magnetic field to align the magnetic dipole moments and use a high-transition temperature superconducting quantum interference device, an extremely sensitive detector of magnetic flux, to measure the magnetic relaxation signal when the field is turned off. Unbound particles randomize direction by Brownian rotation too quickly to be detected. In contrast, particles bound to L. monocytogenes are effectively immobilized and relax in about 1 s by rotation of the internal dipole moment. This Néel relaxation process is detected by the superconducting quantum interference device. The measurements indicate a detection limit of (5.6 ± 1.1) × 106 L. monocytogenes in our sample volume of 20 μl. If the sample volume were reduced to 1 nl, we estimate that the detection limit could be improved to 230 ± 40 L. monocytogenes cells. Time-resolved measurements yield the binding rate between the particles and bacteria.
Antibodies are widely used as biological probes to identify specific microorganisms or molecules (1, 2). The antibodies are linked to a label and introduced into the sample, where they bind to the targets of interest and provide a means of detection. Common labels include enzymes, fluorescent dyes, radioisotopes, or magnetic particles. This general technique has various applications. In an immunoassay, the goal is to detect and quantify specific targets. Tagged antibodies can also be used to separate target antigens selectively or to measure the affinity between antibody and antigen. In this article, we present a sensitive method for detecting magnetically labeled bacteria by using a superconducting quantum interference device (SQUID), a highly sensitive detector of magnetic flux. This assay can be used to monitor bacteria in a liquid sample and to determine the rate of binding between antibody-linked particles and bacteria.
Magnetic particles have several advantages as labels. They are stable and nontoxic and can be manipulated with a magnetic field, making it possible to separate target antigens magnetically (3). Methods have been developed to detect small numbers of such particles by using Hall probes (4), giant magnetoresistance arrays (5), atomic force microscopy (6), force-amplified biological sensors (7), and SQUIDs (8–10).
Weitschies, Kötitz, and colleagues pioneered the use of SQUIDs for magnetic immunoassays (8, 11–16). They developed a magnetic relaxation immunoassay in which magnetic particles bound to targets are distinguished from unbound particles by their different relaxation times. By using a low-critical-temperature (Tc) SQUID, the group implemented a solid-phase magnetic relaxation immunoassay for detecting human IgG. Enpuku et al. (17) used a high-Tc SQUID to detect human IFN-β. They immobilized target antigens on a surface, labeled them with magnetic particles, applied a magnetic field to magnetize the particles, and measured the change in magnetic flux as the sample was passed under the SQUID.
In a previous communication (10), we described the use of a high-Tc SQUID microscope to detect immobilized targets, consisting of liposomes carrying the FLAG epitope, which were labeled with magnetic particles. Here, we demonstrate a new method for detecting targets that are not immobilized but rather are in suspension (Fig. 1). We couple 50-nm-diameter γ-Fe2O3 particles to polyclonal antibodies raised against the bacterial pathogen Listeria monocytogenes and add them to a suspension of that organism. After allowing time for the particles to bind to the targets, we place the sample 130 μm above a high-Tc SQUID and apply a pulsed magnetic field to align the magnetic dipole moments. Each time the field is turned off, the SQUID detects the magnetic relaxation signal. Unbound particles relax in ≈50 μs by Brownian rotation; this time is too short for the SQUID system to measure. Conversely, particles bound to the relatively large bacteria are able to rotate only slowly. These particles undergo Néel relaxation, in which their internal dipole moments relax to the lowest energy state. The resulting magnetic decay, which occurs in ≈1 s, is detected by the SQUID. Because the measured magnetic relaxation is due only to the bound particles, changes in the magnetic relaxation amplitude over time indicate the rate at which particles bind to bacteria.
Fig. 1.

Measurement procedure. A suspension of superparamagnetic particles, coupled to antibodies, is added to the liquid sample. (A) A magnetic field is applied to align the magnetic moments of the particles. (B) At time t ≈ τB after the field is turned off, unbound particles have randomized direction by Brownian rotation, whereas particles bound to bacteria are still aligned. The magnetic moments of the bound particles reorient slowly by means of Néel relaxation.
This straightforward assay format does not require immobilization of the targets or washing away of the unbound particles. It has the potential for improved accuracy over conventional immunoassays because no materials are lost. We demonstrate that this technique can successfully differentiate between bound and unbound particles and present results from titration experiments in which the concentration of either bacteria or particles is varied. We show how the relaxation signal depends on the applied magnetic field and present time-resolved data illustrating that this technique can measure binding reaction rates. Finally, we discuss improvements to the technique and potential applications.
Theory
We differentiate between bound and unbound particles by the different mechanisms by which they relax after the removal of a magnetic field. Brownian relaxation (18) is a physical rotation of the particles, with a relaxation time for a sphere
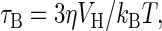 |
[1] |
where η is the viscosity of the medium, VH is the hydrodynamic volume, kB is Boltzmann's constant, and T is the temperature. Taking T = 293 K and η = 10–3 kg·m–1·s–1, we find τB is ≈50 μs for particles with a hydrodynamic diameter of 50 nm.
Néel relaxation (19) originates from the anisotropy of the crystalline lattice. Many magnetic materials have an easy axis of magnetization; when the crystal is magnetized along that axis, the energy is minimized. If an external field rotates the magnetization away from the easy axis, the magnetization eventually returns to its preferred direction on removal of the field. The Néel relaxation time for a single domain particle is
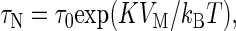 |
[2] |
where τ0 is ≈10–9 s, K is the magnetic anisotropy constant, and VM is the magnetic core volume. We note that τ0 and K depend on the shape of the particle, and values of τ0 vary by up to four orders of magnitude (20). The particles used here were composed of γ-Fe2O3, for which the bulk anisotropy constant is K ≈ 2.5 × 104 J·m–3. The magnetic core of each particle consisted of a cluster of ≈10-nm nanoparticles. Whereas Eq. 2 predicts τN ≈ 25 ns for an individual 10-nm nanoparticle at T = 293 K, magnetic interactions between the nanoparticles within each core slow down the overall relaxation rate. Hence, the Néel relaxation time of these particles fell within the 1-ms to 1-s measurement window of our SQUID system.
Methods
SQUID Microscope. The measurement configuration is shown in Fig. 2. A dc SQUID is a superconducting loop interrupted by two Josephson junctions (21). The voltage across the current-biased SQUID oscillates quasisinusoidally as a function of the magnetic flux Φ threading the loop with a period of the magnetic flux quantum, Φ0 = h/2e ≈ 2 × 10–15 T·m–2. To linearize the flux-to-voltage conversion, we operate the SQUID in a flux-locked loop that maintains the flux through it at a constant value; the output voltage of this feedback circuit is proportional to Φ. The SQUID was made from a 200-nm-thick film of the high-Tc superconductor YBa2Cu3O7-δ (YBCO) laser deposited onto a (100) SrTiO3 bicrystal substrate and patterned by photolithography and argon ion milling. A bicrystal contains an in-plane misorientation of the crystallographic axis; an epitaxially grown film mimics the misorientation, forming a grain boundary that supports only a weak supercurrent. To form Josephson junctions, 2-μm-wide microbridges were patterned in the film across the grain boundary. The SQUID, shown in Fig. 3 Inset, had an effective flux capture area of 0.016 mm2 and a peak-to-peak modulation amplitude of 11 μV. Operated with bias reversal (22) to reduce low-frequency noise, it exhibited a white noise of 22 μΦ0 Hz–1/2 at frequencies down to ≈1 Hz.
Fig. 2.

Top portion of the SQUID microscope. The SQUID, inside a vacuum enclosure, is mounted on a sapphire rod thermally connected to a liquid nitrogen reservoir (not shown). A 75-μm-thick sapphire window separates the vacuum chamber from atmosphere. The sample is contained in a Lucite holder, with a 3-μm-thick Mylar base, aligned against a positioning element.
Fig. 3.

Example of magnetic decay signals. For the traces shown, the concentration of bacteria was 108 per ml, and the concentration of particles was 0.13 relative to the stock suspension. A 0.4-mT field was pulsed on for 1 s and off for 1 s, and data were recorded each time the field was turned off; 100 averages were taken. Because the particle–antibody complexes show little cross-reactivity to E. coli, the “E. coli” and “particles alone” curves overlay each other. (Inset) Configuration of the YBCO SQUID. The slit is 4 μm wide.
The SQUID microscope (23) (Fig. 2) brings a biological sample at room temperature and atmospheric pressure very close to the SQUID, which is maintained at 77 K in a vacuum. Because the magnetic field from a collection of dipoles decreases with distance, it is important to minimize the separation between the sample and the SQUID. The SQUID was mounted on a sapphire rod thermally linked to a liquid-nitrogen reservoir; these components were enclosed in a fiberglass vacuum chamber. Above the SQUID, a 75-μm-thick sapphire window separated the vacuum chamber from the atmosphere. The gap between the SQUID and the window was 55 ± 10 μm, resulting in a total SQUID–sample distance of 130 ± 10 μm. The entire apparatus was enclosed in a triple-layer μ-metal shield to attenuate the earth's magnetic field.
For each measurement, a sample holder was positioned on the window between two coils, which provided a magnetizing field parallel to the plane of the SQUID. The sample holders consisted of 11-mm-wide and 3.2-mm-thick Lucite squares. A 3.2-mm-diameter hole was drilled through each square, and a 3-μm-thick Mylar film, attached to the Lucite with wax, sealed the bottom of the hole. The sample was offset laterally from the SQUID by 1.6 mm, one-half the sample diameter, to maximize the field from the sample coupled to the SQUID.
Bacteria. The target bacteria were the DP-L2161 strain (24) of L. monocytogenes, which has a deletion in the hly gene encoding listeriolysin O and is thus 105 times less virulent than the wild-type strain (25). The L. monocytogenes were grown overnight in 3 ml of brain–heart broth in an incubator-shaker (37°C, 250 rpm). Before the assay, the bacteria were washed three times by centrifugation and resuspended in PBS. We counted the bacteria by measuring their optical density at 600-nm wavelength and multiplying it by 6 × 108 L. monocytogenes per ml to convert to concentration (D. A. Portnoy, personal communication). Because the conversion factor depends on the phase to which the bacteria are grown, we estimate a counting error of ±20%. The K1 strain of Escherichia coli (26) was used as a control. The bacteria were grown overnight in 3 ml of Luria broth (Miller's LB broth) in an incubator-shaker (37°C, 250 rpm), and washed and counted in the same manner as the L. monocytogenes. In this case, the optical density was multiplied by 1 × 109 E. coli per ml to convert to concentration (27).
The purpose of transferring the bacteria to a buffer solution was to improve the counting accuracy and to create a reproducible environment for antibody/particle binding. However, antibody-coated magnetic particles are used to separate cells in a variety of solutions, so we do not anticipate binding problems in other media.
Magnetic Particles. Superparamagnetic particles conjugated to monoclonal mouse anti-biotin antibodies were obtained from Miltenyi Biotec (Auburn, CA). The particles, composed of 55–59% γ-Fe2O3, 35–39% dextran, and 2–10% antibody by weight, were suspended in a buffer containing 0.05% sodium azide. Their hydrodynamic diameters ranged from 20 to 100 nm, with an average of 50 nm. The iron oxide core of each particle consisted of a cluster of ≈10-nm nanoparticles.
Antibodies. The particles were coupled to polyclonal goat anti-Listeria IgG antibodies supplied by OEM Concepts (Toms River, NJ). The antibodies, raised against all serogroups of Listeria species known to cause food-borne illness, were purified by the vendor on an antigen-affinity column with the Listeria bacteria immunogen preparation as antigen. The antibodies were biotinylated by using the FluoReporter MiniBiotin-XX Protein Labeling Kit from Molecular Probes, which uses a 14-atom spacer. The protocol provided by Molecular Probes was modified to achieve ≈1–3 biotin molecules per antibody.
Coupling of Antibodies to Particles. To couple the antibodies to the particles, we mixed the solution of biotinylated antibodies with the undiluted particle suspension, incubated the mixture overnight at 4°C, and filtered it through a 0.22-μm centrifugal filter with a low binding Durapore poly(vinylidene difluoride) microporous membrane (Millipore) to eliminate particle aggregates. Ideally, we would have washed away the unbound antibodies, but we were unable to do this without a concomitant loss of particles. Instead, we varied the volume ratio of antibodies to particles to maximize the binding of the particles to L. monocytogenes. The binding signal increased progressively up to a ratio of 1.3:5, and thereafter remained constant up to 5.3:5. To ensure saturation of binding, a volume ratio of 4:5 was used for all subsequent particle preparations.
Results
Titration Experiments. To determine the sensitivity and specificity of the technique, we carried out a series of bacterial and particle titrations. The particle–antibody complexes and bacteria were prepared as described above and mixed together in various concentrations; the samples contained ≈0.015% sodium azide. We incubated the samples a minimum of 4 h 45 min to ensure that at least 75% of the binding reaction would be complete at the time of the measurement. (The time required was determined from data discussed in Binding Rate Measurements.) After the incubation period, each sample was agitated with a pipet to resuspend any material that had settled, and a sample holder containing 20 μl was placed on the microscope. We pulsed the 0.4-mT magnetic field on for 1 s and off for 1 s and recorded the magnetic decay each time the field was turned off. The data from 100 pulses were averaged. Observation of the samples under a phase-contrast microscope indicated that the bacteria were nonmotile, and thus possibly dead, at the time of the measurement.
Fig. 3 shows typical time traces for an L. monocytogenes sample and associated controls. These data were fit to a sum of logarithmic and exponential functions. The logarithmic decay is characteristic of Néel relaxation for particles with a wide distribution of sizes, and therefore of relaxation times (28). We believe the exponential decay comes from particle aggregates, formed after the filtration step, which are large enough to relax via Brownian rotation on a measurable timescale without being bound to targets. The fitting function is
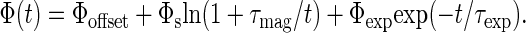 |
[3] |
Here, Φoffset is an offset caused by the fact that the SQUID measures relative, rather than absolute, magnetic flux; Φs, the logarithmic decay amplitude, is proportional to the number of bound particles; τmag = 1 s is the magnetization time; Φexp, the exponential decay amplitude, depends on the number of unbound particle aggregates; and τexp is the exponential decay time constant. By fitting Eq. 3 to measurements of particles alone, we found τexp ≈ 15 ms, corresponding to a hydrodynamic diameter of ≈340 nm for a sphere. Fixing τexp = 15 ms in later experiments improved the reproducibility of Φs and allowed us to use a simpler fit algorithm.
The logarithmic decay amplitudes for the samples measured in the titration experiments are plotted in Fig. 4. For the bacterial titration, the concentration of L. monocytogenes or E. coli was varied, whereas the particle concentration was fixed at 0.05 relative to the stock suspension. For the particle titration, the particle concentration was varied, whereas the density of bacteria was fixed at 108 per ml L. monocytogenes or E. coli.
Fig. 4.

(A) Bacterial titration. The concentration of L. monocytogenes or E. coli was varied, whereas the particle concentration was fixed at 0.05 relative to the stock suspension. (B) Particle titration. The particle concentration was varied, whereas the bacterial concentration was fixed at 108 per ml L. monocytogenes or E. coli. The magnetic relaxation signal of each sample was fit to a combination of logarithmic and exponential functions; the logarithmic amplitude of each sample is displayed. A two-antibody model, described in the text, was used to fit the L. monocytogenes data.
Several sources of error exist. SQUID noise, particularly at low frequencies, is the largest source of random error and was determined by fitting the individual traces to Eq. 3 and calculating the mean and standard deviation of the fit coefficients for 100 averages. The standard deviation, σs, of the logarithmic amplitude was typically ≈10 μΦ0. We estimated other sources of error, such as variability among biological samples and variation in sample position relative to the SQUID by measuring a series of nominally identical samples. The standard deviation was found to be 6.9% of the signal level. Thus, the total error in σs is
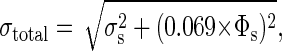 |
[4] |
where the value of Φs is determined separately for each sample measurement.
The logarithmic decay amplitude from the particles alone is very low (Fig. 4). It does not limit the sensitivity because when only a few targets are present, fewer particles are needed to saturate the binding sites. Therefore, for a dilute sample, we can reduce the magnitude of the background decay by using fewer particles.
The assay clearly distinguishes L. monocytogenes from E. coli (Fig. 4). The E. coli signal is only ≈15% of the L. monocytogenes signal, assuming enough particles are present to saturate the L. monocytogenes binding sites. If we subtract out the control (particles alone) signal from the L. monocytogenes and E. coli data, the E. coli signal is <8% of the L. monocytogenes signal. It could be reduced still further by adsorbing the antibodies against E. coli to eliminate cross-reactive groups.
The bacterial titration curve, shown in Fig. 4A, appears to have two distinct slopes, with a crossover at ≈108 L. monocytogenes per ml. This shape may result from the polyclonal nature of the antibodies. We presume that various types of antibodies are present, which are reactive against different epitopes and have different binding affinities. As each type of antibody is depleted, the slope of the curve decreases. Accordingly, we fit the data to a model in which there are two distinct types of antibodies, referred to as “A” and “B,” and two corresponding types of antigenic determinants, also referred to as “A” and “B”; we chose this model for its simplicity and for its consistency with the empirical observation of two nonzero slopes in Fig. 4A. The A and B in the model could represent two broad-affinity classes of antibodies or two dominant antigenic determinants. We made the further assumption that each particle is conjugated exclusively to A or B antibodies. This assumption was based on data indicating that ≈25% of the antibody-linked particles were reactive against this strain of L. monocytogenes.** Thus, the number of particles with more than one type of antibody against these bacteria should be relatively small.
The four independent parameters of the model are the fraction of A antibodies, the fraction of A antigens, the particle concentration required to saturate a given number of bacteria, and the magnetic signal produced by a saturated bacterium. Fitting simultaneously to the bacterial and particle titration curves, we varied the four parameters until
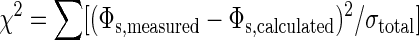 |
[5] |
was minimized. The sum is over all data points; Φs,measured is the measured logarithmic decay amplitude, Φs,calculated is the calculated amplitude with given fit parameters, and σtotal is the uncertainty in the measured amplitudes determined by Eq. 4. The resulting fits are shown in Fig. 4. We find that the fraction of A antibodies is 0.53 ± 0.03; the fraction of A antigens is 0.99 ± 0.01; (2.0 ± 0.4) × 109 L. monocytogenes per ml are saturated by the full-strength particle suspension; and each saturated Listerium per ml produces a (3.6 ± 0.7)× 10–9 mΦ0 logarithmic decay signal. The detection limit, taken as the minimum quantity of L. monocytogenes detectable with 95% confidence, is 2σtotal. Because σtotal≈σs in the limit of small Φs, the detection limit is 2σs ≈ 20 μΦ0. This yields a detection limit of (5.6 ± 1.1) × 106 L. monocytogenes per ml, corresponding to (1.1 ± 0.2) × 105 L. monocytogenes in a 20-μl sample volume.
Because the L. monocytogenes have so few B antigens, 0.0015 concentration particles are sufficient to saturate the B antigens in the particle titration shown in Fig. 4B. Hence, the slope change in this curve is barely visible.
Dependence of Signal on the Applied Magnetic Field. The alignment of the particle dipole moments increases with magnetic field until the field is strong enough to align them completely. To determine how the signal level depends on the applied field, we prepared a test sample to measure at various field strengths. We diluted the original particle suspension by a factor of 12.5, passed it through a 0.22-μm centrifugal filter, placed 20 μl in a sample holder, and allowed the liquid to evaporate. We measured the Néel relaxation signal of this sample in response to fields ranging from 0 to 2.2 mT, recording just one trace for each field value. Measurements were taken for both increasing and decreasing fields to ensure that there was no hysteresis. The results, plotted in Fig. 5, show that the signal can be increased by a factor of up to 3 by increasing the applied field above the 0.4 mT used in the titration experiments. As the field is increased, the slight (≈0.001°) misalignment of the coils couples more flux vortices into the SQUID. When the field is turned off, these vortices leave the SQUID on a timescale of seconds, producing a relaxation signal that contributes a systematic error to the experiment. Although this effect limited the field to 0.4 mT, it is not a fundamental limitation and could be reduced by improving the coil alignment.
Fig. 5.

Dependence of the Néel relaxation signal on the applied magnetic field. The magnetic relaxation signal from a test sample, consisting of particles evaporated onto a sample holder, was measured for different values of applied field. Each decay curve was fit to a combination of logarithmic and exponential functions; the logarithmic amplitudes are shown.
Binding Rate Measurements. In another set of experiments, we measured the time dependence of the Néel relaxation signal. Because only bound particles contribute to the measured relaxation signal, the change in relaxation amplitude over time indicates the rate at which particles bind to bacteria. As in the titration experiments, we prepared the particle–antibody complexes and washed and counted the bacteria. In quick succession, we mixed the particles and L. monocytogenes, vortexed the mixture for 5 s, and transferred 20 μl to a sample holder. We placed the sample on the SQUID microscope and measured the Néel relaxation signal in response to a pulsed 0.4-mT field, taking 100 averages. The time between mixing the sample and obtaining the first measurement was 3–7 min. We repeated the measurement approximately every 4 min.
A typical dependence of SQUID signal on elapsed time is shown in Fig. 6, for 2.5 × 108 L. monocytogenes per ml and particles of concentration 0.05 relative to the stock suspension. The data were fit to an exponential function,
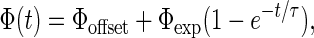 |
[6] |
where Φoffset is the signal from particles that bind to bacteria during the mixing stage, the sum of Φoffset and Φexp is the equilibrium relaxation signal, and τ is the time constant for the postmixing binding process. For the data in Fig. 6, Φoffset = 0.045 ± 0.005 mΦ0, Φexp = 0.24 ± 0.02 mΦ0, and τ = 209 ± 39 min. About 16% of the total binding occurred during the initial mixing stage. The remainder of the binding took place over the course of many hours.
Fig. 6.

Binding rate measurement. L. monocytogenes (concentration, 2.5 × 108 per ml) and particle–antibody complexes (concentration, 0.05 relative to the stock particle suspension) were mixed together, and the magnetic relaxation signal was measured as a function of time. Each decay curve was fit to a combination of logarithmic and exponential functions; the logarithmic amplitudes are shown. The amplitude vs. time data were fit to Φ(t) = Φoffset + Φexp[1 – exp(–t/τ)]. As a control, the signal from particle–antibody complexes alone (concentration, 0.05 relative to the stock particle suspension) was measured over time.
Part of the increase in signal is due to settling of the bacteria and particles. The settling rate depends on the degree of cross-linking of the bacteria and particles – the larger the complexes formed, the faster the settling rate. To measure the settling rate, we prepared four different samples of bacteria and particles and allowed ample time for the binding to reach equilibrium (at least 24 h at 4°C). We then mixed each sample to resuspend any settled material, transferred 20 μl to a sample holder, and measured the time dependence of the signal. We found that the signals increased by 2.4% to 19%/h, depending on the sample, corresponding to settling rates of 0.14 to 1.1 μm/s.
If we correct the data in Fig. 6 for settling by using the largest observed settling rate and fit again to Eq. 6, we find the time constant for binding is lowered to 164 ± 31 min, a reduction of about 22%. Thus, the uncertainty in the settling rate contributes substantially to the uncertainty in the binding rate. The settling problem could be addressed by continuously mixing the sample during the course of the measurement.
The question remains whether the measured binding rates reflect antibody–antigen binding rates or the time it takes for the antibody-coated particles to diffuse toward the bacteria. We calculated (29) that diffusing particle–antibody complexes with a hydrodynamic diameter of 100 nm would bind to 1.5-μm-diameter perfectly sticky spheres of concentration 2.5 × 108 per ml in ≈100 s. Because this diffusion-limited rate is two orders of magnitude faster than the observed reaction rate, the observed binding rate reflects the dynamics of antibody–antigen interactions.
Discussion and Future Directions. The technique presented here has several possible applications. It could be implemented as an immunoassay to test for the presence of specific bacteria. Because it does not require immobilization of the targets or separation of the unbound tags, it could potentially provide more accurate quantification than conventional immunoassays. It could also be used to characterize the reactants in a system, as was done in fitting the titration data to the two-antibody model. A distinctive feature of this technique is its ability to measure binding rates of reactants in suspension. The time series measurements could be expanded to determine how the binding rate depends on various experimental parameters. Another possible application is the measurement of bacterial transport with applications in bioremediation. To implement bioremediation strategies successfully, one needs to predict the transport of bacteria through contaminated media. However, most techniques for studying bacterial transport involve optical detection and cannot be used for opaque media. By replacing the present sample holder with an elongated channel (≈50 mm long, 3 mm wide, and 3 mm high) mounted on a scanning stage, tagging the bacteria with magnetic particles, and using the SQUID to measure the relaxation signal as a function of space and time, one could study the movement of bacteria through any type of medium. Calculations based on the current system indicate a resolution of ≈1 mm.
As an immunoassay, this technique is quantitative, reasonably fast, and moderately sensitive. The specificity depends largely on the antibodies used. For the affinity-purified anti-Listeria antibodies used here, the cross-reactivity to E. coli was <8% of the L. monocytogenes signal, after correcting for background. The quantitative nature of the assay is evident from the titration data. We see in Fig. 4A that the signal increases linearly with bacterial concentration. The slope is constant so long as enough particles are present to saturate the L. monocytogenes binding sites. The speed of the assay is limited by the binding rate of the particles to the bacteria. In these experiments, we incubated the samples several hours before performing measurements on them. However, by increasing the incubation temperature or centrifuging the particles and targets to increase their concentration, one could expect to reduce the incubation time significantly. Further, in applications where high sensitivity or accuracy is not required, measurements could be taken before the binding reaction is complete.
The current detection limit is (5.6 ± 1.1) × 106 L. monocytogenes per ml, equivalent to (1.1 ± 0.2) × 105 L. monocytogenes in a 20-μl sample volume. The sensitivity to absolute number of bacteria could be greatly improved by decreasing the sample volume. For a sample much larger than the SQUID, as is currently the case, a sizeable fraction of the particles lie far away from the SQUID sensing area. Because the magnetic field from a dipole falls off inversely as the cube of the distance, these distant particles contribute little to the signal. By decreasing the sample volume, while holding the concentration of particles and targets fixed, one could increase the signal per particle, and hence improve the sensitivity.
For example, consider scaling down the sample volume to 1 nl by reducing the sample area to 0.01 mm2 and the height to 0.1 mm. Because the SQUID effective area is 0.016 mm2, the SQUID would capture the flux much more efficiently than in the current version. Further, because the new sample height would be similar to the SQUID sample separation, a particle at the top of the sample would contribute about 25% of the flux as one at the bottom. This contrasts sharply with the current configuration, in which particles at the top of the sample are so far away (2.7 mm) that their flux contribution is negligible. We calculate that a 1-nl sample of the above dimensions, located at its optimal position over the SQUID, would have a signal per particle 480 times greater than the current sample. Thus, the sensitivity of the technique would improve to 230 ± 40 L. monocytogenes. Note that whereas the sensitivity to number highly depends on sample volume, the sensitivity to concentration is relatively independent of sample volume.
A further sensitivity improvement could be achieved by immobilizing the bacteria on the Mylar base of the sample holder by means of a second antibody. This is essentially the method adopted in our earlier experiment (10) in which liposome targets were affixed to a Mylar film. If we were to attach the bacteria in the hypothetical 1-nl sample to the bottom of the sample chamber, thereby bringing all the particles to within 130 μm of the SQUID, we calculate that the sensitivity would improve to 120 ± 20 bacteria. Although immobilizing the targets requires an extra step, the return is greater sensitivity and the ability to detect a target of any size. Clearly, if an application requires detection of a few targets, it is highly desirable to immobilize the targets on a substrate or reduce the sample volume by concentration methods. Microfluidics could be used to implement a small volume assay and to provide continuous mixing (30, 31).
Acknowledgments
We thank D. A. Portnoy for providing the L. monocytogenes culture and for helpful discussions, S.-K. Lee and H.-M. Cho for their help with the SQUID fabrication, T. M. Handel and colleagues for their assistance with the biological experiments, Y. R. Chemla for his early work on this assay, C. M. Klapperich and J. Song for technical assistance, and B. Dorval of OEM Concepts for providing the ELISA protocol to test antibodies. This work was supported by the Director, Office of Energy Research, Office of Basic Energy Science, Materials Sciences and Engineering Division of the U.S. Department of Energy under Contract DE-AC03-76SF00098.
Abbreviations: SQUID, superconducting quantum interference device; Tc, critical temperature.
Footnotes
Because the OEM Concepts anti-Listeria antibodies bind to all pathogenic Listeria species, we do not expect all such antibodies to bind to the L. monocytogenes strain used in this experiment. When we bind these antibodies to particles, many particles become coated with only nonbinding antibodies and do not contribute to the detection signal. To measure this effect, we carried out experiments in which we coupled biotinylated anti-Listeria antibodies to L. monocytogenes cells, washed away the unbound antibodies, and added particles with anti-biotin on their surface. Measurements of the Néel relaxation signal yielded the magnetic signal per bacterium and the concentration of particles needed to saturate a given number of bacteria. In these experiments, we assumed that all particles were capable of binding to L. monocytogenes. By comparing these results with those obtained in experiments in which the antibodies were linked to the particles at the outset, we determined that ≈25% of the antibody-linked particles were reactive against L. monocytogenes.
References
- 1.Englebienne, P. (2000) Immune and Receptor Assays in Theory and Practice (CRC, Boca Raton, FL).
- 2.Wild, D., ed. (2001) The Immunoassay Handbook (Nature Publishing Group, London).
- 3.Šafar̆ík, I. & Šafar̆íková, M. (1999) J. Chromatogr. B 722, 33–53. [PubMed] [Google Scholar]
- 4.Besse, P.-A., Boero, G., Demierre, M., Pott, V. & Popovic, R. (2002) Appl. Phys. Lett. 80, 4199–4201. [Google Scholar]
- 5.Miller, M. M., Sheehan, P. E., Edelstein, R. L., Tamanaha, C. R., Zhong, L., Bounnak, S., Whitman, L. J. & Colton, R. J. (2001) J. Magn. Magn. Mater. 225, 138–144. [Google Scholar]
- 6.Baselt, D. R., Lee, G. U., Natesan, M., Metzger S. W., Sheehan, P. E. & Colton, R. J. (1998) Biosens. Bioelectron. 13, 731–739. [DOI] [PubMed] [Google Scholar]
- 7.Baselt, D. R., Lee, G. U., Hansen, K. M., Chrisey, L. A. & Colton, R. J. (1997) Proc. IEEE 85, 672–680. [Google Scholar]
- 8.Weitschies, W., Kötitz, R., Bunte, T. & Trahms, L. (1997) Pharm. Pharmacol. Lett. 7, 1–7. [Google Scholar]
- 9.Enpuku, K, Minotani, T., Gima, T., Kuroki, Y., Itoh, Y., Yamashita, M., Katakura, Y. & Kuhara, S. (1999) Jpn. J. Appl. Phys. 38, L1102–L1105. [Google Scholar]
- 10.Chemla, Y. R., Grossman, H. L., Poon, Y., McDermott, R., Stevens, R., Alper, M. D. & Clarke, J. (2000) Proc. Natl. Acad. Sci. USA 97, 14268–14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kötitz, R., Matz, H., Trahms, L., Koch, H., Weitschies, W., Rheinlander, T., Semmler, W. & Bunte, T. (1997) IEEE Trans. Appl. Superconductivity 7, 3678–3681. [Google Scholar]
- 12.Kötitz, R., Weitschies, W., Trahms, L., Brewer, W. & Semmler, W. (1999) J. Magn. Magn. Mater. 194, 62–68. [Google Scholar]
- 13.Schambach, J., Warzemann, L., Weber, P., Kötitz, R. & Weitschies, W. (1999) IEEE Trans. Appl. Superconductivity 9, 3527–3530. [Google Scholar]
- 14.Kötitz, R., Weitschies, W., Trahms, L. & Semmler, W. (1999) J. Magn. Magn. Mater. 201, 102–104. [Google Scholar]
- 15.Haller, A., Hartwig, S., Matz, H., Lange, J., Rheinlander, T., Kötitz, R., Weitschies, W. & Trahms, L. (1999) Supercond. Sci. Tech. 12, 956–958. [Google Scholar]
- 16.Lange, J., Kötitz, R., Haller, A., Trahms, L., Semmler, W. & Weitschies, W. (2002) J. Magn. Magn. Mater. 252, 381–383. [Google Scholar]
- 17.Enpuku, K., Minotani, T., Hotta, M. & Nakahodo, A. (2001) IEEE Trans. Appl. Superconductivity 11, 661–664. [Google Scholar]
- 18.Debye, P. (1929) Polar Molecules (Chemical Catalog, New York).
- 19.Néel, L. (1949) Ann. Geophys. 5, 99–136. [Google Scholar]
- 20.Fannin, P. C. & Charles, S. W. (1994) J. Phys. D Appl. Phys. 27, 185–188. [Google Scholar]
- 21.Clarke, J. (1996) in SQUID Sensors: Fundamentals, Fabrication and Applications, ed. Weinstock, H. (Kluwer Academic, Dordrecht, The Netherlands), pp. 1–62.
- 22.Koch, R. H., Clarke, J., Goubau, W. M., Martinis, J. M., Pegrum, C. M. & Van Harlingen, D. J. (1983) J. Low Temp. Phys. 51, 207–224. [Google Scholar]
- 23.Lee, T. S., Dantsker, E. & Clarke, J. (1996) Rev. Sci. Instrum. 67, 4208–4215. [Google Scholar]
- 24.Murray, E. G. D., Webb, R. A. & Swann, M. B. R. (1926) J. Pathol. Bacteriol. 29, 407–439. [Google Scholar]
- 25.Jones, S. & Portnoy, D. A. (1994) Infect. Immun. 62, 5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kauffmann, V. F. (1944) Acta Pathol. Microbiol. Scand. 21, 20–45. [Google Scholar]
- 27.Elbing, K. & Brent, R. (2002) in Current Protocols in Molecular Biology, eds. Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A. & Struhl, K. (Wiley, New York), p. 1.2.2.
- 28.Berkov, D. V. & Kötitz, R. (1996) J. Phys. Condens. Matter 8, 1257–1266. [Google Scholar]
- 29.Berg, H. C. (1983) Random Walks in Biology (Princeton Univ. Press, Princeton).
- 30.Weigl, B. H., Bardell, R. L. & Cabrera, C. R. (2003) Adv. Drug Delivery Rev. 55, 349–377. [DOI] [PubMed] [Google Scholar]
- 31.Chován, T. & Guttman, A. (2002) Trends Biotechnol. 20, 116–122. [DOI] [PubMed] [Google Scholar]