

Abstract
The reaction between CO2 and aqueous amines to produce a charged carbamate product plays a crucial role in post-combustion capture chemistry when primary and secondary amines are used. In this paper, we report the low energy negative-ion CID results for several anionic carbamates derived from primary and secondary amines commonly used as post-combustion capture solvents. The study was performed using the modern equivalent of a triple quadrupole instrument equipped with a T-wave collision cell. Deuterium labeling of 2-aminoethanol (1,1,2,2,-d4-2-aminoethanol) and computations at the M06-2X/6-311++G(d,p) level were used to confirm the identity of the fragmentation products for 2-hydroxyethylcarbamate (derived from 2-aminoethanol), in particular the ions CN−, NCO− and facile neutral losses of CO2 and water; there is precedent for the latter in condensed phase isocyanate chemistry. The fragmentations of 2-hydroxyethylcarbamate were generalized for carbamate anions derived from other capture amines, including ethylenediamine, diethanolamine, and piperazine. We also report unequivocal evidence for the existence of carbamate anions derived from sterically hindered amines (Tris(2-hydroxymethyl)aminomethane and 2-methyl-2-aminopropanol). For the suite of carbamates investigated, diagnostic losses include the decarboxylation product (−CO2, 44 mass units), loss of 46 mass units and the fragments NCO− (m/z 42) and CN− (m/z 26). We also report low energy CID results for the dicarbamate dianion (−O2CNHC2H4NHCO−2) commonly encountered in CO2 capture solution utilizing ethylenediamine. Finally, we demonstrate a promising ion chromatography-MS based procedure for the separation and quantitation of aqueous anionic carbamates, which is based on the reported CID findings. The availability of accurate quantitation methods for ionic CO2 capture products could lead to dynamic operational tuning of CO2 capture-plants and, thus, cost-savings via real-time manipulation of solvent regeneration energies.
Electronic supplementary material
The online version of this article (doi:10.1007/s13361-011-0161-5) contains supplementary material, which is available to authorized users.
Key words: Post-combustion capture, Amines, Carbamate anion
Introduction
Research into the chemical capture of CO2 (either via aqueous amine solutions [1–11], ionic liquids [12–17], or metal-organic frameworks [18–27]) has intensified in recent years as a result of the rate at which atmospheric CO2 levels are rising. At present, the chemisorption route to CO2 capture using organic amines at large point sources (e.g., coal-fired power stations) appears to be the most promising immediate technology for the reduction of anthropogenic CO2 emissions [28].
Our understanding of the process chemistry between aqueous amines and CO2 is sufficient for small-scale industrial applications, but improvements are still needed before the scale-up and rollout for post-combustion capture purposes. Several groups have proposed reaction equilibria [29–32] (see Scheme 1), which describe the entire process at any level. On the other hand, the molecular properties that confer excellent capture characteristics for certain amines are not well understood, and novel amine discovery/development is an active area of research. Aside from the base catalysis route (Reactions (1) and (2) in Scheme 1, the only viable pathway for tertiary amines to sequester CO2, [29]), primary (1°) and secondary (2°) amine molecules can react at slightly elevated temperatures (40–60 °C) in an alkaline aqueous environment to form a carbamate derivative R1R2NCO−2, which in turn may undergo hydrolysis to yield HCO−3 and water (Reactions 3 and 4 in Scheme 1).
Scheme 1.
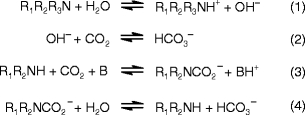
Key equilibria describing aqueous chemistry between amines and CO2
Note that both significant forms of aqueous ‘sequestered’ CO2 are anionic. In principle, all reactions in Scheme 1 are reversible by acidification of the reaction mixture or by heating. Typical process regeneration temperatures range from 100 to 150 °C. The Lewis base ‘B’ in Scheme 1 is either a molecule of water or amine.
A number of mechanisms based on aqueous kinetic data for the initial endoergic activation reaction by 1°, 2° amines have been proposed over the years [5, 29–31, 33]. Arguably the most controversial is the zwitterion mechanism of Danckwerts [33, 34], which postulates the formation of an ion pair intermediate R1R2NH+CO−2. To date, there is no evidence supporting the existence of such a species. It should also be stated that the solvent medium cannot be considered passive during the CO2 capture process, and the mechanism of CO2 capture by aqueous amines may be catalyzed by localized solvation structure.
The group of 1°-amines which possess no hydrogen atoms at the alpha position (Cα) to the reactive nitrogen have very desirable aqueous CO2 capture properties from a regeneration perspective. In industry parlance, this class of CO2 sorbent is referred to collectively as “sterically hindered” [29, 31]. A unique aspect of the capture chemistry of these species is that either (1) their carbamate derivatives are inherently susceptible to hydrolysis, or (2) there is a kinetic constraint along the pathway to carbamate formation. Unstable or hydrolysis-prone carbamates are economically desirable, as it is more cost-effective to strip aqueous CO2 from capture liquors when it is in the form of HCO−3. The performance of sterically-hindered amines has led to speculation that their reactions with CO2 only generate bicarbonate via reactions 1 and 2 (Scheme 1), much the same as for 3°-amines.
The most commonly used methods for examining the dynamic speciation of aqueous CO2 capture liquors have been ATR-FTIR and NMR spectroscopies [35–38]. While these techniques afford the simultaneous detection of neutral and ionic species, the sensitivity of these methods is poor relative to mass spectrometry (MS). MS analysis of degraded capture liquors has recently become popular [39, 40], although there is still no report of the detection of a gas-phase carbamate ion derived from an alkanolamine sorbent using this approach. As both forms of sequestered CO2 in the capture liquors of 1°, 2°-amines are anionic, MS appears to be a natural choice for the quantitation of this process variable. Real-time knowledge of the bicarbonate/carbamate speciation within PCC liquors will enable the determination of accurate heat requirements for solution regeneration, which will afford plant operators the opportunity to dynamically tune regeneration to reduce energy demands and achieve cost savings.
In this paper, we report on the collision-induced dissociation (CID) behavior of carbamate anions derived from functionalized amines; with a few exceptions, these are most commonly alkanolamines. Aside from carbamino protein adducts (proteins with a carboxylated N-terminus) [41] and a recent pre-dissociative vibrational study of piperidine-1-carboxylate [42], there is little information pertaining to hydrolyzed or anionic carbamates in the open literature. The carbamate anions discussed in this work should not be confused with esterified carbamates (NHR-C(=O)OR'), which find widespread application as pesticides and for which there are well-developed techniques for their analysis and quantitation using both GC- and LC-MS [43, 44].
We begin this paper with a detailed examination of the gas-phase chemistry of the carbamate derived from 2-aminoethanol (MEA); both computational and deuterium-labeling CID experiments are used to confirm MS structural assignments. We then proceed to identify similarities between condensed- and gas-phase chemistries, and generalize the 2-hydroxyethylcarbamate results for carbamates derived from more complex alkanolamines. In this context, dissociations common to the carbamate (anion) suite are utilized for the development of a dynamic quantitation method. The method entails separation via ion chromatography (IC) and MS detection using multiple reaction monitoring (MRM). We are particularly interested in developing this method for carbamates derived from sterically-hindered amines, which are typically present at low concentrations and evade detection using NMR and ATR-FTIR. Although sterically-hindered amines are rarely used for CO2 capture as a sole sorbent, they play a role in synergistic amine blends when the rate of absorption by a mixture of amines is greater than the rate of any of the individual components in isolation. Potential limitations of IC-MRM for carbamate quantitation are also discussed.
Experimental
Reagents and Carbamate Synthesis
The carbamate derivatives of capture amines were generated by mixing equimolar amounts (1–2 mmol) of the respective amine and NaHCO3 (Sigma, Sydney, Australia, 99.9%) in 15 mL charcoal-filtered water (R > 18 MΩ). The amines investigated and their structures are presented in Figure 1. Reagent purity: MEA (Sigma, Sydney, Australia, > 99%), EN (Sigma, Sydney, Australia, 99%), PZ (Sigma, Sydney, Australia, 99 %), AMP (Fluka, Sydney, Australia, 99%), DEA (Aldrich, Sydney, Australia, > 99.5%), Tris (Sigma, Sydney, Australia, > 99%). The mixtures were then heated to 60 °C in a water bath for 2 h. The solutions were diluted appropriately before direct infusion into the mass spectrometer for CID experiments. 1,1,2,2-d4-2-Aminoethanol was purchased from CDN Isotopes (Sydney, Australia).
Figure 1.
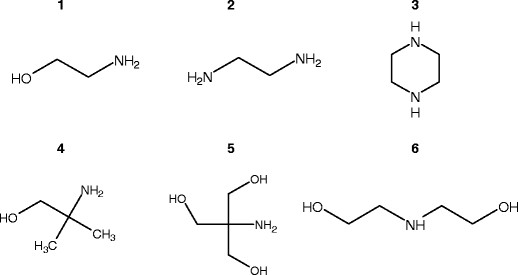
CO2 capture amines for which chemistry with bicarbonate (specifically carbamate formation) was investigated. 1 = 2-aminoethanol (ethanolamine, MEA); 2 = 1,2-diaminoethane (ethylenediamine, EN); 3 = piperazine (PZ); 4 = 2-amino-2-methyl-1-propanol (AMP); 5 = 2-amino-2-(hydroxymethyl)propane-1,3-diol (Tris); 6 = 2,2'-iminodiethanol (diethanolamine, DEA)
MS Conditions
All mass spectrometry (MS) experiments were performed using a Waters Acquity HPLC-MS/MS (Sydney, Australia) equipped with an electrospray ionization (ESI) source and a T-wave collision cell (Q1TQ2). Typical experimental conditions include: capillary voltage = 1.9–2.3 kV; cone voltage = 10–30 V; desolvation gas flow = 400–800 L/h; curtain gas flow = 0–10 L/h; collision cell bias = 1–50 V (MS/MS); infusion flow rate = 30–50 μL/min. CID experiments were performed at unit mass resolution. The collision gas employed was argon (99.999% purity, BOC, Tighes Hill, Australia). The analyzer background pressure was < 1.2 × 10–5 mbar, which rose to 1.2–2.3 × 10–3 mbar upon admission of the collision gas. Spectra were averaged over many scans (in some cases >100), particularly the CID spectra for ions of low abundance.
Theory
Gas-phase density functional theory (DFT) calculations were performed for 2-hydroxyethylcarbamate and various proton shift isomers, plus certain reactions of relevance to the CID process. We have used the Minnesota 2006 meta-density functional [45] with an augmented triple-zeta basis set [M06-2X/6-311++G(d,p)]. It has been demonstrated that the M06 meta-functional outperforms B3LYP across the board, and performs as well as CCSD(T) calculations with similar-sized basis sets for reaction energies and barrier heights [46]. All calculations were performed with the GAMESS rev. 1 software package [47]. The MacMolPlt 7.0 program [48] was used for visualization.
Geometry optimizations were performed using a Newton-Raphson steepest descent algorithm until a stationary point was located, characterized by a gradient less than 0.0001. We have used the recommended density of grid points to reduce integration errors in the DFT exchange-correlation quadrature to less than 1 microhartree per atom, namely NRAD = 96, NTHE = 36 and NPHI = 72, producing 248832 grid points per atom. Vibrational analysis was performed for all stationary points. All transition structures exhibit a single imaginary vibrational frequency (NIMAG =1).
Molar heat capacities (Cp, Cv), entropies, enthalpies and free energies at specified temperatures for both molecules and ions were provided by the GAMESS program. All gas phase reaction free energies were calculated according to standard thermochemical conventions [49].
IC-MS/MS
The IC-MS/MS experiments were performed with the Waters Acquity system described above. The modifications necessary to perform IC separations include: mobile phase A: MeOH, mobile phase B: 10 mM KOH; solvent flow rate 0.3 mL/min; column = Dionex IonPac AS4, 4 mm × 250 mm (Lane Cove, Australia); mobile phase gradient: 95.0:5.0 (A:B), t = 0 min to 50.0:50.0, t = 10 min, to 1.0:99.0 at t = 16 min; isocratic to t = 26 min; to 95.0:5.0 at t = 27 min; isocratic for 3 min. MS settings: ion mode = negative; detection mode = MRM; dwell time = 100 ms; interscan delay = 20 ms, transitions 103.0 → 59.0; 104.0 → 60.0; 129.0 → 85.0. The manufacturer’s setting controlling the resolving power of Q2 was relaxed for the MRM experiments (from 15 to 13.5).
Results and Discussion
Carbamate Derivative of 2-Aminoethanol
Most mass spectrometers in commercial analytical laboratories rarely operate in negative-ion mode, so our investigations were initially concerned with a method of quantifying protonated carbamic acids in positive-ion mode (carbamates are the conjugate bases of carbamic acids). The low energy positive-ion CID spectrum of putative protonated 2-hydroxyethylcarbamic acid (m/z 106, [HOCH2CH2CHCO2H + H]+) is presented in the Supplementary Information (Figure S1). Peaks due to consecutive water loss are evident (m/z 88, 70), together with a peak which may/may not correspond to protonated formic acid (m/z 47). The peak at m/z 23 is due to a sodium adduct with the same mass-to-charge ratio as the ion of interest. Without labeling studies, it is impossible to conclude that the mass-selected ion packet is representative of protonated 2-hydroxyethylcarbamic acid. Further confounding positive identification of this ion is the occurrence of bis-N,N-(2-hydroxyethyl)amine (DEA) at low concentrations in the MEA stock solution (confirmed separately using ion chromatography, result not shown). Protonated DEA, [(HOC2H4)2NH + H]+, has the same mass-to-charge ratio as the protonated carbamic acid derivative of interest. This anomaly could be resolved with elemental composition measurement (accurate mass), but standard triple quadrupole instruments do not possess this capability. For the reasons outlined, quantitation of acid derivatives in positive-ion mode was not pursued further.
The low energy negative-ion CID spectrum of putative 2-hydroxyethylcarbamate (HOC2H4NHCO−2) is presented in Figure 2A. The corresponding spectrum for its isoelectronic analogue, the putative carbamate derivative of 1,2-diaminoethane (H2NC2H4NHCO−2, m/z 103) is presented in Figure 2B. It is clear from Figure 2B that substitution of the hydroxyl group in MEA for an amine group in EN has only a minor effect on the carbamate fragmentation chemistry, which manifests as a smaller water loss peak for 2-hydroxyethylcarbamate. Common neutral losses and fragment ions, and their relative abundances, are presented in Table 1.
Figure 2.
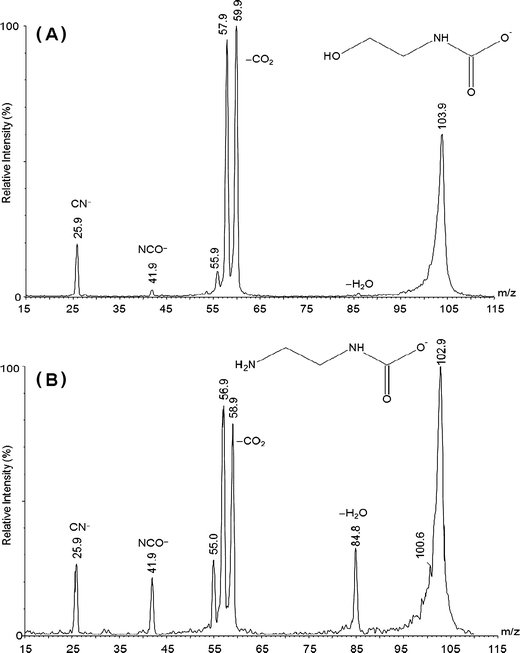
(a) Negative-ion low energy CID spectrum of 2-hydroxyethylcarbamate, HOC2H4NHCO−2; (b) negative-ion low energy CID spectrum of 2-aminoethylcarbamate, H2NC2H4NHCO−2
Table 1.
Normalized CID Neutral Loss/Product Ion Abundances for the Carbamate Derivatives of CO2 Capture Amines Investigated in this Work. T-cell bias = 12 V. Dissociation Products Specific to a Particular Carbamate are not Presented
| Normalized carbamate CID product abundance (%), T-cell bias = 12 V | ||||||
|---|---|---|---|---|---|---|
| R-carbamate, R = | 2-hydroxyethyl | 2-aminoethyl | Bis(2-hydroxyethyl) | Piperazine-1a | (1-hydroxypropan-2-yl) | 1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl |
| Parent amine (see Figure 1) | MEA | EN | DEA | Pz | AMP | Tris |
| Neutral loss/product ion: | ||||||
| -H2O | 1.3 | 32.0 | 4.7 | − | − | 18.9 |
| -2 × H2O | − | − | 0.6 | − | − | 2.0 |
| -CO2 | 76.1 | 40.8 | 72.0 | 84.4 | 89.1 | 54.9 |
| -46 | 19.2 | 20.0 | 12.3 | 13.7 | 10.9 | 2.4 |
| -48 | 2.3 | 2.9 | − | − | − | − |
| NCO– | 0.1 | 3.2 | <0.1 | − | − | − |
| CN– | 1.0 | 1.2 | − | − | − | − |
aPiperazine-1-carboxylate
Overall, the excellent correspondence between the CID spectra in Figure 2A and 2B leaves little doubt that carbamate ions are being sampled, in particular the common losses of 18, 44, and 46 mass units, and the common fragments m/z 26, 42. To further dispel any doubt as to the structural identity of m/z 104 (Figure 2A) and enable us to conclude we are sampling only carbamate derivatives in the gas phase, putative 1,1,2,2-d4-2-hydroxyethylcarbamate (m/z 108) was synthesized from 1,1,2,2,-d4-ethanolamine and NaHCO3 and subjected to CID (see Supplementary Information Figure S2). The dominant peak in the spectrum corresponds to loss of 44 mass units (m/z 64), which can only be due to CO2 expulsion and confirms the diagnostic nature of this loss for carbamate species. M/z 26 and 42 are also present, and can only correspond to CN− and NCO−. Loss of H2O from the parent ion is also observed (m/z 90), but there is no evidence of HDO or D2O loss, confirming that reactive amino- and hydroxyl-group hydrogens are incorporated in the water molecule lost from the unlabelled congener. A follow-up loss from m/z 90 ([C2,D4,N,O2]−) cannot contribute to the product ion measured at m/z 61. This ion could be due to (1) formation of HCO−3 (loss of neutral d4-ethanimine), (2) loss of HDO after hydrogen scrambling, which in turn facilitates CO loss from a meta-stable product ion, (3) loss of HD from either d4-aminoethanolate or d4-(2-hydroxyethyl)azanide, or (4) loss of d3-acetaldehyde to form d1-carbamate, NHD-CO−2. To help resolve the origin of m/z 61, m/z 64 ([C2,D4,H2,O,N]− was generated in-source and subjected to MS/MS (quasi-MS3). Fragments at m/z 61 and m/z 58 were detected (see Supplementary Information Figure S3), however this result simply identifies ‘(iii)’ as a potential pathway to formation of this ion packet. Neighboring substituent effects in the gas phase chemistry of functionalized radical amine cations, as identified by Schwarz and Levsen [50], leads to activation of C–H bonds proximal to ionized heteroatoms, and explains the incorporation of deuterium in the neutral losses giving rise to m/z 61, 60, and 58. As these ions are not integral to our identity assignment, no further comment is necessary.
The negative-ion MS/MS spectra presented in Figure 2 establish that carbamates can be readily synthesized and identified using tandem mass spectrometry. Theoretical studies described in the next section shed light on the energetics of carbamate dissociations. 2-Hydroxyethylcarbamate is chosen as a model system due to its small size (56 electrons, computationally tractable), and because it is the carbamate derivative of MEA, the most studied and widely-used amine for CO2 scrubbing.
2-Hydroxyethylcarbamate Dissociations: M06-2X/6-311++G(d,p)
It is reasonably well established that the lowest energy gas phase conformer for 2-hydroxyethylcarbamate (HOC2H4NHCO−2, Structure 1 in Figure 3) is hydrogen-bonded between the carbamate CO2 moiety and the hydroxyethyl OH group [51, 52]. Other isomeric forms, which are possible intermediates in pre-dissociative rearrangements, include carboxy(2-hydroxyethyl)azanide (syn-HOC2H4NCO2H−, Structure 2 in Figure 3; anti-HOC2H4NCO2H−, Structure 3 in Figure 3), and 2-(carboxyamino)ethanolate (syn-−O-C2H4NHCO2H, Structure 4 in Figure 3 ; anti-−O-C2H4NHCO2H, Structure 5 in Figure 3). The lowest energy conformers of these species were found to be +61.3 kJ and +108.7 kJ less stable than Structure 1, respectively, and will not play a significant role in condensed phase chemistry. In addition, the relative energies of several 2-aminoethylcarbonate conformers were determined (−O2CO-C2H4-NH2, structures not shown); the most stable conformer was found to be +19.0 kJ/mol higher in energy than Structure 1. We conclude that only 2-hydroxyethylcarbamate is being sampled from aqueous amine solutions and studied in the mass spectrometer.
Figure 3.
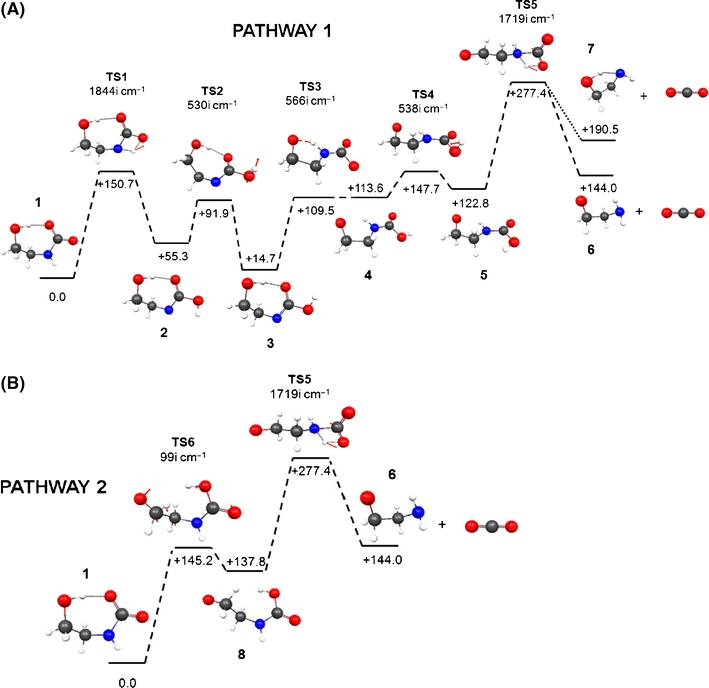
M06-2X/6-311++G(d,p) potential energy surfaces for CO2 loss from 2-hydroxyethylcarbamate (Pathways 1 and 2). All energies are in kJ relative to the most stable conformer of 2-hydroxyethylcarbamate (1, 0.0 kJ). TS = transition structure; carbon = black, hydrogen = white, oxygen = red, nitrogen = blue. Vectors describing reactive atomic motions in TS’s are in red
One important aspect of CO2 loss from 1 is the form of the product ion [C2,H6,O,N]−; direct loss will presumably lead to 2-hydroxyethylazanide (Structure 7 in Figure 3), whereas intramolecular proton migration from the hydroxyl group to the nitrogen atom (either accompanying or immediately following CO2 loss) will lead to 2-aminoethanolate (Structure 6 in Figure 3). The density functional results suggest that the 2-aminoethanolate form is far more stable (by +46.5 kJ/mol) than the 2-hydroxyethylazanide form. To determine whether the migration accompanies or follows carbamate decarboxylation, the 2-hydroxyethylazanide to 2-aminoethanolate proton transfer transition structure (TS) was located. The relative free energy of this TS represents the proton migration activation energy, and was found to have an energy of +41.6 kJ/mol relative to 2-aminoethanolate (confirmed by intrinsic reaction coordinate (IRC) calculations). DFT occasionally performs poorly for barrier heights [53], so coupled cluster calculations with perturbative triples corrections and an augmented correlation-consistent triple-ζ basis set [CCSD(T)/aug-cc-pVTZ//M06-2X/6-311++G(d,p)] were used to determine the single point energies for the three participating structures, i.e., the two minima and the connecting TS. The relative free energies at the two levels of theory are presented in Table 2 for comparison.
Table 2.
Relative Energies of 2-Aminoethanolate, 2-Hydroxyazanide, and the Connecting Transition Structure
| Structure | SCF energies (hartree) | Relative free energies (298 K, kJ/mol) | Relative energies (0 K, kJ/mol) | |||
|---|---|---|---|---|---|---|
| M06-2X/ 6-311++G(d,p) | CCSD(T)/ aug-cc-pVTZ | M06-2X/ 6-311++G(d,p) | CCSD(T)/ aug-cc-pVTZ | M06-2X/ 6-311++G(d,p) | CCSD(T)/ aug-cc-pVTZ | |
| 2-Aminoethanolate | -209.7663371 | -209.4752307 | 00.0 | 00.0 | 00.0 | 00.0 |
| TS | -209.7465832 | -209.4559034 | 41.7 | 40.5 | 41.3 | 40.2 |
| 2-Hydroxyethylazanide | -209.7468252 | -209.4567026 | 46.5 | 43.9 | 41.3 | 44.0 |
Both sets of calculations agree that intramolecular proton transfer is barrierless, and 2-hydroxyethylazanide is inherently unstable. This conclusion is qualified on the basis that the M06-2X/6-311++G(d,p) geometries accurately represent the CCSD(T) geometries at the corresponding potential energy surface points.
Decarboxylation Pathways for 2-Hydroxyethylcarbamate
According to the calculations, the decarboxylation of 1 to produce CO2 and 2-aminoethanolate anion is endoergic by +144.0 kJ/mol. With reference to Figure 3, several hydrogen transfer reactions have a similar energy demand. Two pathways which were identified as resulting in decarboxylation involve migration of the hydroxyethyl group proton to (1) an azanide-imidoate nitrogen, Pathway 1 TS 3 and Structure 4, and (2) an aminocarboxyl oxygen, Pathway 2 TS6 and Structure 8. Pathway 1 begins with an intramolecular proton migration (TS1 and Structure 2) to form 2-hydroxyethylazanide (Structures 2 and 3) before isomerization to an ethanolate form (Structures 4, 5, and TS 4) and then decarboxylation; Pathway 2 results in direct formation of anti-2-(carboxyamino)ethanolate, Structure 8. Decarboxylation proceeds exclusively via TS5 and has an energy demand of +277.4 kJ/mol (+66.3 kcal/mol). IRC calculations confirm that intramolecular proton migration with the carboxyamino group leads to breaking of the N-CO2 bond, generating 2-aminoethanolate (Structure 6) and carbon dioxide.
Attempts were made to locate a TS corresponding to concerted proton-transfer/CO–2loss; this path corresponds to the shift of the hydroxyethyl proton to the amino nitrogen with concerted N–CO2 bond-breaking. A TS was located, but it did not lead to direct dissociation [TS8, see Supplementary Information (S4)]; instead, reaction path following in both directions confirmed that TS8 leads only to hydrogen exchange between the OH and NH groups via a quasi 4-coordinate ammonium structure (N–C bond length = 1.601 Å). In any case, the relative energy of TS8 (+134.2 kJ/mol) lies below the energy of dissociated CO2 and 2-aminoethanolate (+144.0 kJ/mol). All searches for stable ammonium structures (PES minima) were unsuccessful. We conclude that gas-phase decarboxylation is a multi-step process, and proceeds through a carbamic acid intermediate.
Stepwise Loss of H2O and CO Via an Isocyanate Intermediate; Carbamate (NH2-CO−2) Formation from 2-Hydroxyethylcarbamate
All TSs referred to in the following discourse are presented in Supplementary Information (S5). A TS for water loss which is consistent with the results from the isotopologue studies of 2-hydroxyethylcarbamate was also located. That is, the water molecule eliminated possesses the hydrogen atoms originally attached to (1) the amino nitrogen and (2) the oxygen of the hydroxyethyl group. The free energy of activation was found to be +137.5 kJ/mol, and leads to formation of 2-isocyanatoethanolate (−O-C2H4-N = C=O). The calculations also reveal that the extended isocyanatoethanolate structure cyclises with a negligible activation energy to form either an oxazolate or an oxazolidinide (Scheme 2).
Scheme 2.
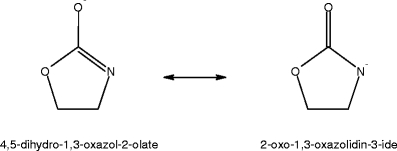
2-Hydroxyethylcarbamate dehydration products
The low intensity of the isocyanatoethanolate product ion in the CID spectrum of 2-hydroxyethylcarbamate (m/z 86, Figure 2A) suggests further decomposition of the dehydration product might be facile. Indeed, the computations indicate that loss of NCO− (isocyanate anion) from the dehydration product requires a free energy of activation of +269.4 kJ/mol, and explains the origin of the peak at m/z 42 in Figure 2A. In contrast, the loss of CO requires an activation energy of +500.8 kJ/mol (almost 5.2 eV). With reference to the CID spectrum of 1,1,2,2-d4-2-hydroxyethylcarbamate (HO-C2D4-NH-CO−2, see Supplementary Information) the absence of a peak at m/z 62 suggests CO loss from the isocyanatoethanolate anion is not operative at the collision energies employed.
One final TS is worth mentioning. Carbamate, or NH2CO−2, is the simplest ion with a carboxy group attached to an amino centre and is readily derived from ammonia and bicarbonate solutions. Decomposition of 2-hydroxyethylcarbamate via fission of the N–Cα bond (with an associated hydrogen migration) could conceivably give rise to carbamate ion. A TS was found which leads to formation of carbamate, accompanied by an Cα–N hydrogen shift, with a relative energy of +319.1 kJ/mol with respect to Structure 1. The energy required for this decomposition is only slightly larger than the decarboxylation activation energy, so it is likely that m/z 61 in the CID spectrum of 1,1,2,2-d4-2-hydroxyethylcarbamate (Supplementary Information, Figure S2) has contributions from NHD-CO−2.
2-Hydroxyethylcarbamate Dissociations In Vacuo: Correspondence with the Condensed-Phase
Amine degradation will be a significant cost factor for large-scale post-combustion CO2 capture operations. The CID experiments reported in this paper are low energy in nature and ion fragmentation results from gentle heat transfer to the ions during several collisional encounters with a neutral target gas (Ar). While there are clear disparities between the environment of an ion in the dilute vacuum and one forming multiple hydrogen bonds within a dielectric protic solution, some correspondence between the decomposition chemistry in the two phases cannot be excluded. Several relevant aspects have been identified in this study, and are discussed below.
A clear parallel can be found in the facile loss of CO2 from carbamate ions in the gas- and condensed phases. One of the many reasons post-combustion CO2 capture using aqueous amines is considered an attractive greenhouse gas abatement option is the low thermal energy of solvent regeneration. Temperatures less than 150 °C are usually sufficient to decompose carbamates. It is doubtful though, that the decomposition proceeds via the same mechanism in vacuo as in the condensed phase (in the presence of a protic solvent).
Dehydration of gas-phase carbamate ions leads to formation of isocyanates. Isocyanates are important polymer intermediates (e.g., polyurethanes) and must be prepared in the laboratory under anhydrous conditions in order to avoid the unwanted generation of carbamates. It is likely that the MEA degradation product 2-oxazolidinone, encountered in degraded capture solutions [54], is not the result of amine oxidation, but rather decomposition of the carbamate during solvent regeneration at elevated temperatures. It appears that a small percentage of MEA degradation via this pathway will be unavoidable during normal CO2 capture plant operations.
Ammonia and organic acids (oxalic, glycolic, acetic and formic) are usually monitored during CO2 capture operations as a metric of solvent performance [54]. Aqueous metal ions or metal surfaces (in which the metal is in a low-oxidation state) are known to contribute to capture amine degradation and, particularly, to the abundance of ammonia as a degradation product. Relative to amine regeneration, a small additional activation energy (41.7 kJ/mol in excess of the decarboxylation energy) is sufficient to induce acetaldehyde loss from 2-hydroxyethylcarbamate. This suggests NH2CO−2 could be formed from HO-C2H4-NH-CO−2 (2-hydroxyethylcarbamate), particularly during solvent regeneration. Acetaldehyde is believed to be a primary solvent degradation product of MEA, and well established oxidation chemistry will produce the C2-acids (given above) from this intermediate. Carbamate should also decompose to NH3 and CO2 under the conditions employed to regenerate loaded or CO2-rich MEA solutions. The computational results indicate that ammonia should also be considered as a carbamate- or thermal-degradation product, and not simply as an oxidative degradation product.
We now turn our attention to the CID spectra of more complex alkanolamines, which are used in post-combustion capture solvents.
CID Spectra of bis(2-Hydroxyethyl)Carbamate and Piperazine-1-Carboxylate
Both bis(2-hydroxyethyl)carbamate (m/z 148) and piperazine-1-carboxylate (m/z 129) are carbamates derived from secondary functionalized amines. They are, respectively, the CO2 capture products of DEA (6, Figure 1) and PZ (3, Figure 1), both of which are promising candidates for post-combustion capture application. The CID spectra of both carbamates exhibit strong CO2-loss peaks [appearing at m/z 104 and 85, respectively, see Supplementary Information (S6)], as well as losses of 46 mass units. Beyond this, there are few spectral similarities. There is a small water loss peak in the spectrum of bis(2-hydroxyethyl)carbamate, however there is no evidence of water loss in the spectrum of piperazine-1-carboxylate; both water and CO2 are lost sequentially from bis(2-hydroxyethyl)carbamate. Instead of a water loss peak at m/z 111, a very small peak at m/z 112 indicates piperazine-1-carboxylate might lose ammonia. Overall, the distinctive peaks corresponding to the losses of 44, 46 mass units leave little doubt that the ions sampled are carbamates. We refrain from further peak assignments as labeling and theoretical studies have not been undertaken.
We now apply the knowledge gained from the study of relatively “stable” carbamates to the putative carbamates of classical “sterically hindered” primary amines and answer the vexing question: Do sterically hindered amines form carbamates? Both the sterically-hindered amines (2-amino-2-methyl-1-propanol (AMP, 4 in Figure 1) and Tris(hydroxymethyl)aminomethane (Tris, 5 in Figure 1) find application in post-combustion capture solvents: the former in promoted amine mixtures, and the latter as a vapor pressure inhibitor for volatile ammonia capture solutions.
Sterically-Hindered Amines Also form Carbamates: Dissociation Spectra of (1-Hydroxypropan-2-yl)Carbamate and [1,3-Dihydroxy-2-(Hydroxymethyl)Propan-2-yl]Carbamate
To date, there have been no reports relating to the detection of carbamates derived from sterically hindered functionalized amines. The CID spectrum of the putative carbamate derivative of Tris(hydroxymethyl)aminomethane is presented in Figure 4. The spectrum presented was averaged over many scans, and the cone voltage and gas were kept low (10–20 V and 0–50 L/min) in order to facilitate carbamate observation and detection. Also shown in this figure, is the broad scan ESI-negative ion spectrum of the aqueous reaction mixture from which the carbamate ion was sampled. It is evident that the peak subsequently mass-selected for interrogation at m/z 164 ([1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]carbamate, the carbamate derivative of Tris) is quite weak; this could be a consequence of the propensity of the carbamate species to hydrolyze, or to a particular kinetic constraint to its formation. The behavior of sterically-hindered amines in CO2 capture solutions was believed to parallel that of tertiary amines, i.e., this class of amine was assumed to act purely in a base-catalytic role, not interacting directly with either aqueous CO2 or bicarbonate. The CID spectra of (1-hydroxypropan-2-yl)carbamate [see Supplementary Information (S7)] and [1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]carbamate demonstrate the existence of carbamates for these species. Diagnostic fragments and ions of Tris-carbamate include peaks at m/z 26 (CN−), m/z 42 (NCO−), and direct loss of 44 mass units (CO2) together with methanol and water loss peaks. The major dissociation pathway for (1-hydroxypropan-2-yl)carbamate is loss of CO2, with smaller peaks corresponding to loss of CO2 followed by (1) H2, (2) H2O, and (3) NH3. A small peak due to CN− at m/z 26 is barely discernable, but nevertheless present. These findings concur with theoretical results, which support the stability of the carbamates for these species [55, 56], and dispels the notion of specific base-catalysis for this class of capture amines.
Figure 4.
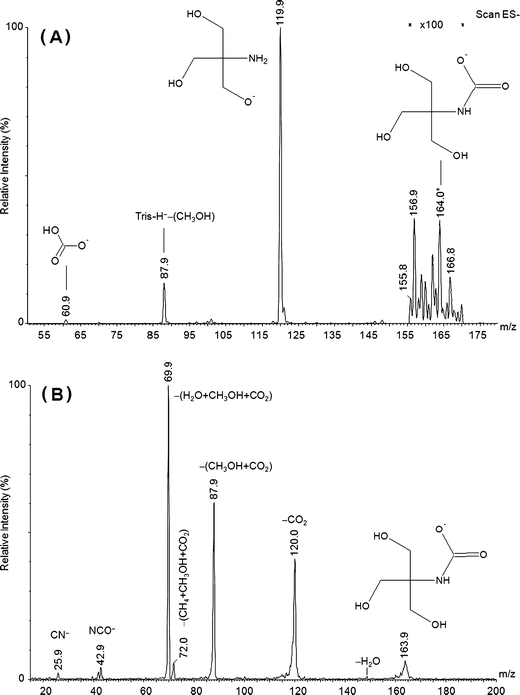
(a) The broad scan negative-ion spectrum of the aqueous reaction mixture from which [1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]carbamate was sampled using ESI-MS; (b) low energy negative-ion CID spectrum of [1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]carbamate
As there are several capture amines, which are functionalized diamines, the dicarbamate (dianion) derivative of ethylenediamine has also been investigated, and the results discussed below.
CID Behavior of a Dicarbamate: Ethane-1,2-Diyldicarbamate
A small peak at m/z 73 observed in the broad scan ESI-negative ion spectrum of the ethylenediamine/NaHCO3 mixture was mass-selected and subjected to CID experiments. The resulting spectrum is presented in the Supplementary Information (S8). Peaks at m/z 129, 102, and 85 indicate the parent ion is doubly-charged. The various fragmentations are rationalized according to Scheme 3.
Scheme 3.
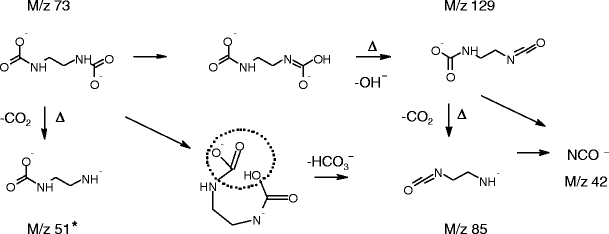
Dissociations observed for m/z 73 derived from an aqueous mixture of NaHCO3 and ethylenediamine. The asterisk denotes a fragmentation that was not observed
All fragmentation pathways of ethane-1,2-diyldicarbamate lead to charge-separation; most result from multiple dissociation processes following the initial loss of OH−.
Carbamate Separation Using Ion-Chromatography
A method capable of separating and quantifying the carbamates which form during CO2 capture could offer unprecedented insight into the synergistic capture performance of amine blends. Blends or mixtures of more than two amines can exhibit CO2 capture rates, which are greater than the sum of the rates of the individual constituents under identical conditions. Such a method would also offer the benefits described earlier concerning regeneration energy cost-savings.
The total ion chromatogram for a mixture of three carbamate derivatives is presented in Figure 5; the ion chromatograms for each of the MRM transitions monitored (one for each carbamate) are presented in the Supplementary Information (S9). The solution analyzed consisted of a 1:1:1:3 mixture of MEA:PZ:EN:HCO−3. Although the IC-method effectively separates the carbamates, which should enable determination of the response of the MS detector with varying carbamate concentration, quantifying each component will require synthesized standards which are currently unavailable. Overall, the effect of the electrospray process on the abundance of the carbamate ions is not yet clear, as the total ion counts for each carbamate are relatively low (<500 counts for each component). Unlike positive-ion mode, the low mass region (m/z 20–200) in negative-ion ES is not as crowded with solvent ions, so good detection limits can be achieved, even with low analyte ion counts. The high selectivity of MRM further enhances analyte sensitivity.
Figure 5.
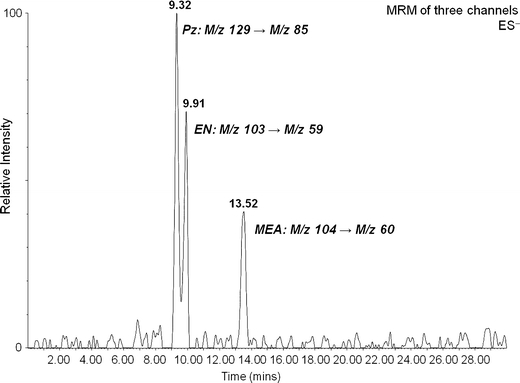
Negative IC-MS/MS (MRM) total ion chromatogram of a 1:1:1:3 molar mixture of 2-aminoethanol, 1,2-diaminoethane, piperazine, and NaHCO3. MRM transitions give rise to decarboxylation peaks (loss of CO2) for each carbamate derivative
Conclusions
The carbamate derivatives of CO2-capture sorbents are readily synthesized via the addition of 1°-, 2°-amines to aqueous solutions containing NaHCO3, followed by gentle heating. Carbamate ions prepared in this way can be transferred directly from the solution phase to the gas phase using electrospray ionization, and subjected to MS/MS experimentation. With the exception of 3°-amines, the results demonstrate both the existence and gas-phase stability of carbamate species for all amines, including those classed as “sterically hindered.” The high abundance of the decarboxylation dissociation product is consistent with condensed-phase amine scrubbing chemistry, as exemplified by the widespread application of this technology for reversible CO2 capture. Although the mechanism of N–CO−2 bond cleavage in aqueous solutions is likely to involve base molecules acting as proton shuttles, the low temperatures required for process solvent regeneration (100–150 °C for alkanolamines) suggests the carbamate group is easily activated. Other aspects of the gas-phase chemistry of carbamates i.e., carbamate dehydration leading to isocyanate formation, and Cα–N bond cleavage generating NH2-CO−2 from more complex alkanolamine-carbamates, were also identified, and appear relevant to condensed-phase process chemistry.
It is inevitable that “sterically-hindered” amines will form carbamates during solvent-based CO2 capture from gas streams, as bicarbonate ions are generated via the reaction of OH−(aq) and CO2(aq) in alkaline solutions, regardless of the choice of amine sorbent. This finding indicates that mechanistically, CO2 activation in aqueous solutions is feasible via both the carbamate and base-catalyzed routes for all 1°-, 2°-amines. It does not explain why equilibrium 4 in Scheme 1 apparently lies far to the right for sterically hindered amines, or if these amines react directly with solvated CO2. We put forward the following proposition: The hydrolysis or conversion of carbamates to bicarbonate can be observed for aqueous sorbents such as MEA and DEA on a timescale of minutes to hours [57]. This process must be rapid for sterically-hindered functionalized amines, so we contend that these amines disrupt the local solution structure to the extent that loss of order at the solute-solvent interface facilitates the hydrolyzing solvent molecule achieving the critical/reactive configuration. The result is almost instantaneous bicarbonate formation. On the other hand, this desirable trait also inhibits amine–CO2 interactions, since the rates of CO2 capture by sterically-hindered amines are usually less than those of unhindered 1°-, 2°-amines, but greater than 3°-amines.
The results presented in this work demonstrate that the separation and sensitive detection of carbamates in CO2 capture process liquors is achievable using IC-MRM. Method development for the quantitation of carbamate derivatives—which were not investigated as part of this work—will need to consider carbamate dissociations on a case-by-case basis. Work investigating the MS detector response to carbamates with variation of the CO2 loading is ongoing.
Electronic supplementary material
CID spectra of protonated 2-hydroxyethylcarbamic acid (S1), 1,1,2,2-d4-2-hydroxyethylcarbamate (S2), de-carboxylated d4-2-hydroxyethylcarbamate (S3), potential energy surface for 2-hydroxyethylcarbamate HO-NH hydrogen scrambling derived from M06-2X/6-311++G(d,p) (S4), transition structures for NH2CO−2 loss from 2-hydroxyethylcarbamate, and NCO− and CO losses from 2-isocyanatoethanolate (S5), CID spectra of bis(2-hydroxyethyl)carbamate and piperazine-1-carboxylate (S6), (1-hydroxypropan-2-yl)carbamate (S7) and ethane-1,2-diyldicarbamate (S8), individual MRM chromatograms for piperazine-1-carboxylate, 2-aminoethylcarbamate and 2-hydroxyethylcarbamate (S9).
Positive-ion CID spectrum of putative protonated 2-hydroxyethylcarbamic acid (M/z 106). (DOC 48 kb)
Negative-ion CID spectrum of 1,1,2,2-d4-2-hydroxyethylcarbamate. (DOC 44 kb)
Negative-ion CID spectrum of de-carboxylated d4-2-hydroxyethylcarbamate generated in-source (quasi-MS3). (DOC 51 kb)
2-hydroxyethylcarbamate HO-NH hydrogen scrambling pathway derived from M06-2X/6-311++G(d,p). Energies are relative to the lowest energy 2-hydroxyethylcarbamate conformer (1, 0.0 kJ/mol). (DOC 294 kb)
Transition structures for NH2CO−2 loss from 2-hydroxyethylcarbamate, and NCO− and CO losses from 2-isocyanatoethanolate. Oxygen = red, Carbon = black, Hydrogen = white, Nitrogen = blue. Reaction coordinate vectors shown in red. (DOC 67 kb)
Negative-ion CID spectrum of bis(2-hydroxyethyl)carbamate (A) and piperazine-1-carboxylate (B). (DOC 90 kb)
Negative-ion CID spectrum of (1-hydroxypropan-2-yl)carbamate. (DOC 54 kb)
Negative-ion CID spectrum of ethane-1,2-diyldicarbamate (the dicarbamate derivative of 1,2-diaminoethane). (DOC 50 kb)
Ion chromatograms for each carbamate in the mixture: top=piperazine -1-carboxylate, centre =2-hydroxyethylcarbamate. (DOC 390 kb)
Acknowledgments
P.J. and M.I.A. thank the Coal Portfolio and the Computational and Simulation Sciences Transformed Capability Platform (CSS-TCP) c/- Dr. John Taylor, Centre for Mathematical and Information Sciences (CMIS, CSIRO) for financial support. The authors also thank the NCI Facility for a generous allocation of computer time.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Special Issue: The capture of carbon dioxide from industrial sources: technological developments and future opportunities. Ind. Eng. Chem. Res.45, 2413–2900 (2006)
- 2.Caplow M. Kinetics of carbamate formation and breakdown. J. Am. Chem. Soc. 1968;90:6795–6803. doi: 10.1021/ja01026a041. [DOI] [Google Scholar]
- 3.Savage DW, Sartori G. Amines as rate promoters for carbon dioxide hydrolysis. Faraday Discuss. Chem. Soc. 1984;77:17–31. doi: 10.1039/dc9847700017. [DOI] [Google Scholar]
- 4.Chakraborty AK, Astarita G, Bischoff KB. CO2 absorption in aqueous solutions of hindered amines. Chem. Eng. Sci. 1986;41:997–1003. doi: 10.1016/0009-2509(86)87185-8. [DOI] [Google Scholar]
- 5.Crooks, J.E., Donnellan, J.P.: Kinetics and Mechanism of the reaction between carbon dioxide and amines in aqueous solution. J. Chem. Soc. Perkin Trans. II 331–333 (1989)
- 6.Little RJ, Versteeg GF, Van Swaaij WPM. Kinetics of CO2 with primary and secondary amines in aqueous solutions—I. Zwitterion deprotonation kinetics for DEA and DIPA in aqueous blends of alkanolamines. Chem. Eng. Sci. 1992;47:2027–2035. doi: 10.1016/0009-2509(92)80319-8. [DOI] [Google Scholar]
- 7.Pacheco MA, Rochelle GT. Rate-based modeling of reactive absorption of CO2 and H2S into aqueous methyldiethanolamine. Ind. Eng. Chem. Res. 1998;37:4107–4117. doi: 10.1021/ie980123g. [DOI] [Google Scholar]
- 8.Freguia S, Rochelle GT. Modeling of CO2 capture by aqueous monoethanolamine. AIChE J. 2003;49:1676–1686. doi: 10.1002/aic.690490708. [DOI] [Google Scholar]
- 9.Ma’mun S, Svendsen HF, Hoff KA, Juliussen O. Selection of new absorbents for carbon dioxide captures. Env. Conv. Man. 2007;48:251–258. doi: 10.1016/j.enconman.2006.04.007. [DOI] [Google Scholar]
- 10.Böttinger W, Maiwald M, Hasse H. Online NMR spectroscopic study of species distribution in MEA–H2O–CO2 and DEA–H2O–CO2. Fluid Phase Equilib. 2008;263:131–143. doi: 10.1016/j.fluid.2007.09.017. [DOI] [Google Scholar]
- 11.Kim I, Hoff KA, Hessen ET, Haug-Warberg T, Svendsen HF. Enthalpy of absorption of CO2 with alkanolamine solutions predicted from reaction equilibrium constants. Chem. Eng. Sci. 2009;64:2027–2038. doi: 10.1016/j.ces.2008.12.037. [DOI] [Google Scholar]
- 12.Bara JE, Carlisle TK, Gabriel CJ, Camper D, Finotello A, Gin DL, Noble RD. Guide to CO2 separations in imidazolium-based room-temperature ionic liquids. Ind. Eng. Chem. Res. 2009;48:2739–2751. doi: 10.1021/ie8016237. [DOI] [Google Scholar]
- 13.Huang J, Rüther T. Why are ionic liquids attractive for CO2 absorption? An Overview. Aust. J. Chem. 2009;62:298–308. doi: 10.1071/CH08559. [DOI] [Google Scholar]
- 14.Gutowski KE, Maginn EJ. Amine-functionalized task-specific ionic liquids: a mechanistic explanation for the dramatic increase in viscosity upon complexation with CO2 from molecular simulation. J. Am. Chem. Soc. 2008;130:14690–14704. doi: 10.1021/ja804654b. [DOI] [PubMed] [Google Scholar]
- 15.Carvalho PJ, Alvarez VH, Schröder B, Gil AM, Marrucho IM, Aznar M, Santos LMNBF, Coutinho JAP. Specific solvation interactions of CO2 on acetate and trifluoroacetate imidazolium based ionic liquids at high pressures. J. Phys. Chem. B. 2009;113:6803–6812. doi: 10.1021/jp901275b. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Zhang S, Lu X, Zhou Q, Fan W, Zhang X. Dual amino-functionalised phosphonium ionic liquids for CO2 capture. Chem. Eur. J. 2009;15:3003–3011. doi: 10.1002/chem.200801184. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Huo F, Liu Z, Wang W, Shi W, Maginn EJ. Absorption of CO2 in the ionic liquid 1-n-hexyl-3-methylimidazolium Tris(Pentafluoroethyl)trifluorophosphate ([hmim][FEP]): a molecular view by computer simulations. J. Phys. Chem. B. 2009;113:7591–7598. doi: 10.1021/jp900403q. [DOI] [PubMed] [Google Scholar]
- 18.Bourelly S, Llewellyn PL, Serre C, Millange F, Loiseau T, Férey G. Different adsorption behaviors of methane and carbon dioxide in the isotypic nanoporous metal terephthalates MIL-53 and MIL-47. J. Am. Chem. Soc. 2005;127:13519–13521. doi: 10.1021/ja054668v. [DOI] [PubMed] [Google Scholar]
- 19.Millward AR, Yaghi OM. Metal-organic frameworks with exceptionally high capacity for storage of carbon dioxide at room temperature. J. Am. Chem. Soc. 2005;127:17998–17999. doi: 10.1021/ja0570032. [DOI] [PubMed] [Google Scholar]
- 20.Yang Q, Xue C, Zhong C, Chen J-F. Molecular simulation of separation of CO2 from flue gases in Cu-BTC metal-organic framework. AIChE J. 2007;53:2832–2840. doi: 10.1002/aic.11298. [DOI] [Google Scholar]
- 21.Vimont, A., Travert, A., Bazin, P., Lavalley, J.C., Daturi, M., Serre, C., Férey, G., Bourrelly, S., Llewellyn, P.L.: Evidence of CO2 molecule acting as an electron acceptor on a nanoporous metal-organic-framework MIL-53 or Cr3+(OH)(O2C–C6H4–CO2). Chem. Commun 3291 (2007) [DOI] [PubMed]
- 22.Bárcia PS, Bastin L, Hurtado EJ, Silva JAC, Rodrigues AE, Chen B. Single and multicomponent sorption of CO2, CH4, and N2 in a microporous metal-organic framework. Sep. Sci. Technol. 2008;43:3494–3521. doi: 10.1080/01496390802282347. [DOI] [Google Scholar]
- 23.Bae YS, Farha OK, Hupp JT, Snurr RQ. Enhancement of CO2/N2 selectivity in a metal-organic framework by cavity modification. J. Mater. Chem. 2009;19:2131. doi: 10.1039/b900390h. [DOI] [Google Scholar]
- 24.Couck S, Denayer JFM, Baron GV, Rémy T, Gascon G, Kapteijn F. An amine-functionalized MIL-53 metal-organic framework with large separation power for CO2 and CH4. J. Am. Chem. Soc. 2009;131:6326–6327. doi: 10.1021/ja900555r. [DOI] [PubMed] [Google Scholar]
- 25.Linag Z, Marshall M, Chaffee AL. CO2 adsorption-based separation by metal-organic framework (Cu-BTC) versus zeolite (13×) Energy Fuels. 2009;23:2785–2789. doi: 10.1021/ef800938e. [DOI] [Google Scholar]
- 26.Bastin L, Bárcia PS, Hurtado EJ, Silva JAC, Rodrigues AE, Chen B. A microporous metal-organic framework for separation of CO2/N2 and CO2/CH4 by fixed-bed adsorption. J. Phys. Chem. C. 2008;112:1575–1581. doi: 10.1021/jp077618g. [DOI] [Google Scholar]
- 27.Demessence A, D’Alessandro DM, Foo ML, Long JR. Strong CO2 binding in a water-stable, triazolate-bridged metal-organic framework functionalized with ethylenediamine. J. Am. Chem. Soc. 2009;131:8784–8786. doi: 10.1021/ja903411w. [DOI] [PubMed] [Google Scholar]
- 28.Rochelle GT. Amine scrubbing for CO2 capture. Science. 2009;325:1652–1654. doi: 10.1126/science.1176731. [DOI] [PubMed] [Google Scholar]
- 29.Sartori G, Savage DW. Sterically hindered amines for carbon dioxide removal from gases. Ind. Eng. Chem. Fundam. 1983;22:239–249. doi: 10.1021/i100010a016. [DOI] [Google Scholar]
- 30.Little RJ, Versteeg GF, Van Swaaij WPM. Kinetics of CO2 with primary and secondary amines in aqueous solutions. II. Influence of temperature on zwitterion formation and deprotonation rates. Chem. Eng. Sci. 1992;47:2037–2045. doi: 10.1016/0009-2509(92)80320-C. [DOI] [Google Scholar]
- 31.Hook RJ. An investigation of some sterically hindered amines as potential carbon dioxide scrubbing compounds. Ind. Eng. Chem. Res. 1997;36:1779–1790. doi: 10.1021/ie9605589. [DOI] [Google Scholar]
- 32.Aboudheir A, Tontiwachwuthikul P, Chakma A, Idem R. Kinetics of the reactive absorption of carbon dioxide in high CO2-Loaded. Concentrated aqueous monoethanolamine solutions. Chem. Eng. Sci. 2003;58:5195. doi: 10.1016/j.ces.2003.08.014. [DOI] [Google Scholar]
- 33.Danckwerts PV. The reaction of CO2 with ethanolamines. Chem. Eng. Sci. 1979;34:443–446. doi: 10.1016/0009-2509(79)85087-3. [DOI] [Google Scholar]
- 34.Ismael M, Sahnoun R, Suzuki A, Koyama M, Tsuboi H, Hatakeyama N, Endou A, Takaba H, Kubo M, Shimizu S, Del Carpio CA, Miyamoto A. A DFT study on the carbamates formation through the absorption of CO2 by AMP. Int. J. Greenhouse Gas Control. 2009;3:612–616. doi: 10.1016/j.ijggc.2009.04.002. [DOI] [Google Scholar]
- 35.Ermatchkov V, Peres-Solado Kamps A, Maurer G. Chemical equilibrium constants for the formation of carbamates in (Carbon Dioxide + Piperazine + Water) from 1H-NMR-spectroscopy. J. Chem. Thermodyn. 2003;35:1277–1289. doi: 10.1016/S0021-9614(03)00076-4. [DOI] [Google Scholar]
- 36.Yang Q, Bown M, Ali A, Winkler D, Puxty G, Attalla M. A carbon-13 NMR study of carbon dioxide absorption and desorption with aqueous amine solutions. Energ. Proc. 2009;1:955–962. doi: 10.1016/j.egypro.2009.01.127. [DOI] [Google Scholar]
- 37.Mani F, Peruzzini M, Stoppioni P. CO2 absorption by aqueous NH3 solutions: speciation of ammonium carbamate, bicarbonate, and carbonate by a 13C NMR study. Green Chem. 2006;8:995–1000. doi: 10.1039/b602051h. [DOI] [Google Scholar]
- 38.Park H, Jung YM, You JK, Hong WH, Kim JN. Analysis of the CO2 and NH3 Reaction in Aqueous Solution by 2D IR COS: Formation of Bicarbonate and Carbamate. J. Phys. Chem. A. 2008;112:6558–6562. doi: 10.1021/jp800991d. [DOI] [PubMed] [Google Scholar]
- 39.Lepaumier H, Dominique Picq D, Carrette L-P. New amines for CO2 capture. I. Mechanisms of amine degradation in the presence of CO2. Ind. Eng. Chem. Res. 2009;48:9061–9067. doi: 10.1021/ie900472x. [DOI] [Google Scholar]
- 40.Lepaumier H, Dominique Picq D, Carrette L-P. New amines for CO2 capture. II. Oxidative degradation mechanisms. Ind. Eng. Chem. Res. 2009;48:9068–9075. doi: 10.1021/ie9004749. [DOI] [Google Scholar]
- 41.Terrier P, Douglas DJ. Carbamino group formation with peptides and proteins studied by mass spectrometry. J. Am. Soc. Mass Spectrom. 2010;21:1500–1505. doi: 10.1016/j.jasms.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Kamrath MZ, Relph RA, Johnson MA. Vibrational predissociation spectrum of the carbamate radical anion, C5H5N-CO2−, generated by reaction of pyridine with (CO2)m−. J. Am. Chem. Soc. 2010;132:15508–15511. doi: 10.1021/ja1073036. [DOI] [PubMed] [Google Scholar]
- 43.Fernández M, Picó Y, Manes J. Determination of carbamate residues in fruits and vegetables by matrix solid-phase dispersion and liquid chromatography-mass spectrometry. J. Chromatog. A. 2000;871:43–56. doi: 10.1016/S0021-9673(99)00907-3. [DOI] [PubMed] [Google Scholar]
- 44.Saraji M, Esteki N. Analysis of carbamate pesticides in water samples using single-drop microextraction and gas chromatography-mass spectrometry. Anal. Bioanal. Chem. 2008;391:1091–1100. doi: 10.1007/s00216-008-2087-8. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06 functionals and twelve other functionals. Theor. Chem. Acc. 2008;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 46.Zhao Y, Truhlar DG. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA. General atomic and molecular electronic structure system. J. Comput. Chem. 1993;14:1347–1363. doi: 10.1002/jcc.540141112. [DOI] [Google Scholar]
- 48.Bode BM, Gordon MS. MacMolPlt: a graphical user interface for GAMESS. J. Mol. Graph. Model. 1998;16:133–138. doi: 10.1016/S1093-3263(99)00002-9. [DOI] [PubMed] [Google Scholar]
- 49.Lias, S.G., Bartmess, J.E.: Gas phase ion thermochemistry, http://webbook.nist.gov/chemistry/ion/
- 50.Schwarz H, Levsen K. The chemistry of ionized amino, nitroso, and nitro compounds in the gas phase. In: Patai S, editor. The Chemistry of Amino, Nitroso and Nitro Compounds and Their Derivatives, Part 1. New York: John Wiley and Sons; 1982. pp. 85–126. [Google Scholar]
- 51.Bouchoux G. Gas phase basicities of polyfunctional molecules. Part 1. Theory and methods. Mass Spectrom. Rev. 2007;26:775–835. doi: 10.1002/mas.20151. [DOI] [PubMed] [Google Scholar]
- 52.Jackson P, Beste A, Attalla MI. Insights into amine-based CO2 capture: an Ab initio self-consistent reaction field investigation. J. Struct. Chem. 2011 [Google Scholar]
- 53.Grimme S, Steinmetz S, Korth M. How to compute isomerization energies of organic molecules with quantum chemical methods. J. Org. Chem. 2007;72:2118–2126. doi: 10.1021/jo062446p. [DOI] [PubMed] [Google Scholar]
- 54.Strazisar BR, Anderson RR, White CM. Degradation pathways for monoethanolamine in a CO2 capture facility. Energy Fuels. 2003;17:1034–1039. doi: 10.1021/ef020272i. [DOI] [Google Scholar]
- 55.Jackson P, Attalla MI. Environmental impacts of post-combustion CO2 capture: new insights. Energy Proc. 2011;4:2277–2284. doi: 10.1016/j.egypro.2011.02.117. [DOI] [Google Scholar]
- 56.daSilva EF, Svendsen HF. Ab initio study of the reaction of carbamate formation from CO2 and alkanolamines. Ind. Eng. Chem. Res. 2004;43:3413–3418. doi: 10.1021/ie030619k. [DOI] [Google Scholar]
- 57.Jackson P, Robinson K, Puxty G, Attalla M. In-situ fourier transform infrared analysis of carbon dioxide absorption and desorption. Energy Proc. 2009;1:985–994. doi: 10.1016/j.egypro.2009.01.131. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Positive-ion CID spectrum of putative protonated 2-hydroxyethylcarbamic acid (M/z 106). (DOC 48 kb)
Negative-ion CID spectrum of 1,1,2,2-d4-2-hydroxyethylcarbamate. (DOC 44 kb)
Negative-ion CID spectrum of de-carboxylated d4-2-hydroxyethylcarbamate generated in-source (quasi-MS3). (DOC 51 kb)
2-hydroxyethylcarbamate HO-NH hydrogen scrambling pathway derived from M06-2X/6-311++G(d,p). Energies are relative to the lowest energy 2-hydroxyethylcarbamate conformer (1, 0.0 kJ/mol). (DOC 294 kb)
Transition structures for NH2CO−2 loss from 2-hydroxyethylcarbamate, and NCO− and CO losses from 2-isocyanatoethanolate. Oxygen = red, Carbon = black, Hydrogen = white, Nitrogen = blue. Reaction coordinate vectors shown in red. (DOC 67 kb)
Negative-ion CID spectrum of bis(2-hydroxyethyl)carbamate (A) and piperazine-1-carboxylate (B). (DOC 90 kb)
Negative-ion CID spectrum of (1-hydroxypropan-2-yl)carbamate. (DOC 54 kb)
Negative-ion CID spectrum of ethane-1,2-diyldicarbamate (the dicarbamate derivative of 1,2-diaminoethane). (DOC 50 kb)
Ion chromatograms for each carbamate in the mixture: top=piperazine -1-carboxylate, centre =2-hydroxyethylcarbamate. (DOC 390 kb)