

Abstract
Vastly stimulated by the discovery of opioid receptors in the early 1970s, preclinical and clinical research was directed at the study of stereoselective neuronal actions of opioids, especially those played in their crucial analgesic role. However, during the past decade, a new appreciation of the non-neuronal actions of opioids has emerged from preclinical research, with specific appreciation for the nonclassic and nonstereoselective sites of action. Opioid activity at Toll-like receptors, newly recognized innate immune pattern recognition receptors, adds substantially to this unfolding story. It is now apparent from molecular and rodent data that these newly identified signaling events significantly modify the pharmacodynamics of opioids by eliciting proinflammatory reactivity from glia, the immunocompetent cells of the central nervous system. These central immune signaling events, including the release of cytokines and chemokines and the associated disruption of glutamate homeostasis, cause elevated neuronal excitability, which subsequently decreases opioid analgesic efficacy and leads to heightened pain states. This review will examine the current preclinical literature of opioid-induced central immune signaling mediated by classic and nonclassic opioid receptors. A unification of the preclinical pharmacology, neuroscience, and immunology of opioids now provides new insights into common mechanisms of chronic pain, naive tolerance, analgesic tolerance, opioid-induced hyperalgesia, and allodynia. Novel pharmacological targets for future drug development are discussed in the hope that disease-modifying chronic pain treatments arising from the appreciation of opioid-induced central immune signaling may become practical.
I. Introduction
Thousands of years ago, opium poppy derivatives were used for a myriad of medical, social, and religious purposes, the relief of pain being reported in texts by Homer (The Iliad and The Odyssey) and Virgil (The Aeneid) as early as 850 BCE. Opioid medications continue to be prescribed for the treatment of acute and chronic pain today, although significant formulation advances and refinement of the physicochemical properties have provided some pharmacokinetic and pharmacodynamic enhancement. Despite their continual clinical use over several millennia, and intense scientific research in the past century, the importance of certain pharmacological actions of opioids has not yet been fully appreciated, including their action on immune signaling within the central nervous system, such as that derived from non-neuronal cells, such as glia. These non-neuronal actions of opioids have been investigated at the preclinical level, in parallel to their neuronal actions, over the past 4 decades, since the identification of endogenous opioid peptides and opioid receptors. Unfortunately, the advance in our understanding of opioid-induced central immune signaling has not yet been capitalized upon and amalgamated with the progress made in the opioid neuroscience and opioid pharmacology fields. For example, no controlled clinical studies have yet been conducted to determine the impact of central immune signaling on human opioid actions in healthy control and/or patient populations. As has been pointed out in the past, it is interesting to ponder what might have occurred if the opioid systems were originally identified by immunologists rather than pharmacologists (Heijnen et al., 1991; Peterson et al., 1998) and what implications this might have had for the refinement and development of opioid analgesics in the clinical management of pain.
Communication within the central nervous system (CNS1) by both neuronal and non-neuronal immune-competent cells, such as glia, contributes to homeostatic and pathological states within the CNS. This “central immune signaling” profoundly affects all types of cells within the CNS, contributing to altered behavioral responses. This happens via the release of factors such as cytokines and chemokines, as well as indirectly via the modification of the extracellular milieu of neuroexcitatory factors (e.g., via inhibition of glial glutamate transporters). It is apparent that central immune signaling induced by endogenous opioid peptides plays a critical role in the development and maintenance of the healthy CNS, modulating both neuronal and non-neuronal systems. At the same time, it is clear from preclinical studies that exogenous clinically prescribed opioids induce central immune signaling events that limit the beneficial and indicated actions of opioids. Of specific focus to this review are the effects of central immune signaling on opioid analgesia. To this end, core principles and critical concepts of opioid-induced central immune signaling are introduced, including an overview of key cell types (such as astrocytes and microglia) and mediators of central immune signaling (such as cytokines and chemokines) and a discussion of how they modify neuronal function leading to altered opioid analgesia. A review of 4 decades of preclinical research on opioid-induced central immune signaling is included to allow a unifying hypothesis of opioid analgesia, incorporating both central immune signaling and neuronal components. Particular attention is paid to the triggering events of opioid-induced central immune signaling, because it is becoming apparent that the role of classic neuronal opioid receptors expressed by either neuronal or non-neuronal cells cannot account for all central immune signaling events that occur after opioid exposure. Comparisons are drawn between opioid actions mediated through classic pathways versus through immune signaling pathways that participate in attenuating opioid analgesic efficacy. The aim of this review is to provide a better understanding of opioid actions within the CNS, by combining our current neuroscience, immunology, and pharmacology knowledge of opioid actions, hopefully enhancing our appreciation of the full complement of opioid actions within the CNS, the neuroimmunopharmacology of opioids. This integration of preclinical data may lead to improved pain relief and enhanced treatment options for sufferers of acute and chronic pain. The present review extends what has been addressed in prior reviews of opioids (e.g., Ossipov et al., 2004, 2005) and glia (e.g., DeLeo et al., 2004; Watkins et al., 2005, 2007a,b) by integrating prior knowledge of opioid actions together with current and rapidly expanding knowledge of opioid effects on glia. Sections III and IV of this review provide the first in-depth analysis of these issues in the context of glial dysregulation of opioid actions and the newly described mechanisms by which this occurs.
II. Central Immune Signaling
A. What Do Immunocompetent Cells Contribute to Central Immune Signaling?
1. Astrocytes.
Several cell types in the CNS are capable of immune signaling (that is, communication via classic immune molecules or peptides), the most abundant of these being astrocytes. Glia were thought of originally as merely structural supports, providing the “glue” that holds the nervous system in place. Evidence generated in the past few decades has demonstrated that this is not the case. Astrocytes are derived from neuronal stem cells and are immunohistochemically identified by labeling for glial fibrillary acidic protein (GFAP), populate all regions of the CNS, and serve numerous functions. These include key structural roles, such as forming the blood-brain barrier; participation in the tripartite synapse (Araque et al., 1999) [also known as the neuronal threesome (Smith, 2010)]; metabolic support of neuronal systems; supplying nutrients and neurotransmitter precursors, as well as regulation of cerebral blood flow (Attwell et al., 2010); maintenance of the extracellular environment, including uptake and release of neurotransmitters (Araque and Navarrete, 2010); regulation of ion concentrations; and detection of instances in which neuronal systems require repair (Araque and Navarrete, 2010; Smith, 2010). Each astrocyte can have as many as 30,000 connections with adjacent cells (Smith, 2010), providing immense modulatory and integrating capability. In these ways, astrocytes play a critical role in neuronal function.
In addition, astrocytes play a role in maintaining glutamate homeostasis, which is of central importance to CNS activity, and to opioid pharmacodynamics. Glutamate is critical for normal CNS function but can also act as a two-edged sword, because excessive extracellular glutamate leads to an influx of calcium ions into neurons, resulting in uncontrolled neuronal signaling and neurotoxicity. Therefore, extracellular glutamate is finely controlled, not only by its natural degradative half-life but also by being actively transported out of the synapse. Two main glutamate transporters involved in this process are EAAT1 (rat homolog glutamate/aspartate transporter; GLAST) and EAAT2 (rat homolog glutamate transporter-1; GLT-1), both of which are primarily expressed by astrocytes (as well as microglia, to a more limited degree), GLT-1 accounting for as much as 1% of the total membrane proteins (Danbolt et al., 1990). As discussed in section III.C.5, changes in astrocyte homeostasis have profound effects on glutamate transporter expression and function, as well as active glutamate release, leading to a rapid dysregulation of glutamate homeostasis with profound acute and chronic signaling and neurotoxic implications for the surrounding neuronal systems. Therefore, although not traditionally an immune signal, glutamate can be thought of as contributing to the collection of central immune signals derived from CNS immunocompetent cells such as astrocytes.
Astrocytes also play key immunological roles within the CNS. Upon activation, astrocytes are capable of expressing the full array of immune signaling and detection systems, such as cytokines and chemokines (Ben Achour and Pascual, 2010). However, the manner in which this pattern of expression changes from basal homeostasis to a fully reactive state remains to be fully described. Moreover, astrocytes display significant regional and temporal phenotypic heterogeneity, adding to the complexity of their functions (Zhang and Barres, 2010). Astrocyte immune signaling can critically affect all neighboring cell types, influencing the diverse astrocyte functions noted above. Therefore, any change in local or general central immune signaling can have a profound impact on astrocyte function (Smith, 2010). Furthermore, if such adaptations were to occur in critical pain and/or opioid analgesia centers, substantial alterations in pain processing would occur.
2. Microglia.
Believed to be the most reactive and mobile cells of the CNS, microglia are the specialized phagocytic representatives of the CNS, playing a role similar to that of peripheral macrophages (Graeber and Streit, 2010). Microglia are less numerous than astrocytes and account for approximately 15% of the total cells of the CNS. Like their peripheral macrophage counterparts, microglia are able to respond to damage or insults and present antigens. Adult microglial cells originate from primitive myeloid precursors and are an ontogenically distinct population in the mononuclear phagocyte system (Ginhoux et al., 2010). This discovery of a lineage of microglia distinct from other hematopoietic progenitor cells may prove critical for future development of microglia-based specific pharmacotherapies, suggesting the possibility of cell-type–specific factors that could be targeted.
Microglia have recently been added to the original tripartite synapse/neuronal threesome concept, so as to form a functional tetrapartite synaptic unit along with astrocytes and the pre- and postsynaptic terminals. Such a construct was recently proposed to account for microglial contributions to neuroplasticity and central sensitization (De Leo et al., 2006), which are likely to be important to opioid pharmacodynamics. Microglia are found in all regions of the brain and spinal cord, and like astrocytes, they display remarkable regional and temporal phenotypic heterogeneity. In contrast to their characterization as a “quiescent” phenotype, it is now known that microglia constantly survey the extracellular space (Nimmerjahn et al., 2005) and synaptic cleft (Wake et al., 2009) and to modulate this surveillance activity in response to insults.
Given their shared roles with peripheral macrophages, microglia possess many of the same immune signaling and response systems. Critical to their surveillance role, microglia express key innate immune receptors and accompanying response pathways, such as the innate immune pattern-recognition Toll-like receptors (TLR) that facilitate early response to insult and damage. Furthermore, and crucial to this review, are the evolving endogenous danger signal (e.g., heat shock proteins), the xenobiotic (e.g., opioids) sensing capacity of these innate immune receptors, and the reactive phenotype that microglia assume upon activation of these systems (Buchanan et al., 2010).
It is now clear that a single microglial cell can exist in a continuum of reactive phenotype states influenced by changes in the microenvironment. Certain immunohistochemical markers have been used both to identify microglia and to infer a reactive phenotype (based on up-regulation of such markers), including phospho-p38 (Svensson et al., 2005b), ionized calcium binding adaptor molecule 1 (Iba1), CD11b (complement receptor 3, identified using the OX42 antibody clone in rats), and major histocompatibility complex II (Graeber, 2010). As discussed in sections III.C.2–4, opioid exposure is capable of producing such microglia-reactive phenotypes.
It is noteworthy that once the original insult has resolved, microglia do not always return to their basal ramified phenotype; rather, they stay in a “primed” state, exhibiting elevated expression of antigen-presenting receptors (Perry et al., 1985) and apparently maintaining intracellular systems capable of faster and greater responses upon reactivation (Rönnbäck and Hansson, 1988). Moreover, like astrocytes, primed microglia display heterologous sensitization of detection and response systems, which leads to cross-sensitization that affects multiple microglial functions (Frank et al., 2007). Consequently, these primed microglia are well positioned to play a critical role in chronic pathological conditions and in situations in which repeated exposure to challenges occurs, thus contributing significantly to central immune signaling. As reviewed in section III.C, in the context of opioid treatment, repeated dosing regimens could be viewed as repeated challenges, thereby highlighting the importance of primed microglia to opioid pharmacodynamics.
3. Oligodendrocytes.
Like other glial cells, oligodendrocytes provide a key support and maintenance role for neurons, acting as the insulators of the CNS, ensheathing up to 50 axons per cell with myelin. Although less well characterized than microglia and astrocytes, oligodendrocytes are also capable of initiating some forms of immune-like signaling (Ramos et al., 2007). They are also very sensitive recipients of immune signaling, because this results in profound demyelination of neuronal axons and profound deficits in neuronal function (Piaton et al., 2010). Therefore, oligodendrocytes are a potential source of, and contributor to, central immune signaling.
4. Blood-Brain Barrier Endothelial Cells.
The blood-brain barrier forms an important boundary between the sensitive microenvironment of the CNS and the relatively volatile environment of the systemic circulation. Key to this barrier are endothelial cells of the CNS blood vessels forming tight junctions, facilitating exclusion of constituents of the systemic circulation from entry into the CNS. It is noteworthy that only recently has it been hypothesized that endothelial cells of the blood-brain barrier might contribute to immune signaling within the CNS (Quan et al., 2003; Simka, 2009). After a tissue insult and the ensuing release of peripheral or central immune signals, the tight junctions become leaky, exposing the CNS to peripheral immune signals (Stamatovic et al., 2003; Song and Pachter, 2004; Bennett et al., 2010). Although long thought to occur only as a result of insults to the CNS, it has recently been demonstrated that disruption of the blood-spinal cord barrier also occurs after peripheral nerve injuries (Gordh et al., 2006; Beggs et al., 2010). Consequently, blood-brain barrier endothelial cells remain a significant yet largely uncharacterized source of central immune signaling and contributor to altered neuronal function.
5. Peripheral Immune Cells of the Central Nervous System.
The CNS is no longer considered an entirely peripheral immune-privileged organ (Hickey, 1999), and peripheral immune cell infiltration has been implicated in many diseases (Wilson et al., 2010). This increased CNS accessibility is due largely to modifications of blood-brain barrier endothelial permeability by the induction of endothelial expression of receptors and tethering proteins, which facilitate immune cell transendothelial migration (Muller, 2009). Once in the CNS, infiltrating immune cells are able to interact with central immune signaling events, facilitating and supporting their propagation and differentiation of the immune response and eventual modification of neuronal actions.
6. Neurons.
Traditionally thought of as responding to “merely” neuronally derived neurotransmitters, it is now clear that neurons express a myriad of classic immune signaling receptors and ligands that can be up-regulated under certain conditions (Adler and Rogers, 2005). Although the consequences of neuronal immune signaling is less clear, it is apparent that neuronal immune signaling events possess the capacity to act, either directly or indirectly, via actions on nearby glia to consequently modify neuronal physiology and behavior. Neurons are known to express a variety of chemokines (e.g., monocyte chemoattractant protein-1/CCL-2, fractalkine/CX3CL1) that, upon release, activate glia (Dansereau et al., 2008; Milligan et al., 2008; Abbadie et al., 2009; Clark et al., 2009; Thacker et al., 2009; Staniland et al., 2010). Release of these chemokines from neurons in the pain pathway, for example, has been causally linked to the proinflammatory activation of spinal cord glia and release of proinflammatory cytokines leading to pain amplification (Milligan et al., 2008; White and Wilson, 2008). Recent discoveries have also uncovered the possibility of direct pathogen-to-neuron signaling, owing to neuronal TLR expression under some conditions (Wadachi and Hargreaves, 2006; Li et al., 2009). The behavioral consequences of such neuronal innate immune signaling are tantalizing, especially given the topics of opioid-induced TLR signaling to be reviewed in section III.B.3, but remain largely uninvestigated.
Although neurons can release classic immune molecules, they can also be the target of such glial-derived immune signals. For example, the following neuronal changes have been reported in response to proinflammatory cytokines: up-regulation of cell surface expression of calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Ogoshi et al., 2005); up-regulation of cell surface expression of N-methyl-d-aspartic acid (NMDA) receptors (Stellwagen et al., 2005); expression of phosphorylated NMDA receptors that exhibit increased calcium conductance (Viviani et al., 2003); down-regulation of GABA (Stellwagen et al., 2005); an increase in presynaptic release of neurotransmitters (Morioka et al., 2002); and down-regulation of G protein-coupled receptor kinase 1 (Kleibeuker et al., 2007), which when expressed at basal levels constrains neuronal excitability. Taken together, such changes in neuronal excitability and synaptic strength can clearly amplify neuronal reactivity to incoming signals.
B. What Are the Key Soluble Released Factors in Central Immune Signaling?
1. Cytokines.
Cytokines are, in general, soluble low-molecular-weight polypeptide signaling molecules/hormones (proteins or glycoproteins) that bind to specific receptors on the surface of target cells, which are coupled to intracellular signaling and second messenger pathways. Cytokines have been grouped into families based on homology of the amino acid sequence, chromosomal location, and in some cases functional homology (Borish and Steinke, 2003). Cytokine receptors exhibit high affinity, dissociation constants ranging from 10−10 to 10−12 M. Because of this high affinity, very low concentrations of cytokines can cause a biological effect, often at levels undetectable by current quantification techniques. Within the CNS, cytokines can bidirectionally communicate with each of the cell populations outlined above, performing critical roles in homeostasis, physiological responses to stress and immunological challenges, and pathological conditions. These actions occur via specific cytokine receptors and via direct cytokine modulation of other receptor functions, such as NMDA receptor cell surface expression and phosphorylation (Viviani et al., 2003; Zhang et al., 2008). As outlined in this review, cytokines are a critical component of opioid-induced central immune signaling with profound behavioral significance.
2. Chemokines.
Chemokines make up a large family of small proteins, initially characterized as chemotactic cytokines, that control the trafficking of cells expressing chemokine receptors and are now known to be involved in cellular migration and intercellular communication. Chemokine receptors are members of the G protein-coupled receptor superfamily. Chemokine distribution in the CNS, like that of neurotransmitters, is heterogeneous, theoretically allowing them to perform specific functions within specific circuitries of the CNS (Adler and Rogers, 2005). This potential for specific action is exemplified by the fact that chemokines are capable of bidirectional communication with neuronal cells and non-neuronal cells alike. A clear role for chemokines has been established in modulation of pain and opioid responses as discussed below.
3. Innate Immune Pattern Recognition Systems and Endogenous Danger Signals.
In recent years, the participation of pattern recognition receptors in normal and pathological CNS functioning has gained increasing attention (Buchanan et al., 2010). These receptors are expressed by the innate immune system cells of the CNS, including endothelial cells, microglia, and some astrocytes, but rarely by adult neurons under nonpathological conditions (Rivest, 2009; Buchanan et al., 2010; Chen and Nuñez, 2010). However, in some cases, functional neuronal TLR expression has been reported (Wadachi and Hargreaves, 2006; Li et al., 2009), although the behavioral role of such expression is yet to be clarified. Unlike traditional receptors for neurotransmitters, which display ligand selectivity and specificity, pattern recognition receptors have explicitly evolved to recognize multiple diverse conserved pathogen-associated molecular patterns that are associated with microbial pathogens or cellular stress. This multiligand recognition ability is derived from their common leucine-rich repeat containing trait (Kim et al., 2007b; Kumar et al., 2009; McGreal, 2009).
Of particular importance to the CNS is the TLR family, a collection of ∼12 single-transmembrane receptors, some found on cell surface, others inside the cell. Of these receptors, several have been extensively characterized, especially TLR4, the receptor that recognizes endotoxin [lipopolysaccharide (LPS)] and can generate sepsis (Hoshino et al., 1999). TLR4 is also activated in response to endogenous danger signals (“alarmins”), such as heat shock proteins and cell membrane components released from stressed/damaged cells (Buchanan et al., 2010). Binding of agonist ligands to TLR4 and its accessory molecules such as MD-2 and CD14 activates downstream intracellular signaling pathways similar to those activated by interleukin-1β (IL-1β) binding to its cognate receptor, resulting in a powerful proinflammatory signal (O'Neill, 2008). Indeed, the striking similarity of these pathways is reflected by the term Toll/IL-1 receptor signaling cascade (Muzio and Mantovani, 2001). A few other TLRs have also been reported to play pivotal roles in pathological CNS conditions, such as TLR2 (Mallard et al., 2009; Hong et al., 2010) and TLR3 (Obata et al., 2008). Both TLR2 and TLR4 signal through two adaptor proteins, myeloid differentiation primary response gene 88 (MyD88) and Toll/IL-1 receptor domain-containing adapter-inducing interferon-β (TRIF) (Iwasaki and Medzhitov, 2004), inducing phosphorylation of mitogen-activated protein kinases (MAPK), such as extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38. Parallel activation of inositol trisphosphate (IP3)/Akt signaling together with the previously outlined kinase activation culminates in proinflammatory transcription and eventual translational formation of cytokines such as IL-1β in its immature form (Buchanan et al., 2010) (see Fig. 1 for summary).
Fig. 1.

TLR4 signaling cascade and evidence for modulation by neuropathic pain and opioids. TLR4 signaling occurs via a cascade of events. The classic TLR4 ligand is Gram-negative bacterial LPS (hexagon), which is transported to the cell via LPS-binding protein and transfers LPS to CD14 on the cell membrane. This leads to intracellular activation of acid sphingomyelinase, which generates ceramide. Ceramide induces the generation of a lipid raft containing the coreceptor MD2, TLR4, and the 70- and 90-kDa heat shock proteins (HSP), among other elements. Ceramide also activates NADPH oxidase, which leads to peroxynitrite formation. CD14 transfers LPS to MD2, leading to both MD2-TLR4 heterodimerization and then homodimerization of MD2-TLR4 pairs. Recent evidence also suggests that opioids, such as morphine, can interact with MD2, causing a similar activation of a functional TLR4 signaling unit. Ensuing intracellular signaling occurs through toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) to at least three parallel pathways: cell motility and cell survival/apoptosis occur through the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, and proinflammatory products such as cytokines result from activation of the NF-κB and MAPK pathways. The gray boxes provide summaries of the converging evidence that neuropathic pain and opioids interact with the TLR4 signaling cascade. IRAK, IL-1-receptor-associated kinase; UEV1A, ubiquitin-conjugating enzyme E2 variant 1A; RIP2, receptor-interacting protein 2; TAK1, transforming growth factor-β-activated kinase-1; TAB3, transforming growth factor-β-activated kinase 1/MAP3K7 binding protein 3; MKK, MAP kinase kinase; IKK, IκB kinase complex; IκBα, inhibitor of nuclear factor-κB α; NEMO, NF-κB essential modifier. [Adapted from Watkins LR, Hutchinson MR, Rice KC, and Maier SF (2009) The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30:581–591. Copyright © 2009 Elsevier Science. Used with permission.]
Recent preclinical findings have demonstrated that small-molecule xenobiotics (molecules with a molecular mass of less than ∼800 kDa that are not found endogenously within the organism) are also capable of activating TLR signaling, one class of these xenobiotics being opioids. Early evidence suggests that opioid-induced TLR-dependent central immune signaling significantly modifies opioid analgesic efficacy. Therefore, TLRs may play a pivotal sensing role for the innate immune system within the CNS by detecting the presence of drugs, such as opioids, and translating their presence into a central immune signal.
Among the other pattern-recognition receptors, of which there are several types, the nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs) are also key players in immune signaling. These cytoplasmic proteins serve a variety of functions in the regulation of inflammatory and apoptotic responses. Of particular note (of the more than 20 NLRs) is NLRP3, which recognizes a diverse range of molecular patterns, ranging from endogenous compounds (such as ATP) or microbial molecules (such as bacterial DNA) to patterns associated with stress responses (such as toxins or uric acid crystals). Upon activation of NLRP3 in the cytosol, a complex assembly process is initiated, clustering the large caspase-1-activating complex called inflammasome (Bauernfeind et al., 2011). The formation of an active inflammasome is critical for the maturation of TLR signaling products because it leads to the cleavage of immature cytokines such as the conversion of IL-1β and IL-18 to their mature forms. Activation of NLRs is a key step in initiating several forms of central immune signaling.
C. What Is the Impact of the Central Immune Signaling on Pain?
1. Proinflammatory Central Immune Signaling Leads to Hyperalgesia and Allodynia.
Disease or trauma affecting the peripheral or central neuronal sensory pathways can produce a form of chronic pain known as neuropathic pain. This may occur with CNS disorders, such as stroke and multiple sclerosis, or with conditions associated with peripheral nerve damage, such as diabetic neuropathy or herpes zoster infections (shingles). It can also be induced by mechanical trauma or by neurotoxic chemicals (including drugs such as chemotherapeutics) (Goucke, 2003). It is a debilitating disease causing immeasurable suffering and reduced quality of life.
The pathophysiological mechanisms underlying the generation and maintenance of this kind of pain are intensely investigated and progressively better understood. Knowledge of the neuronal underpinnings of neuropathic pain has been complemented in the last 2 decades by heightened appreciation of the role played by central immune signaling originating from glia (and possibly resident and recruited immune cells) and communicated via proinflammatory cytokines and chemokines (Milligan and Watkins, 2009). This role is corroborated by selective blockade of glial transformation into their reactive phenotypes, using either fluorocitrate, which disrupts the Krebs cycle of glia by inhibiting the glia-specific enzyme aconitase (Hassel et al., 1992; Berg-Johnsen et al., 1993), or minocycline, which disrupts the activation of microglia and is generally considered to be devoid of direct effect on neurons or astrocytes (Ledeboer et al., 2005). Both interventions have been shown to effectively prevent allodynia and hyperalgesia in a wide range of pain models (Meller et al., 1994; Watkins et al., 1997; Milligan et al., 2000, 2003; Raghavendra et al., 2003a; Ledeboer et al., 2005).
Microglia and astrocytes are not likely to be the only glial cells involved in pain enhancement, but because they are most accessible to study, most research has focused on these cells (Watkins et al., 2007b; Milligan and Watkins, 2009). It is believed that, until activated, glia have little to no role in pain transmission (Meller et al., 1994; Watkins et al., 1997; Song and Zhao, 2001), but central immune signaling may contribute to “setting” the basal nociceptive threshold (Wolf et al., 2003; Shavit et al., 2005b). However, both astrocytes and microglia in the spinal cord assume a reactive phenotype (defined immunohistochemically by increased expression of cell-type-specific activation markers) in response to inflammation and/or damage to peripheral tissues, peripheral nerves, spinal nerves, and spinal cord (Watkins et al., 2001; Watkins and Maier, 2003; Milligan and Watkins, 2009). Enhanced pain associated with nearly every clinically relevant animal model of chronic pain examined to date is blocked by disruption of glial activation and spinal cord proinflammatory cytokine actions (Meller et al., 1994; Watkins et al., 1997; Milligan et al., 2000, 2003; Sweitzer et al., 2001; Chacur et al., 2004; see also Ledeboer et al., 2006b).
The mechanism(s) by which glially derived central immune signaling alters neuronal excitability is the focus of ongoing investigation. Neurons, including those in spinal cord dorsal horn and in the trigeminal nucleus, express receptors for proinflammatory cytokines and chemokines (Dame and Juul, 2000; Holmes et al., 2004; Ohtori et al., 2004) and exhibit increased neuronal excitability in response to these immune signals (Oka et al., 1994; Reeve et al., 2000). For example, IL-1β has been demonstrated to enhance neuronal NMDA conductance, including in spinal cord dorsal horn (Viviani et al., 2003), although IL-1β action at the IL-1 receptor is crucial to the enhanced pain states (Wolf et al., 2006). Tumor necrosis factor-α (TNF-α) rapidly up-regulates membrane expression of neuronal AMPA receptors (Beattie et al., 2002) and increases AMPA conductance (De et al., 2003). TNF-α also enhances neuroexcitability in response to glutamate (Emch et al., 2001), and IL-1β induces the release of the neuroexcitant ATP via an NMDA-mediated mechanism (Sperlágh et al., 2004). In addition, proinflammatory cytokines can induce the production of a variety of neuroexcitatory substances, including nitric oxide, prostaglandins, and reactive oxygen species (Watkins et al., 1999; Samad et al., 2001). Thus, central immune signaling can increase neuronal excitation, via various mechanisms, and thus enhance pain intensity, thereby contributing causally to the pathology of neuropathic pain.
2. Molecular Mediators Triggering Central Immune Signaling That Create and Maintain Pathologic Pain-Inducing Conditions.
TLR signaling was implicated (even though not explicitly discussed) in initiating central immune signaling associated with enhanced pain in earlier studies, in which the effect of injecting the immunogenic portions of bacteria and viruses over the spinal cord was examined (Meller et al., 1994; Reeve et al., 2000; Kehl et al., 2004). This use of bacteria and viruses without placing the results into the context of TLR signaling most likely occurs because TLR4 was not discovered to be the LPS receptor until 1998 (Poltorak et al., 1998), and TLR2 was not defined as a receptor for viruses until 2002 (Bieback et al., 2002; Düesberg et al., 2002).
The early spinal cord studies noted above demonstrated that central immune signaling initiated by TLR4 is causal to allodynia and hyperalgesia. Tanga et al. (2004) showed a correlation between increased spinal microglial TLR4 activation and the onset of behavioral hypersensitivity. In a later study, Tanga et al. (2005) demonstrated that TLR4-null mutant mice had significantly attenuated behavioral hypersensitivity and decreased expression of spinal microglial markers and proinflammatory cytokines after L5 spinal nerve transection, a standard rodent model of neuropathic pain. It is noteworthy that up-regulation of central TLR4 was associated with hypernociception as shown in diverse pain models (Raghavendra et al., 2004b) (see Fig. 1 for summary). As discussed in section II.B.3, in addition to detecting molecular patterns associated with invading pathogens, TLR4 detects host cell stress and damage (Miyake, 2007; Hutchinson et al., 2009b). In the case of neuropathic pain, Tanga et al. (2005) demonstrated that sensory nerve damage led to the activation of TLR4-dependent central immune signaling, resulting from the production of endogenous danger signals. These signals, in turn, induce nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) activation and subsequent induction of proinflammatory cytokines (Vabulas et al., 2002; Tsan and Gao, 2004) (see Fig. 1 for summary). Furthermore, TLR4 blockade not only prevents the initial development of neuropathic pain (Tanga et al., 2005) but also reverses established neuropathic pain (Bettoni et al., 2008; Hutchinson et al., 2008c; Cao et al., 2009; Lan et al., 2010; Liu et al., 2010a; Saito et al., 2010; Wu et al., 2010). The nociceptive consequences of TLR4-induced central immune signaling are due, at least in part, to p38-dependent activation of reactive glial phenotypes (Liu et al., 2010a) and up-regulation of prostaglandin E2 and TNF-α (Saito et al., 2010). Roles for other TLRs in initiating chronic pain states have also been demonstrated for TLR2 (Kim et al., 2007a; Shi et al., 2011) and TLR3 (Obata et al., 2008). Recent data suggest that the involvement of TLR2 in pain may be at peripheral, rather than central, sites of action (Shi et al., 2011).
Other direct neuron-to-glia signals have been implicated in enhanced nociception. For example, fractalkine (CX3CL1) is a protein tethered to the extracellular surface of neurons that can be released and diffuse away in response to strong neuronal activation, such as pathological pain conditions (Harrison et al., 1998; Chapman et al., 2000). It is also considered to be a putative neuron-to-glia signal, because spinal cord neurons alone express CX3CL1 and microglia alone express the respective CX3CR1 receptors, a (Harrison et al., 1998; Hughes et al., 2002; Watkins et al., 2003; Verge et al., 2004; Milligan et al., 2008). Indeed, under conditions of neuropathic pain, the expression of CX3CR1 on microglia is up-regulated (Verge et al., 2004), induced by an IL-6/p38 MAPK signaling cascade (Lee et al., 2010). The administration of exogenous CX3CL1 induces exaggerated pain states, whereas the blockade of endogenous CX3CL1 or knockout of CX3CR1 attenuates these symptoms in animal models of neuropathic pain (Watkins et al., 2003; Milligan et al., 2004, 2008; Staniland et al., 2010). This suggests that peripheral nerve injury leads to the release of CX3CL1 from neurons in the dorsal spinal cord. Furthermore, administration of a CX3CR1 antagonist after establishment of a neuropathic pain model reduces nociceptive responses, suggesting prolonged release of CX3CL1 and a role in the maintenance of neuropathic pain (Milligan et al., 2004). Although the release of CX3CL1 is probably not the earliest initiating neuron-glia activation signal in neuropathic pain, it is clearly critical for the maintenance of central immune signaling associated with chronic pain.
Another likely important chemokine, CCL2 (monocyte chemotactic protein-1), is up-regulated in dorsal root ganglion (DRG) neurons by chronic constriction injury, transported to the dorsal horn, and released in response to neuronal impulses (Tanaka et al., 2004; Zhang and De Koninck, 2006; Thacker et al., 2009). Microglial CCR2 (the receptor for CCL2) is up-regulated by peripheral nerve injury (Abbadie et al., 2003; Zhang and De Koninck, 2006; Thacker et al., 2009), and intrathecal CCL2 administration induces microglial activation, which is abolished in CCR2 knockout mice (Zhang et al., 2007). Given that CCL2 up-regulation in the spinal cord closely precedes microglial reactivity (Zhang and De Koninck, 2006), its secretion by primary afferents seems to be an initiating neuron-glial central immune signaling process causing microglial reactivity and heightened pain sensitivity (Dansereau et al., 2008).
An additional pivotal signal involved in pain transmission and hypersensitivity is ATP acting via P2 purinoceptors (Salter et al., 1993; Liu and Salter, 2005; Burnstock, 2006). Microglia express P2X4 (Tsuda et al., 2003) and P2X7 receptors (Ferrari et al., 1996; Collo et al., 1997; Möller et al., 2000; Chakfe et al., 2002; Ulmann et al., 2008). The involvement of P2X4 was demonstrated by the attenuation of established exaggerated pain responses induced by nerve injury after administration of a P2X4 antagonist (Tsuda et al., 2003). Microglial P2X7 is also involved in inflammatory pain; it has been reported that disruption of the P2X7 receptor gene prevents both chronic inflammatory and neuropathic pain hypersensitivity (Chessell et al., 2005; McGaraughty et al., 2007). Actions of P2X7 receptors may be also mediated by cells other than microglia, given that it is also expressed on macrophages, astrocytes, and the presynaptic terminals of neurons (Trang et al., 2006). Although the exact source of ATP is unknown, it may be actively released from injured or degenerating primary afferents and dorsal horn neurons. Given the role of TLR4 in pain sensitization and the requirement of the ATP-P2X-sensitive NLRP3 inflammasome to complete the cleavage of immature IL-1β to mature IL-1β, it also seems likely that TLR4 and P2 receptor signals may converge to elicit a complete central immune signal pathway. Finally, relating P2X7 back to CX3CL1 (see preceding paragraph), P2X7 signaling to microglia has recently been implicated as a causal factor in the release of cathepsin S that then leads to neuronal release of CX3CL1 (Clark et al., 2010).
Several other central immune signaling factors also play a critical role in hypernociception. For example, microglia and astrocytes express substance P receptors that are critical to short-term actions of this peptide. Intrathecal substance P administration is associated with hyperalgesia and proinflammatory reactivity of glia with selective glial p38 MAPK activation (Palma et al., 1997; Lai et al., 2000; Svensson et al., 2003). Glutamate can cause glial reactivity via either receptor mediated signaling or generation of reactive oxygen species. Under basal conditions, microglia express AMPA receptors that may act to inhibit proinflammatory cytokine release (Hagino et al., 2004), whereas astrocytes express ionotropic non-NMDA and NMDA receptors as well as metabotropic glutamate receptors that result in proinflammatory cytokine secretion via intracellular signaling pathways (Porter and McCarthy, 1997; Milligan and Watkins, 2009). However, it is likely that the combination of direct and indirect actions of glutamate lead to proinflammatory reactive glial phenotypes. Prostaglandins can contribute to central immune signaling because microglia and astrocytes both express prostaglandin receptors, facilitating glial reactivity by prostaglandin E2 secreted from other glial cells, and neurons (Palma et al., 1997; Zhao et al., 2007; Cimino et al., 2008; Telleria-Diaz et al., 2010). Further glial activation occurs via reactive oxygen species, including nitric oxide (Meller and Gebhart, 1993; Freeman et al., 2008).
Finally, the role of the κ opioid peptide dynorphin A in allodynia has recently drawn attention in the central immune signaling literature. Previously it was demonstrated that intrathecal administration of dynorphin A can induce profound allodynia via a non–opioid receptor-dependent mechanism that involved NMDA receptor activity (Vanderah et al., 1996; Laughlin et al., 1997). It is clear that dynorphin is up-regulated during nerve injury and contributes to exaggerated pain states (Zhu et al., 2006). Furthermore, central immune signaling contributes significantly to dynorphin-induced allodynia, because pretreatment with IL-1 receptor antagonist (IL-1ra), or the anti-inflammatory cytokine IL-10, inhibited dynorphin-induced allodynia (Laughlin et al., 2000). It is noteworthy that Svensson et al. (2005a) demonstrated that the degraded dynorphin A(2–17) peptide, which lacks the first amino acid, rendering it opioid-inactive, was able to induce p38 MAPK activation in microglia and increase prostaglandin E2 release. This presumably led to enhanced nociceptive signaling. Completing the evidence of the critical role of central immune signaling in the hypernociceptive actions of dynorphin is the work of Mika et al. (2010), demonstrating that minocycline treatment of neuropathic animals significantly decreased the expression of prodynorphin mRNA. It is noteworthy that this also suggests that minocycline may have some neuronal activity, although the cellular location of the prodynorphin changes was not confirmed. Therefore, dynorphin seems to be another neuron-to-glial signal that results in central immune signaling and enhanced pain; critically, however, this does not involve classic opioid receptors.
III. Opioid Analgesics and Central Immune Signaling
A. What Have We Learned from Several Millennia of Opioid Analgesic Use?
1. Neuronal Opioid Receptors, Characteristics, and Impact of Metabolism.
Despite the use of opiate analgesics derived from the opium poppy for several millennia and the comparatively more recent development of partially and fully synthetic opioids, it was not until the 1970s that research of the opioid system gained significant ground, after the isolation of endogenous compounds that bind to opioid receptors (competing with morphine binding) in brain homogenate preparations (Hughes et al., 1975). These data suggested that specific receptors for opioid compounds must be present. Indeed evidence for the presence of such receptors came from binding studies conducted using brain and selective opioid ligands and nervous tissue (Pert and Snyder, 1973; Simon et al., 1973; Terenius, 1973), which eventually led to the identification and characterization of three opioid receptor types: μ, κ, and δ (Evans et al., 1992; Satoh and Minami, 1995).
As opioid research developed, it became apparent that opioid receptors display several characteristics that distinguish them from other classes of receptors: 1) peptide opioid ligands require an intact NH2 terminus; 2) increased concentrations of sodium ion or the presence of GTP reduce opioid agonist binding but have minimal effect on antagonist affinity; 3) the binding of a ligand to an opioid receptor is often of high affinity (i.e., dissociation constant in the low nanomolar or subnanomolar concentration); 4) binding is stereoselective; 5) binding is blocked by classic opioid antagonists such as naloxone; 6) binding to novel sites has a similar rank order of affinity or effect compared with previously characterized opioid receptors; and 7) responses are pertussis-toxin sensitive, implicating receptor coupling to a G-protein (Sibinga and Goldstein, 1988). These characteristics can be used to ascertain whether novel responses are mediated via a classic opioid receptor. That is, to infer classic opioid receptor involvement, several of these characteristics must hold true, as other receptor classes share some but not all of these properties. In the infancy of opioid research much attention was clearly directed toward the stereoselective receptors that seemed to be critical for the opioid analgesic responses, whereas the nonstereoselective binding sites were considered to be experimental noise in the assays (Goldstein et al., 1971).
It is noteworthy that early opioid binding studies conducted by Goldstein et al. (1971) (see Fig. 2) demonstrated that binding to nonstereoselective but saturable specific binding sites (A minus B) were 30-fold more prevalent than the stereoselective opioid sites (B minus C), which constituted approximately only 2% of the total binding. Although the significance of opioid stereoselective sites has been well characterized, the significance of the nonstereoselective, but highly abundant, sites has not. Moreover, many of the principals outlined by Goldstein et al. (1971) to examine opioid binding have not been followed in subsequent studies, leading to the absence of further data on these abundant but specific nonstereoselective binding sites. Even studies in mice with triple knockout of the opioid receptors were conducted in a way that did not allow examination of these nonstereoselective but specific saturable binding sites because of the nature of the control methods (Simonin et al., 2001; Clarke et al., 2002; Kieffer and Gavériaux-Ruff, 2002). Even subsequent studies conducted in Goldstein's laboratory ceased to report the saturable but nonstereoselective binding sites (Lowney et al., 1974). Delfs et al. (1994) conducted one of the only studies that provided an opportunity to examine the nonclassic opioid binding sites by using parallel in situ labeling of μ opioid receptor mRNA and the binding of tritiated naloxone. The distribution of μ opioid mRNA expression mostly paralleled that of naloxone binding except for some notable brain regions. These include the cerebral cortex, area postrema, dorsal raphe nucleus, layer IV of the somatosensory cortex, anterior cingulated cortex I, frontal parietal I and III-IV, piriform, entopeduncular nucleus, and superficial gray of the superior colliculus, which contained numerous naloxone binding sites but very few labeled neurons. The possible significance and cellular location of these or similar binding sites is discussed in detail below.
Fig. 2.

Stereoselective and nonstereoselective binding of a labeled opioid active (−)-ligand (filled reverse L) and the displacement of its binding from various types of sites in tissue by excess unlabeled opioid inactive (+)-ligand (open L) or excess unlabeled (−)-ligand (open reverse L). A, the binding of the labeled (−)-ligand that will participate in all the possible kinds of binding interactions. B, binding of excess unlabeled (+)-ligand and labeled (−)-ligand will result in blockade of labeled (−)-ligand entering the nonstereoselective but saturable binding sites. Thus, the difference of A minus B represents nonstereoselective but saturable binding. C, binding of excess unlabeled (−)-ligand will result in blockade of labeled (−)-ligand entering the saturable by nonstereoselective and stereoselective binding sites. Thus, the difference of B minus C represents saturable stereoselective binding. [Adapted from Goldstein A, Lowney LI, and Pal BK (1971) Stereospecific and nonspecific interactions of the morphine congener levorphanol in subcellular fractions of mouse brain. Proc Natl Acad Sci USA 68:1742–1747. Copyright © 1971 United States National Academy of Sciences. Used with permission.]
A key feature of opioid ligands is that they show varying affinity for the different opioid receptors and their subtypes. This characteristic has been used experimentally to determine the function of specific opioid receptor subtypes, and clinically to elicit enhanced analgesia. Examination of opioid chemical structures and binding affinities to the receptors allows predictions of structure-effect relationships. For example, Chen et al. (1991) demonstrated, using rat brain homogenates, that N-demethylation of 4,5-epoxymorphinan at position 17, such as in morphine, gives rise to compounds with reduced binding affinity to the μ opioid receptor. The functional group at position 6 has little effect on binding affinity; however, the functional group at position 3 is vital to this interaction, the presence of a hydroxyl group conferring the highest affinity. It is important to note that in vivo metabolism can result in the conversion of a poor analgesic to a potent analgesic metabolite by revealing functional groups that allow high-affinity binding to opioid receptors. An example of this is the CYP2D6 conversion of codeine to morphine as well as conversion of oxycodone to oxymorphone. Therefore, poor in vitro binding does not necessarily imply poor in vivo analgesic activity because of this in vivo bioactivation of pro-drugs. The converse, however, is also true; for example, the modification of the functional group at position 3, often by conjugation, results in a profound loss of opioid receptor activity (e.g., morphine-3-glucuronide) (Chen et al., 1991). However, although this loss of opioid function may signify the end of this molecule's direct opioid receptor activity, it is apparent that metabolites such as morphine-3-glucuronide are capable of having long-term indirect consequences for opioid analgesia and nociceptive hypersensitivity that involve aspects of central immune signaling (Komatsu et al., 2009; Hutchinson et al., 2010c; Lewis et al., 2010).
2. Central Anatomical Locations of Opioid Analgesic Action and Pain Processing.
Opioids produce an analgesic effect when microinjected into CNS sites such as the periaqueductal gray, rostral ventromedial medulla, amygdala, insula, or spinal cord (Yaksh and Rudy, 1978). Furthermore, microinjection of opioid antagonists into these sites, or their inactivation by other means, reduces the analgesic effect of systemically administered opioids. With the cloning of the three classic opioid receptors (μ, δ, and κ) and a fourth related receptor (opioid-like receptor 1) and the generation of selective antibodies for each, it became possible to map their CNS distributions. Each is present in the insula, amygdala, hypothalamus, hippocampus, rostral ventromedial medulla, and spinal cord dorsal horn (Mansour et al., 1995; Darland et al., 1998). At spinal sites, ligands for the classic opioid receptors can produce an analgesic effect, in part by reducing excitatory amino acid and neuropeptide release from primary afferents (Suarez-Roca et al., 1992; Glaum et al., 1994; Grudt and Williams, 1994) and in part by a direct postsynaptic inhibition of central neurons that are activated by noxious stimulation.
If central immune signaling modifies the neuronal activity in any key center directly related to opioid action, or in the ascending or descending nociceptive pathways, then this may have a profound impact on the pharmacodynamic actions of opioids. Three areas of particular interest in this regard are the periaqueductal gray, rostral ventromedial medulla, and the dorsal root ganglia; they are reviewed in more detail here. This knowledge will be built upon later in this review to demonstrate that central immune signaling in opioid rich sites and/or in pain-processing sites is able to impact opioid analgesia.
a. Periaqueductal gray.
The periaqueductal gray of the midbrain is a small area of gray matter surrounding the central canal of the brainstem and densely packed with heterogeneous neurons (Mayer and Price, 1976; Bandler and Shipley, 1994). The periaqueductal gray integrates input from the limbic forebrain and diencephalon and is thought to represent the mechanism whereby cortical and other inputs act to modulate the nociceptive transmission from the spinal cord dorsal horn (Bandler and Keay, 1996). Analgesia resulting from microinjection of opioid agonists into the amygdala is blocked by lignocaine inactivation of, or opioid antagonist injection into, the periaqueductal gray (Pavlovic et al., 1996; Helmstetter et al., 1998). Given that few periaqueductal gray efferents project directly to the dorsal horn (Kuypers and Maisky, 1975; Castiglioni et al., 1978; Mantyh and Peschanski, 1982), it has been shown that the modulatory effect of the periaqueductal gray is exerted indirectly through efferent connections with a variety of brainstem structures, such as the rostral ventromedial medulla.
b. Rostral ventromedial medulla.
The rostral ventromedial medulla has been studied at length and is recognized as a major component of the pain modulatory circuitry, exerting its own effects in addition to relaying modulatory effects from higher brainstem sites (Fields and Basbaum, 1978; Behbehani and Fields, 1979; Pomeroy and Behbehani, 1979). There are three distinct populations of neurons in the rostral ventromedial medulla: those that discharge just before the occurrence of withdrawal from noxious heat (on-cells); those that stop firing just before a withdrawal reflex (off-cells); and those that show no consistent changes in activity when withdrawal reflexes occur (neutral cells) (Fields and Heinricher, 1985; Fields et al., 1991). Off-cells exert a net inhibitory effect on nociception, because their activation is sufficient to produce behavioral antinociception (Heinricher et al., 1994). Conversely, on-cells exert a net facilitatory effect on nociception. Periods of ongoing on-cell discharge are associated with enhanced nociception (Heinricher et al., 1989; Ramírez and Vanegas, 1989; Bederson et al., 1990; Foo and Mason, 2003); similarly, direct selective activation of on-cells produces hyperalgesia in lightly anesthetized rats (Neubert et al., 2004). The role of neutral cells remains to be elucidated (Potrebic et al., 1994; Mason, 1997). Although all components of the descending modulatory network are important, the periaqueductal gray and rostral ventromedial medulla play key roles in the underlying mechanisms of pain modulation (Fields and Basbaum, 1978; Pomeroy and Behbehani, 1979). In addition, the projection from the periaqueductal gray into the rostral ventromedial medulla is critical for the execution of its descending modulatory effect on dorsal horn nociceptive neurons (Behbehani and Fields, 1979; Fields et al., 1991; Urban and Smith, 1994; Cameron et al., 1995; Mason, 1999).
The descending modulation of nociception is not only inhibitory. Several lines of evidence demonstrate time-dependent biphasic effects of the pain modulatory system, which can inhibit or facilitate nociceptive transmission (Fields, 1988, 1992; Schaible et al., 1991; Zhuo and Gebhart, 1992, 1997; Ren and Dubner, 1996; Hurley and Hammond, 2000; Terayama et al., 2000). Descending facilitatory pathways from the rostral ventromedial medulla are involved in the maintenance of, but do not seem to be involved in the initiation of, neuropathic pain in animal models (Kovelowski et al., 2000; Burgess et al., 2002; Vera-Portocarrero et al., 2006). Injection of the local anesthetic lidocaine into the rostral ventromedial medulla reverses established behavioral hypersensitivity in nerve-injured animals but does not prevent the expression of this hypersensitivity (Burgess et al., 2002). These opposing modulatory effects indicate the rostral ventromedial medulla as a crucial site for balancing descending modulation.
c. Dorsal root ganglia.
The DRG consists of cell bodies, or soma, of sensory neurons, satellite glia, dendritic cells, macrophages, and endothelial cells (Olsson, 1990). Each neuronal cell body in the DRG is encapsulated by a layer of satellite cells separating neighboring soma (Pannese, 1981; Olsson, 1990; Shinder et al., 1998). Almost every soma is bordered by a rich network of blood vessels that is far denser than the peripheral nerve or dorsal root and is outside of the blood-brain barrier (Olsson, 1990). This cellular organization of the DRG makes processing of pain signals subject to the influence of central immune signaling.
3. Tolerance to Opioid Analgesia.
Opioid tolerance is a phenomenon in which repeated exposure to an opioid results in decreased therapeutic effect of the drug or a need for a higher dose to maintain the same effect, reflected in a rightward shift of the dose-response curve. Although opioid tolerance is a well established and characterized phenomenon in the preclinical literature, it is far less obvious in the available clinical trials data (for review, see Ballantyne and Shin, 2008) with definitive clinical evidence surprisingly lacking. However, there is evidence that prior opioid exposure results in elevated opioid requirements in some patient populations (Rapp et al., 1995; Mitra and Sinatra, 2004). Several early theories were proposed for the mechanism behind opioid tolerance, from the notion of a “silent receptor” (Collier, 1968) to more recent molecular hypotheses discussing specific signaling pathways (Toda et al., 2009). Tolerance to adverse effects of opioids does not develop at the same rate as the analgesic tolerance (Trescot et al., 2008), producing a clinical predicament, because adverse effects may limit the administration of the maximum analgesic dose. Opioid tolerance can be divided into several types. Innate or naive tolerance (in some cases this has been referred to as antianalgesia) is a genetically or environmentally determined insensitivity that is observed during the first administration (Chang et al., 2007; Watkins et al., 2007a). Acquired tolerance can be further divided into pharmacokinetic and pharmacodynamic tolerance; pharmacokinetic tolerance refers to changes in the distribution or metabolism of an opioid drug, resulting in reduced concentrations of the opioid drug in the blood or at the sites of drug action (Chang et al., 2007), whereas pharmacodynamic tolerance refers to adaptive changes in opioid receptor sensitization and/or density. Christie (2008) has further characterized this pharmacodynamic tolerance: 1) at the μ opioid receptor: partial loss of capacity to signal to intracellular effectors over time due to decreased expression and/or reduced coupling efficacy; 2) at the cell: homeostatic adaptations to the signaling system as a result of continued μ opioid receptor activation, such as cAMP up-regulation; 3) at the system: adaptations in networks linked to the μ opioid receptor, such as ORL1-receptor-nociceptin/OFQ systems, cholecystokinin, NK1 signaling, etc., which may function as antiopioids. As will be discussed later, several central immune signaling systems have also been implicated in contributing substantially to all forms of tolerance.
4. Hyperalgesia and Allodynia Induced by Opioid Analgesics.
Opioid-induced hyperalgesia and opioid withdrawal-induced hyperalgesia are paradoxical increases in pain sensitivity that develop after short- and/or long-term opioid exposure, which have clearly been demonstrated in preclinical studies (Ossipov et al., 2004, 2005) and have also been reported to occur in several patient populations (Doverty et al., 2001; Angst and Clark, 2006; Pud et al., 2006; Singla et al., 2007; Hay et al., 2009, 2010), although this is not without controversy (for review, see Fishbain et al. (2009)). Suggested mechanisms include glutamate-associated NMDA receptor activation, causing spinal neuron sensitization, which is supported by blockade of opioid-induced hyperalgesia after NMDA receptor antagonism (King et al., 2005; Ossipov et al., 2005; Mao, 2006). Other studies have documented that opioid-induced hyperalgesia results from increased excitatory neurotransmitters such as cholecystokinin, which are released by neurons from the rostral ventromedial medulla and in turn activate spinal pathways that up-regulate spinal dynorphin. Both cholecystokinin and dynorphin act as pronociceptive agents (Dourish et al., 1988; Xu et al., 1992; Vanderah et al., 2000, 2001; Gardell et al., 2002). Crain and Shen (2000) have investigated mechanisms whereby the neuronally bound GM1 ganglioside may induce a switch from the inhibitory Gi/o to stimulatory Gs coupling of the μ opioid receptor. Their in vitro data suggest that ultra-low doses of opioid antagonists may selectively block Gs-coupled μ opioid receptors because of lower activation thresholds (Crain and Shen, 2000). In a similar fashion to tolerance, recent advances in central immune signaling research have implicated several similar systems in contributing to opioid-induced hyperalgesia and allodynia, lending support to the hypothesis that hyperalgesia/allodynia and tolerance share similar mechanisms associated with opioid exposure (Ossipov et al., 2003).
5. Non-Neuronal Expressions of Opioid Receptors within the Central Nervous System.
As highlighted previously, opioid receptors are expressed in several non-neuronal organs and cell types throughout the body, including the CNS. An understanding of the cellular and temporal expression of these opioid receptors is critical to understanding the actions that classic opioids may have on central immune signaling by these non-neuronal cells.
Astrocyte opioid receptor expression is by far the best defined, including expression of μ (Dobrenis et al., 1995; Hauser et al., 1996; Ruzicka et al., 1996; Festa et al., 2002; Burbassi et al., 2010), κ (Bunn et al., 1985; Gorodinsky et al., 1995; Maderspach et al., 1995), and δ receptors (Thorlin et al., 1998a). However, this opioid receptor expression is highly varied. For example, Ruzicka et al. (1995), using primary cultures of astrocytes from various rat brain regions, generally found lower levels of μ receptor mRNA and much more δ and κ in cortical, striatal, cerebellar, hippocampal, and hypothalamic astrocytes. When expressed, μ opioid receptor mRNA was most abundantly expressed in cortical cultures, whereas the greatest levels of δ receptor mRNA were found in the cortical and hypothalamic cultures, and significant κ receptor mRNA levels were produced by the cortical, hypothalamic and cerebellar cultures. The rank order of total opioid receptor mRNA expression across different astrocyte cultures was cortex > hypothalamus > cerebellum = hippocampus > striatum (Ruzicka et al., 1995).
Another key feature of astrocyte opioid receptor expression is that it is highly dependent on cell maturity and cell cycle. Immature astrocytes display significantly greater κ (Gurwell et al., 1996; Tryoen-Toth et al., 1998) and δ (Thorlin et al., 1998b) opioid receptor expression. Persson et al. (2000) demonstrated that astrocyte δ opioid receptor protein levels increased 2-fold during mitosis and mRNA increased 3-fold during the G1/S transition. Others have found similar results for μ and κ receptor expression (Thorlin et al., 1999).
Glial μ opioid receptor expression accounts for 2 to 3% of the total opioid receptor expression observed in the rat spinal cord (Cheng et al., 1997), nucleus tractus solitarius (Glass and Pickel, 2002), dentate gyrus (Drake et al., 2002), caudate putamen (Rodriguez et al., 2001), and up to 9% in the nucleus accumbens (Svingos et al., 1996). It is noteworthy that in the spinal cord, this μ opioid receptor expression was mainly observed along the plasma membrane and sometimes near astrocytic gap junctions (Cheng et al., 1997). μ Opioid receptor-positive astrocytes have also been observed in the striatum, but the proportion of positive cells was not reported (El-Hage et al., 2006). Delta opioid receptor expression has been observed on glial-like cells in the dentate gyrus (Commons and Milner, 1996) and rat spinal cord (Cheng et al., 1997), and this accounted for up to 5% of the total δ opioid receptor expression observed. The greatest expression of κ opioid receptors is in glial cells of the posterior pituitary (pituicyte) (Bunn et al., 1985; Burnard et al., 1991). It is noteworthy that glial κ opioid receptor expression is rarely observed in the medial prefrontal cortex (Svingos and Colago, 2002) and spinal cord (Harris et al., 2004). Such low-level–to–complete absence of opioid receptor expression (Delfs et al., 1994) has led some to hypothesize that opioids modulate astrocyte function via some other nonopioid receptor mechanism (Schwartz et al., 1994).
Surprisingly few opioid receptor expression data are available for microglia. Horvath et al. (2010b) recently reported small punctate staining of the μ opioid receptor on some processes and cell bodies of some CD11b-positive microglia in the mouse spinal cord. In vitro culture data demonstrate that fetal human and mouse microglia express μ (Chao et al., 1997; Bokhari et al., 2009) and κ opioid receptor mRNA (Chao et al., 1996) and protein (Chang et al., 1996). Neonatal rat cortical microglial cultures express the complement of μ, κ, and δ opioid receptors (Turchan-Cholewo et al., 2008); however, how the expression changes with maturation in these cells is unclear. In addition, μ3 opioid receptor subtype expression by microglia has also been reported (Dobrenis et al., 1995).
Oligodendrocyte expression of opioid receptors also depends on the maturity status of the developing cell, and opioid regulation seems to be critical to oligodendrocyte maturation; μ (Knapp and Hauser, 1996; Knapp et al., 1998; Tryoen-Toth et al., 2000) and κ (Tryoen-Toth et al., 1998) opioid receptor expression is highest in immature cells and decreased in mature cells. δ Opioid receptor expression is apparently absent from this cell type (Knapp et al., 1998).
B. Why Look Beyond Neuronal Opioid Analgesic Actions?
1. The Clinical Predicament of Ineffective Opioid Analgesia and Adverse Effects.
If opioid medications provided a completely effective, safe, and predictable treatment for the management of acute and chronic pain, with no unwanted adverse effects, then there would be only an academic interest in pain research and the associated study of analgesic mechanisms, with little to no urgency in the pace of clinical and molecular pain research. Clearly this is not the case. The treatment of pain is a complex biopsychosocial process that requires a multidisciplinary approach, combining pharmacological and nonpharmacological interventions. Moreover, as outlined above, there are several nociceptive complications of opioid exposure, let alone the myriad of other potential clinical issues, including physical dependence, cognitive disorders, dysfunction of the immune and reproductive systems, respiratory depression, nausea and vomiting, pruritus, miosis, constipation, and sedation, that may be experienced by some clinical populations (Schug et al., 1992; Højsted and Sjøgren, 2007). Unfortunately, adverse effects associated with opioid treatment may limit its clinical benefit (Slatkin and Rhiner, 2003). This is borne out by data showing that long-term opioid treatment achieves the key goals of pain relief (i.e., improved functional capacity and quality of life) in only a small proportion of patients, leaving the majority poorly maintained with chronic nonmalignant pain conditions (Højsted and Sjøgren, 2007).
Therefore, there is a need to re-evaluate the actions of opioids, including those that directly modify non-neuronal systems, such as central immune signaling, incorporating advances in parallel research fields that may shed new light on how these apparently disparate opioid-induced modulations can alter opioid analgesia. Patient care must be the foremost concern; therefore, providing safe and effective pain relief is critical. Advances in the understanding of opioid action that allow only an opioid-sparing effect may have minimal clinical benefits. However, discoveries that facilitate a paradigm shift in our appreciation of optimal analgesic strategies for the management of acute and chronic pain are imperative. Hope for such a step forward in opioid pharmacology may lie in the knowledge of opioid-induced central immune signaling.
2. Mechanistic Similarities between Pathological Pain and Opioid-Induced Abnormal Pain.
It is obvious that the initial etiologies of neuropathic pain and opioid tolerance/hyperalgesia are distinct, because neuropathic pain can originate from diverse molecular and macrostructure insults in the absence of exogenous opioids (Milligan and Watkins, 2009). However, as discussed in detail in sections II.C and III, there are striking similarities between the broad mechanisms that contribute to opioid tolerance and hyperalgesia (Mao et al., 1995b; Ossipov et al., 2003; King et al., 2005), as well as with those contributing to neuropathic pain (Mao et al., 1995a; Mayer et al., 1999; Raghavendra et al., 2002). Therefore, several years ago, researchers began to look into both the neuropathic pain and opioid literature in search for common mechanisms underlying the nociceptive hyperexcitability and possibly for a better treatment of neuropathic pain, which may circumvent opioid-induced hyperexcitability. It is noteworthy that these common adaptations also occur in overlapping pain processing anatomical locations, such as the rostral ventromedial medulla, spinal cord dorsal horn, and dorsal root ganglion. Of the several common pathways implicated, central immune signaling continues to be an area of exciting and intense research and has recently been the subject of several excellent reviews (DeLeo et al., 2004; Watkins et al., 2005, 2007a, 2009; Scholz and Woolf, 2007; Mika, 2008; Ren and Dubner, 2008; Inoue, 2009; Milligan and Watkins, 2009; Austin and Moalem-Taylor, 2010; Gao and Ji, 2010).
Unlike neuropathic pain, in which the exact exogenous or endogenous molecular trigger(s) of the chronic pain state remains unknown, it is clear that opioid-induced tolerance and hyperalgesia rely on the presence of opioids and/or their metabolites. One might assume, therefore, that direct opioid-induced receptor activation and ensuing downstream adaptations may entirely account for the presentation of opioid tolerance and hyperalgesia. However, this is not the case because triple opioid receptor knockout mice are not protected against opioid-induced hyperalgesia (Juni et al., 2007; Waxman et al., 2009). These data suggest that opioid administration results in the activation of a parallel pronociceptive and/or counter opioid analgesia regulatory system. This hypothesis is supported by the ability of the classic TLR4 ligand LPS or the opioid-inactive (+)-isomer of morphine to create naive tolerance/antianalgesia (Takagi et al., 1960; Johnston and Westbrook, 2005; Wu et al., 2005, 2006a,b,c). A review of the literature discussing opioid actions mediated by nonclassic opioid receptors follows.
3. Opioid Actions That Are Not Caused by Classic Opioid Receptors.
As outlined previously, opioid receptors share several key characteristics that distinguish them from other possible binding sites (Sibinga and Goldstein, 1988). The most pertinent of these for this discussion are the following: peptide ligands require an intact NH2 terminus; binding is stereoselective; and classic opioid antagonists (such as naloxone) block binding (Sibinga and Goldstein, 1988). It is noteworthy that these classic opioid binding sites are apparently not as abundant as the saturable nonstereoselective locations (Goldstein et al., 1971), therefore providing plentiful opportunities for other opioid actions if these ligand-receptor interactions result in a functional cellular response.
Nonclassic opioid actions were observed in one of the first studies that used synthesized unnatural opioid inactive stereoisomers (Takagi et al., 1960). Takagi and colleagues (1960) observed the induction of naive tolerance/antianalgesia by administration of (+)-morphine. In the same publication, little to no stereoselectivity was observed in the antitussive activity of morphine or codeine. From then on, the classic actions of opioids were continuously examined and reported, whereas the nonclassic sites received minimal attention, probably because of the unknown site(s) of action.
For the hypothesis of nonclassic opioid action to persist in the literature, there should be examples of nonclassic opioid actions that fall loosely into three groups: 1) responses elicited by opioid ligands in the apparent absence of classic opioid receptor expression; 2) responses elicited by opioid ligands in a nonclassic opioid fashion; and 3) similar actions of nonclassic opioid ligands clearly in a nonclassic opioid fashion. In some cases, both classic and nonclassic opioid ligands may act at the same site. This does not mean, however, that a nonclassic opioid ligand must have the same nonclassic opioid site of action as an opioid ligand. For example, opioid antagonists may not act directly to antagonize the actions of a nonclassic opioid ligand; instead, functional antagonism may be the result of indirect activation of an opposing effect. Given the aim of this review, the nonclassic opioid actions discussed here focus primarily on opioid-induced central immune signaling.
Whether the actions of opioids at CNS non-neuronal cells are mediated by opioid receptors has been a topic of discussion as early as the 1980s. For example, Hansson and Rönnbäck (1983) reported significant opioid effects on protein synthesis in astrocyte cultures in the absence of classic morphine binding sites. Rönnbäck and Hansson (1985) and Hansson and Rönnbäck (1985) continued this discussion of the role of astrocytes in the development of opioid tolerance, suggesting that the synthesis of proteins that is required for the development of tolerance and physical dependence are not related to the opioid receptor actions. Despite the fact that negligible specific binding of opioids was observed in the cerebellum (Pert et al., 1974), Rönnbäck and Hansson (1985) and Hansson and Rönnbäck (1985) found significant changes in opioid-induced protein synthesis, and others observed protein metabolic changes in the cerebellum after morphine treatment (Loh and Hitzemann, 1974; Lang et al., 1975). These findings represent a clear dissociation between opioid action and opioid receptor expression. As mentioned previously, triple opioid receptor knockout mice, with the deletion of opioid receptor genes and hence expression, still maintain hyperalgesic responses to opioid exposure (Juni et al., 2007; Waxman et al., 2009; van Dorp et al., 2009).
Owing to the use of more elaborate techniques, as discussed previously, there is now evidence that the resident immunocompetent cells of the CNS possess some opioid receptor expression, albeit not homogeneous or plentiful, and this expression is highly dependent on cell maturation status. In studies examining opioid-induced changes in central immune signaling, conducted in vitro or in vivo using adult rodents, whether the responses elicited are mediated via direct action of the opioid ligand at a classic opioid receptor remains in question. These data raise the need to re-examine the abundant nonstereoselective but saturable binding sites introduced by Goldstein et al. (1971) (see Fig. 2).
Another open question is the specificity of the “selective” opioid agonist and/or antagonists that have been used to elicit and/or block “specific” opioid actions. That is, can an opioid ligand also behave in a nonclassic opioid fashion? As previously outlined, there are plentiful binding sites for some of these ligands in the binding data reported by Goldstein et al. (1971) (see Fig. 2). The opioid active antagonist (−)-naloxone is an example of such an antagonist that has been used in isolation to conclude opioid receptor involvement. There is now significant evidence that (−)-naloxone possesses substantial potent nonclassic opioid receptor activity, in addition to its opioid receptor actions, and that these actions have behavioral significance. Dunwiddie et al. (1982) established that spontaneous activity of hippocampal pyramidal cells in the CA1 region, but not neurons in the frontal cortex, were nonstereoselectively sensitive to naloxone. Whether these were direct or indirect actions on the neuron was not characterized.
Since then it has been established that the opioid receptor active isomer (−)-naloxone and the inactive isomer (+)-naloxone (Iijima et al., 1978) are capable of protecting neurons against NMDA- and quinolinate-induced neurotoxicity (Kim et al., 1987; Choi and Viseskul, 1988), blocking tritiated-LPS binding to microglia (Liu et al., 2000a,), and protecting dopamine neurons against LPS-induced neurotoxicity in vitro or in vivo by multiple actions, including decreasing microglial proinflammatory cytokines IL-1β and TNF-α expression, decreasing expression of reactive phenotype markers, decreasing inducible nitric oxide synthase transcriptional events, and decreasing superoxide production (Das et al., 1995; Kong et al., 1997; Chang et al., 2000; Liu et al., 2000a,b,c; Lu et al., 2000; Liu et al., 2002a; Qin et al., 2005). Furthermore, these neuroprotective actions of naloxone were not limited to LPS, as (+)-naloxone, (−)-naloxone, and naloxone methiodide all protected against β-amyloid-induced neurotoxicity and superoxide formation by microglia (Liu et al., 2002b). It is noteworthy that there is significant evidence that β-amyloid can activate microglia via TLR4-dependent pathways (Tang et al., 2008; Reed-Geaghan et al., 2009; Vollmar et al., 2010), suggesting, given the previously discussed evidence, that naloxone has nonstereoselective cross-ligand sensitivity of action at TLR4.
(−)-Naloxone has also been found to block LPS potentiation of serotonin-induced calcium signaling in cultured astrocytes (Hansson et al., 2008). (+)-Naloxone however failed to protect against cortical ischemia reperfusion injury (Liao et al., 2003) and myocardial ischemia (Chien and Van Winkle, 1996), in which its hypothesized anti-inflammatory and neuroprotection properties would have predicted success, although variables such as escalation of dosing regimens remain to be explored. It is noteworthy that (−)-naloxone was protective in each of these cases at equimolar doses (Chien and Van Winkle, 1996; Liao et al., 2003). These examples of nonstereoselectivity clearly preclude the role of classic opioid receptors in these responses and thus raise the question of the validity of the classic opioid receptor involvement in other studies in which (−)-naloxone was employed in isolation. It is noteworthy that this question is not restricted to (−)-naloxone; the selective μ opioid receptor antagonist β-funaltrexamine possesses properties similar to those of (+)-naloxone, raising the question of the opioid receptor requirement for these actions (Davis et al., 2007, 2008). The clear nonclassic actions of morphine were also demonstrated by Li et al. (2010, 2009) who reported neuronal apoptosis was dependent on TLR2 expression via glycogen synthase kinase 3β- and β arrestin-sensitive mechanisms. Consequently, additional care needs to be taken when assessing classic opioid receptor involvement in future research.
The complexity of the biological systems modulated by nonstereoselective nonopioid binding sites is typified by the work of Wu et al. (2006a) which highlighted the (+)- and (−)-naloxone sensitivity of LPS-induced naive tolerance/antianalgesia to morphine. We have extended these studies by demonstrating that coadministration of morphine together with (+)-naloxone or (+)-naltrexone can potentiate acute morphine analgesia, slow the development of analgesic tolerance, protect against the development of opioid dependence and hyperalgesia (Hutchinson et al., 2010c), and reverse chronic constriction-induced allodynia (Hutchinson et al., 2008c), likely via reduced glial proinflammatory responses. Others have shown actions of (+)-naloxone in scenarios as diverse as blocking cocaine- and amphetamine-induced hyperactivity (Chatterjie et al., 1996, 1998) and reducing superoxide production by human neutrophils (Simpkins et al., 1985). Important to this review are the clear consequences of nonclassic opioid signaling in decreasing opioid analgesia and contributing to unwanted adverse effects, as discussed below.
Parallel to these studies of (+)-naloxone are the nonclassic actions of inactive stereoisomers of opioid agonists. As mentioned earlier, Takagi et al. (1960) first observed the induction of naive tolerance/antianalgesia by administration of (+)-morphine. This antianalgesic action of (+)-morphine has been extended by Wu et al. (2005, 2006a,b,c, 2007), who found, in addition to extensive characterization of the induction of naive tolerance (discussed in detail in section III.B.3), that (+)-morphine decreased the acquisition of conditioned place preference, a behavioral measure of drug reward. Our work has demonstrated that the (+)-isomers of morphine and methadone are capable of inducing allodynia and hyperalgesia accompanied by expression of spinal proinflammatory cytokines (Hutchinson et al., 2010a). (+)-Morphine has also been demonstrated to induce hyper-responsivity when injected in the periaqueductal gray area (Jacquet et al., 1977), to bind nonstereoselectively to mouse thymocytes and a macrophage cell line (Roy et al., 1991, 1992, 1996), and, surprisingly, to protect dopamine neurons from toxicity (Qian et al., 2007). These later neuroprotective actions of (+)-morphine at first seem contrary to the hypothesized detrimental effects of inducing a proinflammatory central immune signal. However, the temporal association of the (+)-morphine challenge with the neurotoxic insult may lead to protection owing to the challenge coinciding with the resolution phase of the initial (+)-morphine insult. Thus (+)-morphine and similar conditioning challenges may indirectly facilitate protection from neurotoxic insults. Again, given the nonclassic actions of morphine reported here, the justification of an opioid receptor involvement based on the results of morphine alone is clearly insufficient.
Although unrelated to the above responses, it is noteworthy that the opioid-inactive isomer of methadone [(+)-methadone] has established NMDA receptor antagonist capacity, thereby demonstrating the clear potential for nonopioid actions of opioid-inactive isomers (Ebert et al., 1995), although this property is not shared by (+)-morphine or (+)-naloxone. (+)-Methadone has also been shown to decrease natural killer cell activity (Ochshorn et al., 1990), decrease human peripheral blood mononuclear cell proliferation (Singh, 1980; Thomas et al., 1995), and decrease cortical neuron neurotoxicity (Choi and Viseskul, 1988). The influence of methadone on the growth and survival of cancerous cells has revealed nonstereoselective binding (Maneckjee and Minna, 1997) and inhibition of growth (Maneckjee and Minna, 1992); (−)-naltrexone is capable of antagonizing this effect (Maneckjee and Minna, 1992) and therefore also displays cross-sensitivity.
It is noteworthy that cross-sensitivity has also been reported with (−)-naloxone [able to block the actions of (+)-morphine] and (+)-naloxone [able to block the actions of (−)-morphine] (Wu et al., 2006a; Hutchinson et al., 2010a). Given that at least some of these responses are cross-sensitive, two alternative hypotheses to explain such responses have been suggested. The first holds that there are true nonstereoselective sites via which both opioid active and opioid inactive isomers act in a competitive and/or noncompetitive fashion. Alternatively, the ligands may act at two different sites, the actions of which nullify each other, thereby creating a state of indirect, but functional, antagonism or blockade of the response. Further evidence for both cases is discussed in detail below.
The nonclassic actions of opioid ligands are not limited to small-molecule analogs; endogenous opioid peptides also possess such nonclassic actions. As mentioned earlier, the κ opioid agonist dynorphin possesses potent pronociceptive actions that are not mediated via classic opioid receptors (Svensson et al., 2005a). This nonclassic action is not limited to pronociceptive consequences, because both dynorphin 1 to 18 and 2 to 17 were able to protect neurons from LPS-induced neurotoxicity (Kong et al., 1997, 2000; Liu et al., 2001) in a κ opioid receptor antagonist-insensitive fashion (Liu et al., 2001). It is noteworthy, however, that both dynorphins were also capable of inducing a TNF-α response (Liu et al., 2001), which would be contrary to a pure blockade of the LPS toxicity, indicating that parallel processing systems may be involved here.
Collectively, these data provide significant evidence for the importance of nonclassic opioid actions in opioid pharmacodynamics. Moreover, given the clear implications of nonclassic opioid responses for opioid analgesia and the association with central immune signaling, this provides a fascinating, potentially clinically relevant application for the involvement of such signaling pathways. Accordingly, a detailed review and characterization of the immune signaling consequences of opioid exposure and the possible role of classic and nonclassic opioid responses follows.
C. What Is the Impact of Opioid Exposure on Central Immune Signaling?
Upon in vivo or in vitro opioid exposure, several aspects of central immune signaling are altered in a way that can modify neuronal homeostasis and in turn lead to changes in behavioral responses. Given the breadth of research in this area over the past 40 years, an attempt has been made to cluster these responses into five principal aspects: cell proliferation/death/survival; cell marker/phenotype (receptor expression); cell function; intracellular signaling; and extracellular signaling. A review of each of these five aspects follows. Figures summarizing the opioid-induced central immune signaling events in astrocytes (Fig. 3), microglia (Fig. 4), and neurons (Fig. 5) are also presented.
Fig. 3.

Consequences of opioid-induced central immune signaling in astrocytes. Opioid-induced central immune signaling can directly and indirectly modify astrocyte function. Opioid exposure is associated with increased expression of astrocyte chemokine receptors and activation markers, as well as the release of inflammatory mediators. It is noteworthy that there is also disruption of the astrocyte control of extracellular glutamate homeostasis owing to decreased glutamate uptake by astrocyte glutamate transporters, creating an environment of neuroexcitability (see section III.C for details).
Fig. 4.

Consequences of opioid-induced central immune signaling in microglia. Opioid-induced microglial responses occur via opioid engagement of classic opioid receptors expressed by microglia and via activation of TLR4-dependent signaling. The activation of these signaling events leads to the release of many inflammatory mediators, increased cellular motility and the up-regulation of cell surface receptors that enhance the microglial sentinel role. These classic and nonclassic actions of opioids at microglia result in a profound proinflammatory central immune signaling response that can influence neuronal and non-neuronal cells. If these microglial-dependent events occur in nociceptive control centers of the CNS, such as the dorsal spinal cord, a profound effect on the pharmacodynamics of the administered opioid can be expected (see section III.C for details).
Fig. 5.

Consequences of opioid-induced central immune signaling in neurons. Neuronal systems can be profoundly modulated and can substantially contribute to central immune signaling. Opioids can act to induce central immune signaling from neurons via direct activation of classic opioid receptors resulting in the release of a myriad of factors that can contribute to neuroexcitability. Moreover, opioids engage with neuronally expressed TLR2 in a nonclassic opioid fashion to induce neuronal apoptosis. Central immune signaling via chemokine receptor induced heterologous desensitization can inhibit classic neuronal opioid signaling. Central immune signals, such as interleukin-1β, from either neuronal or non-neuronal sources can directly and indirectly increase NMDA receptor activity, also contributing to neuroexcitation (see section III.C for details).
1. Changes in Non-Neuronal Cell Proliferation and Survival in the Central Nervous System.
The earliest opioid-induced adaptations of non-neuronal cells observed in the CNS were alterations in cellular proliferation and survival. Roth-Schechter and Mandel (1976) published one of the earliest reports of morphine decreasing glial culture growth rates. Subsequent studies demonstrated similar morphine-induced decreases in astrocyte cell proliferation (Hauser and Stiene-Martin, 1991; Zagon and McLaughlin, 1991; Hauser et al., 1996); this effect was found to be naloxone-sensitive (Stiene-Martin et al., 1991) and generalizable to astrocytes across multiple brain regions (striatum, hippocampus, and cerebral cortex) (Stiene-Martin and Hauser, 1993). Endogenous opioids have also been demonstrated to decrease astrocyte proliferation in vitro (Stiene-Martin and Hauser, 1990; Zagon and McLaughlin, 1991) and thus the endogenous opioid system was linked to embryonic brain development. The inhibitory effect of μ opioid receptors on epidermal growth factor-induced astrocyte proliferation has been further examined in vitro and demonstrated to be linked to up-regulation of ERK phosphorylation (Belcheva et al., 2003, 2005; McLennan et al., 2008). Protein kinase C (Belcheva et al., 2005), β arrestin, and calmodulin pathways seem to be involved in the antiproliferative effects of opioids on astrocytes (Miyatake et al., 2009) (see Fig. 3 for summary).
The developmental hypothesis is supported by the actions of κ opioid agonists demonstrating that dynorphin A and B (Gorodinsky et al., 1995) and synthetic κ opioid agonists (Gurwell et al., 1996) induced inhibition of cerebral astrocyte proliferation in a κ opioid antagonist-sensitive fashion (Gorodinsky et al., 1995; Gurwell et al., 1996); this effect was highly dependent on the cells maturity, immature cells being more κ opioid-responsive (Gurwell et al., 1996). It is noteworthy that this maturity-dependent change correlates well with the κ opioid expression of these cells (Gurwell et al., 1996) as discussed earlier, and raises concerns for extrapolation of results derived in primary cultures from embryonic/early postnatal cells to adults of any species. In contrast, κ opioid agonism causes proliferation in pituitary astrocytes (Bunn et al., 1985; Burnard et al., 1991) and spinal astrocytes (Xu et al., 2007). These opioid-induced cell proliferation responses are not consistently reported in the literature. For example, δ opioid-selective actions on astrocyte proliferation have been observed, in the absence of any effect of μ and κ ligands (Stiene-Martin and Hauser, 1991). Likewise, in vitro to in vivo discrepancies also exist; in vivo morphine administration has been reported to cause no change in astrocyte numbers in the nucleus accumbens, caudate nucleus, and lateral septal nucleus (Lazriev et al., 2001). Thus, the CNS region seems to be a key factor in determining opioid proliferative responses in astrocytes.
The reports of opioid actions on oligodendrocytes are mixed; Stokes and Lee (1976) reported decreased mRNA synthesis after morphine exposure, but other reports show μ opioid receptor agonism to increase proliferation in a naloxone-sensitive fashion (Knapp and Hauser, 1996; Knapp et al., 1998). Once again, the opioid effect was found to be more pronounced in immature cells (Knapp and Hauser, 1996; Knapp et al., 1998). In contrast, κ opioid receptor antagonism increased oligodendrocyte death (Knapp et al., 2001), and opioid antagonism during development was found to have profound inhibitory effects on astrocyte and oligodendrocyte proliferation (Persson et al., 2003).
Morphine has also been shown to induce microglial apoptosis, although astrocytes were resistant to such effects (Hu et al., 2002). The role of morphine-induced caspase-3 has been implicated in the apoptotic death of microglia (Hu et al., 2002) via glycogen synthase kinase-3β and p38 MAPK signaling pathways (Xie et al., 2010); morphine also interacts with HIV Tat protein-induced death (Khurdayan et al., 2004) (see Fig. 4 for summary). It is noteworthy that the resistance of astrocytes to morphine-induced apoptosis remains unclear, because it is not due to lack of apoptotic machinery or apparent lack of opioid receptor expression (Hu et al., 2002), thus the variable expression of other nonclassic sites of opioid action may account for these results.
2. Adaptations in Non-Neuronal Cell Marker and Reactivity Phenotype in the Central Nervous System.
Early studies have also demonstrated profound opioid-induced changes in cellular morphology and phenotypic receptor/immunohistological marker expression. Variable opioid-induced changes in expression of the astrocytic GFAP were first reported in the early 1990s. After long-term systemic morphine administration, a significant increase in GFAP expression was observed in the ventral tegmental area but not in the substantia nigra, locus ceruleus, cerebral cortex, or spinal cord, thereby displaying significant regional heterogeneity (Beitner-Johnson et al., 1993). Moreover, these drug-naive and opioid-induced differences in GFAP expression were strain specific (Beitner-Johnson et al., 1993). It is noteworthy that Beitner-Johnson et al. (1993) hypothesized a possible role for these opioid-induced GFAP adaptations in responses to drugs of abuse, because they occurred primarily in the mesolimbic dopamine system and strain differences were associated with preference for drugs of abuse.
Opioid-induced elevations in GFAP expression by astrocytes after long-term drug exposure has since been observed repeatedly using mRNA (Marie-Claire et al., 2004; Raghavendra et al., 2004a; Tawfik et al., 2005), protein (Tawfik et al., 2005; Wen et al., 2008; Mika et al., 2009; Ramos et al., 2010), and immunohistochemistry quantification of protein (Alonso et al., 2007). However, some studies have failed to show opioid-induced GFAP up-regulation for reasons that are unclear (Horvath et al., 2010a). Regional heterogeneity is apparent, with no morphine-induced changes observed in the dorsal raphe nucleus, hypothalamus, prefrontal cortex, substantia nigra, and thalamus (Beitner-Johnson et al., 1993; Ferrer-Alcón et al., 2000; Song and Zhao, 2001; Hutchinson et al., 2009a). Important for opioid analgesia, GFAP up-regulation has been observed in key pain centers in the CNS, including the dorsal spinal cord, trigeminal nucleus, rostral ventromedial medulla, and periaqueductal gray (Song and Zhao, 2001; Hutchinson et al., 2009a). Increased astrocyte expression of cell surface receptors, such as the chemokine receptors CCR2, CCR3, CCR5, and CXCR2 (Mahajan et al., 2002) and μ, κ, and δ opioid receptors (Turchan-Cholewo et al., 2008), have also been reported after in vitro exposure to morphine (see Fig. 3 for summary).
The mechanisms of morphine-induced elevations in GFAP expression have also been examined, implicating NMDA receptor signaling (Wen et al., 2008), CCL5 (El-Hage et al., 2008a), CCR2 (El-Hage et al., 2006), protein kinase C (Narita et al., 2005), ceramide synthase (Ndengele et al., 2009), sphingosine kinase (Muscoli et al., 2010), matrix metalloproteinase-9 (Liu et al., 2010b), P2X4 (Horvath et al., 2010b), α2-adrenergic receptor (yohimbine) (Garrido et al., 2005; Alonso et al., 2007), and alterations in glutamate homeostasis (ceftriaxone) (Ramos et al., 2010) (see Fig. 3 for summary). Morphine-induced GFAP elevations were naltrexone-sensitive (Beitner-Johnson et al., 1993; Bruce-Keller et al., 2008), and could be blocked even by an ultra-low dose of naltrexone (Mattioli et al., 2010). Several general proinflammatory attenuators have been shown to block opioid-induced GFAP up-regulation, such as antioxidant cis-7-methyl-9-methoxy-5,5a,6,10b-tetrahydroindeno[2,1-b]indole (H-290/51) (Sharma et al., 2010), ibudilast (Hutchinson et al., 2009a), fluorocitrate (Song and Zhao, 2001), propentofylline (Raghavendra et al., 2004a; Narita et al., 2006a,b; Holdridge et al., 2007), pentoxifylline, and minocycline (Mika et al., 2009).
Opioid-induced elevations in microglial reactive phenotype cell surface marker expression (see Fig. 4 for summary) have also been observed using immunohistochemistry for CD11b (Holdridge et al., 2007; Hutchinson et al., 2009a; Agostini et al., 2010; Mattioli et al., 2010) and ionized calcium binding adaptor molecule-1 (Iba1) (Ndengele et al., 2009; Horvath et al., 2010b; Zhou et al., 2010) and confirmed with Western (Mika et al., 2009) and mRNA analysis (Raghavendra et al., 2004a; Tawfik et al., 2005; Cao et al., 2010). Again, significant regional heterogeneity was observed; morphine-induced decreases in CD11b expression were reported in the cornu ammonis of the hippocampus, nucleus accumbens, and substantia nigra, and no change was reported in the dorsal raphe nucleus and medial prefrontal cortex (Hutchinson et al., 2009a). It is noteworthy that morphine-induced elevation in CD11b expression was observed in the dorsal spinal cord, rostral ventromedial medulla and periaqueductal gray (Raghavendra et al., 2004a; Hutchinson et al., 2009a). Such opioid-induced adaptations, in both microglial and astrocytic reactive phenotypes, in these CNS locations indicate sensitivity of pain systems to central immune signaling alterations in opioid responses. It is noteworthy that nonclassic opioids, such as (+)-morphine and morphine-3-glucuronide, are also capable of inducing similar microglial reactivity, as demonstrated by up-regulation of CD11b (Hutchinson et al., 2010a; Lewis et al., 2010) or other microglial activation markers such as TLR4 (Hutchinson et al., 2010a; Lewis et al., 2010).
The mechanisms by which these opioid-induced microglial phenotypic changes occur have been characterized pharmacologically (see Fig. 4 for summary). As with opioid-induced GFAP elevations in astrocytes, they were found to involve ceramide synthase (Ndengele et al., 2009), sphingosine kinase (Muscoli et al., 2010), P2X4 (Horvath et al., 2010b), and P2X7 (Zhou et al., 2010) and were sensitive to naltrexone (Bokhari et al., 2009), naloxone, and d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (Horvath and DeLeo, 2009). The microglial responses were also sensitive to general anti-inflammatory treatments such as ultra-low-dose naltrexone (Mattioli et al., 2010), minocycline (Mika et al., 2009), ibudilast (Hutchinson et al., 2009a), pentoxifylline (Mika et al., 2009), and propentofylline (Raghavendra et al., 2004a; Holdridge et al., 2007). It is noteworthy that a recent report suggests a role for TLR2 in morphine-induced microglial reactivity, another nonclassic site for opioid action (Zhang et al., 2010).
Increased microglial expression of other cell surface receptors such as CCR5 (Li et al., 2002; Bokhari et al., 2009), TLR2 (Zhang et al., 2010), TLR4 (Hutchinson et al., 2008a), P2X4 (Horvath and DeLeo, 2009), and P2X7 (Zhou et al., 2010) have also been reported after opioid exposure. These opioid-induced adaptations in CNS-immune cell receptor expression are accompanied by profound changes in astrocyte and microglial cellular morphology (Holdridge et al., 2007), with the total length of astrocytes altered in a regional specific fashion (Lazriev et al., 2001). Moreover, these preclinical data are confirmed by postmortem clinical evidence of opioid-induced microglial cell surface marker expression phenotypic reactivity (Weber et al., 2006). Such changes in cell morphology are also accompanied by alterations in the blood-brain barrier permeability and hence tight gap junction protein expression and function (Weber et al., 2006). These macrostructural changes have clear implications for drug and metabolite access into the CNS, as well as peripheral cell migration. Morphine alone (Mahajan et al., 2008) or morphine withdrawal (Sharma and Ali, 2006; Sharma et al., 2010) has been found to increase the leakiness of the blood-brain barrier, but this finding is not consistent in the literature (Mantz et al., 1993).
3. Alterations in Non-Neuronal Cell Function in the Central Nervous System.
Adaptations in other microglial and astrocyte functions have also been observed after opioid exposure. For example, phagocytosis in human microglia can be stimulated by morphine (Peterson et al., 1995; Lipovsky et al., 1998), but responses in swine microglia are inhibited (Sowa et al., 1997), demonstrating that microglial cells may be differentially influenced by animal species. In both examples, μ opioid receptors were concluded to be responsible for the effects using classic pharmacological antagonists (Peterson et al., 1995; Sowa et al., 1997; Lipovsky et al., 1998).
Another gross CNS immune cell function altered by opioid exposure is that of chemotaxis. Early studies demonstrated that morphine was able to decrease microglial chemotaxis toward complement protein 5a (Chao et al., 1997) and CCL5 (Hu et al., 2000). Such inhibitory responses by at least CCL5 could be explained by heterologous desensitization of chemokine receptors by opioid receptors (Grimm et al., 1998; Rogers et al., 2000). In contrast, microglial chemotaxis toward ATP was potentiated by morphine in a P2X4-dependent fashion (Horvath and DeLeo, 2009), and morphine itself has been found to induce chemotaxis (Takayama and Ueda, 2005a). These chemotactic responses were concluded to be induced by the classic μ opioid receptor (Takayama and Ueda, 2005a; Horvath and DeLeo, 2009) and Akt/IP3 pathways (Takayama and Ueda, 2005a; Horvath and DeLeo, 2009) (see Fig. 4 for summary). The nociceptive and analgesic consequences of such cellular adaptations are more apparent for chemotaxis than for phagocytosis. Chemotactic recruitment of glia along an ATP concentration gradient toward nociceptive dorsal horn fibers has been hypothesized and may lead to increased presence of reactive glia at the tetrapartite synapse (Horvath and DeLeo, 2009; Horvath et al., 2010a). However, the heterologous desensitization of chemokine receptors by opioids, and hence their chemotactic ability, would theoretically attenuate such recruitment. Obviously, further research is required to establish the impact of opioid-induced changes in chemotaxis on opioid analgesia.
4. Initiation of Non-Neuronal Cell Intracellular Signaling in the Central Nervous System.
The ability to examine opioid-induced activation of general and specific intracellular signaling pathways, as well as the development of pharmacological or genetic interventions, has allowed the identification of several key signaling pathways in the non-neuronal cells of the CNS and the ensuing consequences of this signaling. The first to be examined was the opioid-induced modulation of stimulated cAMP levels. Opioids alone failed to induce a cAMP response, but κ and δ agonists were found to successfully inhibit both forskolin-induced (Eriksson et al., 1990) and prostaglandin E1-induced cAMP elevations (Eriksson et al., 1991). It is noteworthy that in both cases, morphine failed to influence the cAMP response of glial cultures, demonstrating the lack of functional classic μ opioid receptors (Eriksson et al., 1990). Opioid-induced calcium signaling within astrocytes was subsequently found to respond broadly to several μ (Hauser et al., 1998; Hansson et al., 2008), κ (Hauser et al., 1998; Hansson et al., 2008), and δ (Thorlin et al., 1998b) opioid receptor agonists in a selective classic opioid receptor in an antagonist sensitive fashion.
In more recent times, one of the most prominently reported cascades influenced by opioid exposure is the MAPK pathway. The MAPK pathway comprises a collection of serine/threonine-specific protein kinases that are recruited by cell surface receptors and respond to extracellular stimuli and communicate this to elicit various cellular responses, such as gene expression. Three key kinases of this response system are p38, JNK, and ERK, the phosphorylation of which results in an active functional signaling complex (see Fig. 1 for summary). Morphine causes phosphorylation of p38 within microglia (Cui et al., 2006, 2008; Liu et al., 2006; Wang et al., 2009, 2010a, b; Agostini et al., 2010; Horvath et al., 2010a; Xie et al., 2010; Zhou et al., 2010), in a P2X7- (Zhou et al., 2010), calcitonin gene-regulated peptide (CGRP)- (Wang et al., 2010a), and neuronal nitric oxide- (Liu et al., 2006) dependent fashion; this effect could be blocked by naloxone (Xie et al., 2010) and minocycline (Cui et al., 2008). The κ opioid-selective ligand 2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide (U50488) also causes p38 activation by a classic κ opioid receptor, which depends on arrestin3 (Bruchas et al., 2006) and G-protein-coupled receptor kinase 3 (Xu et al., 2007). It is noteworthy that the non-κ opioid, dynorphin 2-17, has also been shown to induce p38 signaling independent of classic κ opioid receptor mechanisms (Svensson et al., 2005a) (see Figs. 3, 4, and 5 for summaries).
Likewise, morphine induces ERK activation in both microglia (Takayama and Ueda, 2005a; Horvath et al., 2010a) and astrocytes (Moulédous et al., 2004) that depends on MEK (Wang et al., 2010a), CGRP (Wang et al., 2010a), matrix metalloproteinase 9 (Liu et al., 2010b), pertussis toxin, calmodulin, β-arrestin2 (Miyatake et al., 2009), and IP3/Akt kinase (Takayama and Ueda, 2005a); this effect is also naloxone-sensitive (Takayama and Ueda, 2005a) (see Figs. 3 and 4 for summary). κ opioid-selective activation of ERK in a β arrestin2 manner is also possible in astrocytes (McLennan et al., 2008).
The role of JNK is less clear, in that it has been reported to be phosphorylated by morphine in astrocytes in a NDMA receptor-dependent fashion (Guo et al., 2009), whereas others have reported it to be unaffected (Wang et al., 2009). Parallel to the MAPK pathway is the IP3/Akt pathway, which is also activated by opioid exposure and seems to be involved in activation of microglial ERK (Takayama and Ueda, 2005a; Horvath and DeLeo, 2009). The Janus tyrosine kinase/signal transducer and activator of transcription pathway in astrocytes is also activated by opioid exposure in a CGRP- and MEK-dependent fashion (Wang et al., 2010a). The common downstream signaling consequence of these MAPK and related pathways is activation of NF-κB. Morphine causes activation of NF-κB within CNS non-neuronal cells in a fashion that depends on ceramide synthase (Ndengele et al., 2009), opioid receptor, p38, neuronal nitric oxide (Sawaya et al., 2009), calcium (El-Hage et al., 2008b), and CGRP (Wang et al., 2010a).
The ceramide/sphingosine signaling system has recently been highlighted as participating in opioid-induced central immune signaling (Ndengele et al., 2009; Muscoli et al., 2010). Ceramide, ceramide synthase, and insoluble acid sphingomyelinase activity are all elevated after morphine (Ndengele et al., 2009; Muscoli et al., 2010). As reviewed in section III.C.5, it is clear that the parallel opioid-induced activation of several intracellular signaling systems outlined here is capable of substantial gene transcription and translation, which profoundly modifies neuronal responses to opioid analgesics. It is clear that classic opioid receptors are involved in some of these central immune signaling responses; however, as discussed below, a key role for nonclassic opioid sites has been for the most part overlooked.
5. Opioid-Induced Changes in Non-Neuronal Cells Contribute to the Extracellular Environment of the Central Nervous System.
The culmination of the intracellular events reviewed above result in the release of a myriad of factors into the extracellular space in the CNS that can act on neuronal and non-neuronal cells alike (see Figs. 3 and 4 for summary). Best known of the immune signaling proteins are the cytokines, especially the proinflammatory cytokines IL-1β, IL-6, and TNF-α, all of which show up-regulation of transcription, translation, and release after morphine administration. IL-1β is elevated by morphine (Johnston et al., 2004; Bokhari et al., 2009; Cao et al., 2010), in a ceramide synthase- (Ndengele et al., 2009), sphingosine kinase- (Muscoli et al., 2010), MEK-, and CGRP- (Wang et al., 2009) dependent manner; this effect could be blocked by ibudilast (Hutchinson et al., 2009a), propentofylline (Raghavendra et al., 2004a), and amitriptyline (Tai et al., 2006; Tai et al., 2009), as well as by the blockade of TNF-α (etanercept) (Shen et al., 2011). IL-6 elevation after morphine (Johnston et al., 2004; Dave and Khalili, 2010) is dependent on sphingosine kinase (Muscoli et al., 2010), p38, CGRP (Wang et al., 2009), and TLR2 (Zhang et al., 2010) and could also be blocked by etanercept (Shen et al., 2011), naltrexone (Bokhari et al., 2009), amitriptyline (Tai et al., 2006; Tai et al., 2009), and propentofylline (Raghavendra et al., 2004a). Finally, morphine elevation of TNF-α (Johnston et al., 2004) is dependent on TLR2 (Zhang et al., 2010), ceramide synthase (Ndengele et al., 2009), p38, and CGRP (Wang et al., 2009) and is sensitive to amitriptyline (Tai et al., 2006; Tai et al., 2009), naltrexone (Bokhari et al., 2009), naloxone, and β-funaltrexamine (Sawaya et al., 2009). Other nonclassic opioid ligands such as (+)-morphine, (+)-methadone and morphine-3-glucuronide are also capable of inducing up-regulation of IL-1β, IL-6, and TNF-α (Hutchinson et al., 2010a; Lewis et al., 2010), suggesting a possible role for nonclassic opioid pathways in inducing opioid proinflammatory responses.
Chemokines are also modulated by morphine exposure within the CNS, with significant attention paid to the role opioids play in modulating HIV-associated protein-induced chemokine expression (Sheng et al., 2003; El-Hage et al., 2006; Turchan-Cholewo et al., 2009). Opioid action alone has been demonstrated to decrease (Mahajan et al., 2005) or increase (Rock et al., 2006) CCL2, CCL3 (Mahajan et al., 2005), CCL5 (Avdoshina et al., 2010), CCL20 (Hutchinson et al., 2009a), and CXCL1 (Hutchinson et al., 2009a).
Opioid-activated non-neuronal cells release additional molecules to the extracellular space that contribute to CNS immune signaling, including elevated complement component C3 (Maranto et al., 2008), nociceptin/orphanin FQ (Takayama and Ueda, 2005b), 27-kDa heat shock protein (Suder et al., 2009), prostaglandin E (resulting from transcriptional up-regulation of cyclooxygenase-1) (Hutchinson et al., 2008b), and prostaglandin E synthase-1 (Wang et al., 2009). It is noteworthy that each of these opioid-induced events directs the CNS toward a proinflammatory reactive phenotype.
Finally, opioid exposure causes alterations in non-neuronal cell control over glutamate homeostasis (see Fig. 3 for summary). It is noteworthy that altered glutamate homeostasis due to decreased glial glutamate transporter expression has been demonstrated in several CNS disturbances in which glia assume a reactive phenotype (Tilleux and Hermans, 2007). Long-term morphine administration results in an elevation of cerebrospinal fluid levels of aspartate and glutamate (Wen et al., 2005; Tai et al., 2007; Wu et al., 2008), most likely resulting from down-regulation of glial GLT-1 and GLAST glutamate transporters (Ozawa et al., 2001; Mao et al., 2002; Nakagawa and Satoh, 2004; Wen et al., 2005, 2008; Tai et al., 2006, 2007; Lin et al., 2010a; Rawls et al., 2010). This elevation of aspartate and glutamate seems to involve aquaporin 4 (Wu et al., 2008) and is sensitive to coadministration with dexamethasone (Wen et al., 2005), ceftriaxone (Rawls et al., 2010), naloxone (Mao et al., 2002), and amitriptyline (Tai et al., 2006, 2007). In addition, the GLT-1 that is expressed exhibits reduced function owing to increased protein damage caused by morphine-induced nitration (Muscoli et al., 2007). Such changes in glutamate expression have profound implications for neuroexcitability, including increased NMDA receptor signaling, as demonstrated by increased phosphorylation of the NR1 subunit (Lin et al., 2010a). It is noteworthy that these changes could be blocked by ultra-low-dose naloxone, which induced anti-inflammatory central immune signaling (discussed above) (Lin et al., 2010a).
D. What Is the Impact of Proinflammatory Central Immune Signaling on Opioid Analgesia?
As reviewed in detail above, opioid exposure induces profound short- and long-term modulations of central immune signaling after opioid exposure. If opioid-induced central immune signaling alters direct opioid effects on dorsal horn neurons, the opioid-induced inhibition of ascending nociceptive transmission, and the modulation of descending inhibitory and/or excitatory pathways, then it could be expected to modify opioid analgesic pharmacodynamics. It is noteworthy that this hypothesis is not a new one, as it was postulated more than 20 years ago that central immune signaling might contribute to altered opioid response (Rönnbäck and Hansson, 1988). However, only in the past decade has the behavioral significance of this non-neuronal opioid action been grasped. Indeed, as discussed throughout section III.D, opioid-induced central immune signaling is reported to detrimentally affect every aspect of the pharmacodynamics of opioid analgesic action.
The realization of new players in opioid analgesia is a major development in opioid pharmacology and thus has been the focus of several excellent recent reviews (Mika, 2008; Romero-Sandoval et al., 2008; Ueda and Ueda, 2009; Watkins et al., 2009; Ji, 2010; O'Callaghan and Miller, 2010; White and Wilson, 2010; Sweitzer and De Leo, 2011). Here, we will highlight and extend previous reviews to discuss the evidence that acute opioid analgesia is substantially modified by the rapid opioid-induced initiation of central immune signaling and that upon repeated opioid exposure, continued central immune signaling leads to analgesic tolerance and enhanced pain states.
1. Acute Opioid Analgesia and Central Immune Signaling.
For the hypothesis that central immune signaling affects acute opioid analgesia to hold true, it is expected that pharmacological and/or genetic manipulations of central immune signaling pathways would lead to changes in analgesic potency and possibly even efficacy. Examples of such effects are evident in the literature but not always observed consistently (Fairbanks and Wilcox, 2000). Acute systemic and intrathecal morphine analgesia is potentiated by the blockade of the key inflammatory cytokine IL-1β, using exogenous IL-1ra (Johnston et al., 2004; Shavit et al., 2005b; Hutchinson et al., 2008a). These results have been confirmed using three separate genetically modified strains: a transgenic knock-in of IL-1ra such that IL-1ra is over-expressed; IL-1 receptor knockout leaving IL-1β without its cognate receptor; and IL-1 receptor accessory protein knockout rendering an IL-1 receptor signal mute because of the lack of an intracellular link to the associated Toll/IL-1 receptor signaling cascade (Shavit et al., 2005b). In each case, morphine analgesia was significantly potentiated and prolonged (Shavit et al., 2005b). The quantitative decrease in morphine analgesic potency caused by the acute induction of IL-1β signaling is nearly 8-fold, as evidenced by a profound leftward shift in the morphine dose response (Hutchinson et al., 2008a). The IL-1β-induced premature curtailing of the full analgesic potential of an acute dose of morphine is evidenced by the ability of IL-1ra to unmask continuing μ opioid analgesia when given after the normal analgesic response has returned to predrug baseline (Shavit et al., 2005b; Hutchinson et al., 2008a).
This antiopioid analgesic central immune signaling response is not limited to IL-1β, as unmasking and/or potentiation of morphine analgesia is also observed by blocking the action of IL-6, TNF-α, or CX3CL1 (Johnston et al., 2004; Hutchinson et al., 2008a). Similar effects can be produced using less-specific pharmacological tools such as minocycline (Hutchinson et al., 2008b) or ibudilast (Hutchinson et al., 2009a; Lilius et al., 2009). Attenuation of glial activation with ibudilast resulted in a 5-fold leftward shift in analgesic potency (Hutchinson et al., 2009a). This induction of antianalgesic central immune signaling is not a phenomenon limited to morphine alone, because oxycodone analgesia was also potentiated 3-fold by ibudilast (Hutchinson et al., 2009a). Activation of endogenous anti-inflammatory systems, such as that elicited by elevations of IL-10, was also capable of potentiating acute morphine analgesia (Johnston et al., 2004). Thus, acute opioid-induced proinflammatory central immune signaling can be pharmacologically modified to enhance acute opioid analgesia.
To date, repeated attempts to quantify short-term transcriptional and/or translational events of these proinflammatory central immune signals, after acute in vivo opioid administration have failed (Johnston et al., 2004; Hutchinson et al., 2008a): despite the demonstration by several in vitro studies and organotypic culture techniques of clear induction of central immune signaling responses (Johnston et al., 2004; Hutchinson et al., 2008a). However, as previously discussed, the cytokine receptors and their cytokine ligands exhibit high affinity and potency, thus very low molar quantities of opioid-induced cytokine release can potentially cause a biological effect, at levels undetectable by current quantification techniques. Moreover, it is possible that these short-term effects result from the activation of stored immature protein and therefore do not require transcription and translation.
The signaling pathways that mediate this acute opposition of opioid analgesia involve, at minimum, p38 and nitric oxide (Hutchinson et al., 2008a). As discussed above, evidence suggests both classic and nonclassic opioid receptor involvement in the induction of such central immune signals. Therefore, it is specifically important to examine the classic opioid nature of these acute opioid responses. For a response to be mediated by a classic opioid receptor, the response must be stereoselective [i.e., mediated only by the (−)-isomers of opioid agonists and antagonists]. Therefore, if the response to an (−)-isomer were modified by a (+)-isomer, this would indicate a possible role for a nonclassic opioid receptor. Indeed, this is the case. Acute morphine analgesia is potentiated by coadministration of the opioid inactive (+)-isomer of either naloxone or naltrexone, but not (+)-nalmefene, implicating a ligand-selective, nonclassic opioid site(s) of action in initiating central immune signaling that attenuates the full analgesic potential of acute morphine (Hutchinson et al., 2010c). The identity of this site(s) continues to be the center of ongoing research, with recent efforts direct at the Toll-like receptors.
In vitro, transfected human cell line data demonstrate that (+)-naloxone and (+)-naltrexone, but not (+)-nalmefene, are able to noncompetitively inhibit either (−)-morphine- or LPS-induced activation of TLR4-dependent NF-κB signaling (Hutchinson et al., 2010c), thereby agreeing with the in vivo data of ligand selectivity (Hutchinson et al., 2010c). Moreover, it is apparent that a wide range of 4,5-epoxymorphinans and fully synthetic opioid analgesics possess TLR4 agonist activity, including opioid inactive stereoisomers and long-lived opioid inactive metabolites, such as morphine-3-glucuronide, which explains their hitherto unknown ability to amplify pain while lacking a neuronal site of action (Hutchinson et al., 2010c; Lewis et al., 2010). In silico, docking simulations suggest this nonclassic binding interaction may reside in the LPS binding cleft of the Toll-like receptor accessory protein MD-2, showing that (+)-naloxone docking in silico is capable of competitively antagonizing the opioid agonist docking (Hutchinson et al., 2010a,b,c) (see Figs. 6 and 7).
Fig. 6.

In silico MD2 docking of opioid ligands predicts in vitro TLR4 signaling activity. A, Hutchinson et al. (2010c) examined the in silico docking results of several opioid ligands to human MD2. These results were used to build an in silico docking to in vitro TLR4 signaling response prediction model (gray circles). This was subsequently tested on seven opioid and three nonopioid ligands, with the predicted (open squares) and actual (closed squares) in vitro scores displayed. The data indicate that the simulated interactions of these ligands with MD2 represented an excellent predictor of in vitro TLR4 signaling activity. It is noteworthy that in this case, not only the efficacy but also the direction of response (agonist versus antagonist) was modeled. B, Hutchinson et al. (2010c) subsequently compiled a modified complete 20-ligand in-silico-to-in-vitro model and retested the prediction ability of the model on the structurally disparate glial attenuator ibudilast. The data suggest that the actions of ibudilast in this human embryonic kidney 293-TLR4 in vitro model were due to ibudilast action at MD2. [Reproduced from Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, and Watkins LR (2010c) Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun 24:83–95. Copyright © 2010 Academic Press. Used with permission.]
Fig. 7.
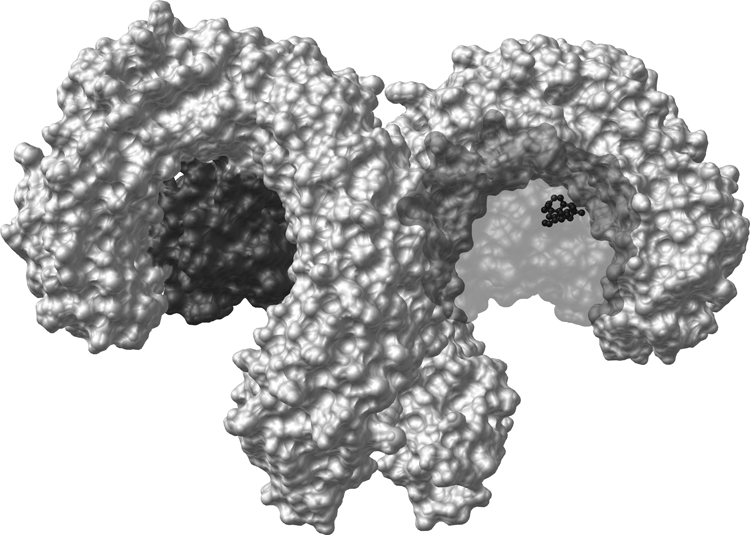
Computer modeling of opioid interaction with the dimerized TLR4-MD2 complex. In silico docking of (−)-morphine (black stick and ball) to the 3D crystalline structure of the human MD2 (dark gray) and TLR4 (light) complex (Protein Data Bank ID 3FXI) demonstrates that the preferred docking location of (−)-morphine is to the LPS binding domain of MD2.
The hypothesis of opioid-induced TLR4 signaling is further supported by the potentiation of acute morphine analgesia after blockade of TLR4 activity by 1) coadministration of competitive TLR4 inhibitors with nonsignaling mutant LPS antagonists (e.g., LPS-RS), 2) peptide inhibition of the key TLR4 accessory molecule toll-interleukin 1 receptor domain-containing adaptor protein, 3) genetic knockout of TLR4 (Hutchinson et al., 2010c), or 4) by the pharmacological blockade of the 90-kDa heat shock protein, which is critical for TLR4 trafficking (Hutchinson et al., 2009b) (see Fig. 1 for summary). (−)-Morphine-induced TLR4 signaling opposes acute (−)-morphine analgesic potency by nearly 3-fold, as evidenced by dose-response analysis (Hutchinson et al., 2010c). Further supporting this TLR4 hypothesis is the use of MyD88 knockout mice that would render a Toll/interleukin-1 receptor signal mute. Knockout of MyD88 significantly enhances short-term morphine analgesia (Hutchinson et al., 2010b). Moreover, (+)-naloxone and minocycline, which potentiate acute morphine analgesia in wild-type mice and rats, fail to provide potentiation of analgesia in TLR4 knockout mice, demonstrating the specificity and selectivity of these TLR4- and glial-targeted treatments (Hutchinson et al., 2010c). Additional compounds shown to have TLR4 inhibitory properties, such as certain tricyclics, have also demonstrated significant three-way correlations between their in vitro TLR4 inhibitory potency, with in vivo potentiation of acute morphine analgesia, which also correlates highly with the in silico docking simulation results (Hutchinson et al., 2010b). The selectivity of amitriptyline was examined in vivo using TLR4 or MyD88 knockout strains and was found to potentiate morphine analgesia only in wild-type mice but not the knockout strains (Hutchinson et al., 2010b). Collectively, these data suggest that opposition of opioid analgesia by acute central immune signaling is initiated, at least in part, by an opioid-induced TLR4 response.
2. Naive Opioid Tolerance/Opioid-Antianalgesia and Central Immune Signaling.
Given that opioid analgesia is profoundly modified by acute opioid-induced central immune signaling, it stands to reason that already established changes in proinflammatory central immune signaling, produced by nonopioid factors may also modify opioid analgesic potency, producing a state akin to “naive tolerance.” Such evidence exists in the literature and has sometimes been referred to as antianalgesia. Once again, IL-1β and innate immune receptors, such as TLR4, are key participants in mediating such a response. The role for IL-1β-induced naive tolerance was established by Shavit et al. (2005b) and Gul et al. (2000), who demonstrated that administration of exogenous IL-1β was sufficient to oppose acute morphine analgesia in an IL-1ra-sensitive fashion (Shavit et al., 2005b).
Treatments that specifically induced a TLR4-dependent central immune signaling response, such as LPS administration, also resulted in attenuation of acute morphine analgesia (Johnston and Westbrook, 2005; Wu et al., 2006a); and this effect could be blocked by (+)-naloxone and (−)-naloxone (Wu et al., 2006a), MK801 (Johnston and Westbrook, 2005), and the astrocyte inhibitor fluorocitrate (Johnston and Westbrook, 2005). Prior administration of nonclassic opioid agonists, such as (+)-morphine, was also capable of inducing naive tolerance (Takagi et al., 1960; Wu et al., 2005), in a p38- (Wu et al., 2006b) and glial- (propentofylline) (Wu et al., 2005) dependent fashion. It is noteworthy that cross-sensitivity of the response was also observed, as (+)- and (−)-morphine-induced naive tolerance were sensitive to either (+)- or (−)-naloxone (Wu et al., 2005, 2006a). These results suggest that naive tolerance can be induced by TLR4-dependent activation of central immune signaling, which via p38-dependent mechanisms induces IL-1β release. This IL-1β release would then result in enhanced neuroexcitability via well characterized IL-1β actions, including up-regulation of NMDA and calcium-permeable AMPA receptor membrane expression, up-regulation of NMDA phosphorylation, and up-regulation of NMDA NR1 subunit expression as reviewed above, collectively contributing to opposition of acute morphine analgesia (see Fig. 5 for summary).
A parallel and/or downstream mechanism by which acute morphine analgesia can be attenuated is via heterologous desensitization of opioid receptors, as discussed earlier. Administration of the chemokines CCL5 (binding to CCR1), CXCL12 (binding to CXCR4) (Szabo et al., 2002; Chen et al., 2007b), and CX3CL1 (binding to CX3CR1) (Chen et al., 2007a) into the periaqueductal gray area significantly attenuated acute opioid analgesia. In addition, electrophysiology studies in single neurons have reported decreased opioid action after exposure to CCL2, CCL3, CCL5, or CXCL8 chemokines (Zhang et al., 2004). Whether the chemokine actions noted above are directly mediated by neurons, or by acting on nearby glia to induce release of IL-1β, for example, remains unknown. Either way, such modulation of opioid receptor function resulting from central immune signaling can also profoundly affect acute opioid analgesic efficacy (see Fig. 5 for summary).
Pathological conditions associated with priming of central immune signaling in the principal nociceptive centers of the CNS are also susceptible to naive opioid tolerance via similar mechanisms. As discussed above, chronic pain states result in the elevation of the same central immune signaling pathways outlined here, undesirably opposing acute opioid analgesia. It is clear that in animal studies of neuropathic pain, opioids show significantly reduced acute analgesic potency (Raghavendra et al., 2002; Raghavendra et al., 2003b; Narita et al., 2004a; Tawfik et al., 2005; Mika et al., 2007; Mika et al., 2009). Central immune signaling and glia contribute significantly to this, because blockade of IL-1β, TNF-α, and IL-6 (Raghavendra et al., 2002) or administration of minocycline, propentofylline, or pentoxifylline was able to reinstate acute opioid analgesic potency (Raghavendra et al., 2002, 2003b; Mika et al., 2007, 2009).
Other pathological conditions also implicate central immune signaling in altered acute opioid response. There is mounting evidence for surveillance of the CNS by peripheral immune cells, resulting in significant central immune signaling. For example, Grace et al. (2011) have reported that transfer of splenocytes from a donor with nerve injury-induced allodynia to a recipient animal is sufficient to produce significant allodynia. Moreover, this transfer increased CNS immune cell trafficking to the dorsal spinal cord by apparent effector cells capable of instating enhanced pain. A similar conclusion could be drawn for the reduced antinociceptive effect of morphine in diabetic mice (Kamei et al., 1992, 1993; Gul et al., 2000). In these animals, splenectomy restored morphine analgesia, whereas transfer of splenocytes from diabetic mice to healthy control mice produces an opioid nonresponsive animal (Kamei et al., 1992). Similar results were found in the morphine-tolerant Beige-J mouse strain, including adoptive transfer of tolerance with splenocytes (Raffa et al., 1988; Kimball and Raffa, 1989). It is noteworthy that the response in diabetic mice was dependent on elevated levels of IL-1β (Gul et al., 2000). Therefore, modifications in peripheral immunology may lead to changes in CNS trafficking and cause adaptations in central immune signaling that are sufficient to produce behavioral changes.
Central immune signaling can act to opose short-term opioid analgesia in several anatomical locations. Intrathecal administration (Raghavendra et al., 2002) and heterologous desensitization (Zhang et al., 2004) studies demonstrate that central immune signaling at the levels of the dorsal spinal cord and/or the dorsal root ganglion can oppose acute opioid analgesia. Brain areas such as the periaqueductal gray are also involved in this effect (Chen et al., 2007a,b; Szabo et al., 2002). Finally, whether central immune signaling in the rostral ventromedial medulla can also attenuate acute opioid analgesia remains to be definitively tested. However, if the hypotheses and mechanisms developed in other key nociceptive centers hold true, then recent evidence of central immune signaling in the rostral ventromedial medulla in neuropathic pain (Wei et al., 2008) and after long-term morphine administration (Hutchinson et al., 2009a) would suggest that descending facilitation of pronociceptive antianalgesic mechanisms would also oppose acute opioid analgesia. Therefore, it seems likely that acute opioid analgesia is susceptible to the ongoing pathology of clinically relevant disease states.
Finally, environmental and/or genetic influences that alter central immune signaling may also contribute to altered acute opioid analgesia. For example, single-nucleotide polymorphisms in the genes encoding IL-6 (Reyes-Gibby et al., 2008) or IL-1ra (Bessler et al., 2006), which are associated with increased proinflammation, lead to increased opioid requirements after surgery, suggesting a possibly reduced opioid analgesic efficacy combined with or separate from increased pain. Therefore, central immune signaling may be a previously unrecognized variable that contributes substantially to heterogeneity in acute opioid analgesic responses.
3. Tolerance to Opioid Analgesics and Central Immune Signaling.
After repeated opioid exposure, significant decrease in opioid analgesia occurs as a result of several parallel and sequential adaptations, some of which involve opioid-induced central immune signaling. It is important to articulate that, even under opioid-tolerant states, opioid analgesia still results from the combined effects of opioid, including direct opioid effects on dorsal horn neurons, the opioid-induced inhibition of ascending nociceptive transmission, and the modulation of descending inhibitory and/or excitatory pathways, albeit with decreased analgesic output. Therefore, as with acute opioid analgesia, similar alterations in central immune signaling in the periaqueductal gray, rostral ventromedial medulla, or dorsal root ganglia have the potential to lead to opioid tolerance. To date, however, the majority of the preclinical research on the contribution of opioid-induced immune signaling to opioid tolerance has focused at the level of the dorsal spinal cord.
Although central immune signaling had been postulated to be involved in opioid tolerance for several decades (Rönnbäck and Hansson, 1988), it was not until the dawn of this millennium that Fairbanks and Wilcox (2000) first implicated spinal proinflammatory immune signaling in opioid tolerance. It is noteworthy that this response was compared with that observed after dynorphin exposure, which had also recently been found to involve proinflammatory non–opioid-mediated central immune signaling (Laughlin et al., 1997, 2000). It is noteworthy that this research established that central immune signaling was also involved in the cross-tolerance to different opioids (Fairbanks and Wilcox, 2000). Soon afterward, Song and Zhao (2001) implicated opioid-induced glial reactivity as critical to morphine tolerance. These studies triggered a profound shift in opioid tolerance research to include a central immune signaling hypothesis as part of the several systems involved in decreased analgesia.
It is clear from the preclinical literature than opioid tolerance involves several aspects of central immune signaling, because tolerance can be prevented, attenuated, and/or reversed by blockade of the action or formation of the proinflammatory cytokines IL-1β, TNF-α, and/or IL-6 (Raghavendra et al., 2002; Shavit et al., 2005a; Hutchinson et al., 2008a; Shen et al., 2011) or chemokines such as CX3CL1 (Johnston et al., 2004); inhibition of MEK activity (and hence blockade of ERK activation) (Wang et al., 2009, 2010a); selective reductions in microglial p38 (Wang et al., 2009, 2010a) or JNK activation (Guo et al., 2009); decreased P2X4 (Horvath et al., 2010b) or P2X7 signaling (Zhou et al., 2010); reduced ceramide/sphingosine signaling (Ndengele et al., 2009; Muscoli et al., 2010); inhibition of matrix metalloproteinase 9 (Liu et al., 2010b); nitric oxide and related superoxide protein damage (Muscoli et al., 2007; Batinić-Haberle et al., 2009); or by general anti-inflammatory treatments such as IL-10 (Johnston et al., 2004), fluorocitrate (Song and Zhao, 2001), minocycline (Mika et al., 2007; Cui et al., 2008; Mika et al., 2009), pentoxifylline (Mika et al., 2007), propentofylline (Raghavendra et al., 2003b; Raghavendra et al., 2004a), ibudilast (Ledeboer et al., 2006a; Lilius et al., 2009), and, interestingly, also by ultra-low-dose naloxone, which acts via elevations of IL-10 expression (Lin et al., 2010b). It is noteworthy that blockade of TLR4 using (+)-naloxone also attenuates the development of morphine tolerance, suggesting that one of the initiating triggers of opioid tolerance is direct activation of TLR4 signaling by opioids (Hutchinson et al., 2010c).
Another critical modulation in glial function related to opioid-induced central immune signaling that contributes to the development of tolerance is down-regulation of glial GLT-1 and GLAST glutamate transporters (Ozawa et al., 2001; Mao et al., 2002; Nakagawa and Satoh, 2004; Wen et al., 2005; Tai et al., 2006, 2007; Wu et al., 2008; Lin et al., 2010a; Rawls et al., 2010). The effects of such down-regulation are blocked by coadministration of any of the following substances: synthetic glucocorticoid dexamethasone (Wen et al., 2005); the glutamate transporter up-regulator ceftriaxone (Rawls et al., 2010); (−)-naloxone (Mao et al., 2002); ultra-low-dose naloxone (Lin et al., 2010a); or amitriptyline, which produces its effect via induction of IL-10 expression and hence indirect induction of an anti-inflammatory response (Tai et al., 2006, 2007). In addition to these pharmacological treatments that restore glutamate transporter homeostasis, tolerance can also be reduced by the NMDA receptor antagonist dizocilpine maleate (MK801) (Wen et al., 2008; Guo et al., 2009), which can in turn reduce the opioid-induced reactivity of astrocytes (Wen et al., 2008). Therefore, the activation of central immune signaling that contributes to morphine tolerance is somewhat cyclical, and interventions at multiple points can act to reduce opioid tolerance. Collectively, these central immune signaling events result in increased neuroexcitability, which counter-regulates opioid analgesia.
Several aspects of central immune events seem to depend on neuronal adaptations that initiate, contribute, and/or maintain the activation of central immune signaling. Neuronal nitric-oxide synthase (leading to p38 activation) (Liu et al., 2006) elevates neuronal CGRP which, in turn, leads to elevations in IL-1β, TNF-α, IL-6, prostaglandin E synthase-1, NMDA receptor subunit expression, and activation of ERK, signal transducer and activator of transcription 3, p38, and NF-κB (Wang et al., 2009, 2010a,b); neuronal NMDA receptor subunit expression favored for neuroexcitability (Guo et al., 2009; Lin et al., 2010a; Wang et al., 2010a); neuronal protein kinase Cγ expression (leading to astrocyte reactivity) (Narita et al., 2004b; Lin et al., 2010a); and cAMP response element-binding and calmodulin-dependent protein kinase II activation (Wang et al., 2010a) (see Figs. 3–5 for summaries). Each can contribute to some aspects of central immune signaling and the development of tolerance. In vitro and in vivo data seem to indicate that both opioid-neuron-glial and opioid-glial-neuron signaling can occur, as well as parallel opioid-neuronal adaptations that decrease continued neuronal responses to continued opioids combined with opioid-glial adaptations that increase continued glial responses to continued opioids. Adding an additional layer of complexity is whether the opioid action is mediated by classic and/or nonclassic opioid receptors at both neuronal and glial sites, given the evidence of (+)-isomer activity and the implied role of TLR4. The role of CGRP was suggested by Wang et al. (2009, 2010a,b) to be a direct action on neurons and non-neuronal cells alike. However, Liu et al. (2010b) proposed that morphine-induced neuronal elevations of CGRP are dependent on neuronal matrix metalloproteinase 9 activity, the associated morphine-induced astrocyte GFAP up-regulation also being dependent on matrix metalloproteinase 9. This suggests, therefore, that additional neuron-to-glial signals, such as CX3CL1, are required for non-neuronal cell proinflammatory reactivity. Further complicating the mechanism are the apparent temporal adaptations that occur during the development of tolerance leading to feed-forward effects of signals, such as amplified NMDA receptor signaling. These questions will continue to be answered by ongoing research investigating the critical role played by central immune signaling in opioid tolerance.
Finally, the evidence of increased rates of opioid tolerance development in neuropathic pain states has been examined and found to involve an opioid-induced increase in central immune signaling (Raghavendra et al., 2002, 2003b; Narita et al., 2004a; Tawfik et al., 2005; Mika et al., 2007, 2009). In a fashion similar to that described above for naive tolerance, basal and/or primed central immune signaling leads to a more rapid and/or greater response via the mechanisms outlined above, leading to a faster counter-regulation of opioid analgesia and hence a reduced efficacy in pain management.
4. Hyperalgesia and Allodynia Induced by Opioids and Central Immune Signaling.
Central immune signaling and the associated neuronal dysfunction are key participants and mediators of chronic pain conditions (Milligan and Watkins, 2009). Likewise, after opioid exposure, when the pronociceptive mechanisms, including central immune signaling, outweigh the combined antinociceptive actions, a concerning exaggerated pain state is observed, presenting itself as allodynia and/or hyperalgesia reported in several disparate patient populations (Doverty et al., 2001; Angst and Clark, 2006; Pud et al., 2006; Singla et al., 2007; Hay et al., 2009, 2010). Proinflammatory central immune signaling (Johnston et al., 2004; Hutchinson et al., 2008a; White and Wilson, 2010) induced by glial reactivity (Raghavendra et al., 2004a; Agostini et al., 2010), causing alterations in glutamate homeostasis (Mao et al., 2002; Ramos et al., 2010) and heterologous desensitization (White and Wilson, 2010), are implicated in this response as well as several other key neuronal adaptations (Ossipov et al., 2004, 2005). It is noteworthy that the temporal association of the heightened pain in relationship to the last opioid administration is crucial, because both opioid withdrawal and classic opioid-induced pain states are reported.
If the same mechanisms are involved in naive tolerance as in tolerance induced by long-term opioid exposure, then both classic and nonclassic opioid-mediated pain systems are likely. The evidence for neuronal adaptations has been reviewed widely, indicating involvement of classic opioid responses (Ossipov et al., 2004, 2005). However, as discussed earlier, there is also evidence for nonclassic opioid-induced pain states (Juni et al., 2007; Waxman et al., 2009), with TLR4 serving as an initiating receptor, as indicated by the use of opioid inactive isomers and pharmacological TLR4 inhibition (Hutchinson et al., 2010a,c; Lewis et al., 2010).
Also key to these nonclassic mechanisms of opioid-induced pain are the apparent metabolic bioactivation of opioids into ligands capable of initiating central immune signaling (Lewis et al., 2010) and the opioid-induced up-regulation of pronociceptive systems that mediate their effect by central immune signaling (Watkins et al., 2005). After administration, morphine is converted into various metabolites, one of which is morphine-3-glucuronide, the opioid-inactive metabolite that is capable of inducing neuroexcitability and pain (Yaksh et al., 1986). Examination of its central immune signaling properties demonstrates that it is capable of inducing glial reactivity, nitric oxide formation, and proinflammatory cytokine release in a TLR4-dependent fashion, leading to the development of hyperalgesia and allodynia (Komatsu et al., 2009; Hutchinson et al., 2010a,c; Lewis et al., 2010). Likewise, dynorphin expression is induced by opioid exposure (Agostini et al., 2010) and is associated with exaggerated pain states, albeit via a non classic opioid-dependent mechanism (Laughlin et al., 1997). Svensson et al. (2005a) demonstrated that the opioid-inactive metabolite dynorphin 2-17 caused microglial activation and elevated prostaglandin production, thereby providing an additional link to its pronociceptive actions and a possible link to opioid-induced pain. In this fashion, the underlying mechanisms of the allodynia induced by morphine-3-glucuronide and dynorphin are very similar to those proposed for the initiation of neuropathic pain by endogenous danger signals, providing an intriguing closure to the mechanistic loop of neuropathic pain, naive tolerance and long-term opioid tolerance.
IV. Unifying the Neuronal and Central Immune Signaling Opioid Hypotheses
The aim of opioid research at every level, from the lab bench to the clinic, from neuroscience to pharmacogenomics, is to better understand the beneficial and adverse actions of opioids and how they occur, with the ultimate goal of improving patient care, in terms of both analgesic efficacy and drug safety. It is apparent that the research of opioid pharmacology and opioid neuroscience is now complemented by the exciting discoveries indicating the importance of central immune signaling. Our understanding of opioid-induced central immune signaling can contribute to the wealth of knowledge of opioid actions at several broad levels: discernment of opioid action in disease where central immune signaling contributes to the pathological condition; appreciation of the significance presented by the nonclassic opioid actions of opioids; identification of novel systems that may contribute to adverse events or side effects of other therapies; and identification of new pharmacological targets and/or therapies for disease. In light of the research reviewed here, a discussion of these topics in more specific terms follows.
A. How Does Opioid-Induced Central Immune Signaling Integrate with the Wealth of Neuronal Opioid Knowledge?
Central immune signaling cannot be thought of as a parallel system separate from that of neuronal synaptic transmission and neuronal communication. These two systems are intertwined in a complex fashion that has yet to be fully appreciated (Adler and Rogers, 2005). Therefore, opioid action in a mixed cell network, such as that found in vivo, can no longer be viewed in terms of neuronal functioning alone but rather as the composite result of both neuronal and central immune signaling events reviewed above. After opioid exposure, both classic and nonclassic opioid receptors are engaged in neuronal and non-neuronal cells. Basal central immune signaling is able to counter the analgesic action of opioid agonists, and together with basal variability in neuronal opioid systems, this leads to significant heterogeneity in acute opioid analgesia. In some cases, this heterogeneity is so profound that there is a complete lack of an analgesic response, hence naive tolerance. If an opioid agonist is administered in a condition in which central immune signaling is active or the machinery for such activity is primed as found in neuropathic pain, for example, attenuated opioid analgesia can be expected, owing in part to the neuronal consequences of central immune signaling.
After repeated opioid administration, both neuronal and central immune signaling adaptations occur simultaneously, in a feed-forward fashion. This creates, in multiple CNS regions, an environment in which opioid agonists lose their analgesic efficacy, hence contributing to opioid tolerance. If adaptations in neuronal and central immune signaling processes become so pronociceptive, even the actions induced by an opioid agonist through classic opioid receptors can no longer achieve sufficient antinociceptive effect to overcome this counter-regulating signaling. This leads to the presentation of opioid-induced hyperalgesia and allodynia. Therefore, the transition from the benefits of short-term opioid analgesics to the detrimental opioid-induced atypical pain states can be thought of as a response continuum, engaging neuronal and central immune signaling systems to varying degrees at different stages to elicit these pharmacodynamic diverse responses. The mechanisms of opioid-induced enhanced pain states are very similar to those operating in neuropathic pain states. Thus, the understanding of this unifying hypothesis provides excellent opportunities to capture advances made, mostly in preclinical studies, in both the neuroscience and pharmacology fields. However, if correct, such a hypothesis raises significant concerns regarding the ever-increasing use of long-term opioid therapy for chronic pain states. This is because the two systems influence each other and consequently may offer only a short-term symptomatic pain relief while in the long-term exacerbating the neuronal and central immune signaling disturbances underlying the condition.
B. Why Is Toll/Interleukin-1 Receptor Signaling Pivotal to Opioid-Induced Central Immune Signaling?
The mechanism(s) by which opioid-induced signaling is initiated has been assumed, and in some cases shown, to involve classic opioid receptors. However, it is noteworthy, as reviewed and highlighted here, that such a conclusion has sometimes been drawn based on the effects of antagonists now known to block TLR4 signaling in addition to blocking opioid receptors. Hence such a conclusion is tenuous until validated with antagonists truly selective for classic opioid receptors and/or until the nonclassic opioid receptor involvement is directly examined. There is now substantial evidence to demonstrate that central immune signaling can also be induced by nonclassic opioid receptor mechanisms. As discussed previously and reviewed here, Toll-like receptors are one such promising type of nonclassic opioid receptor that may trigger central immune signaling. Signaling via the Toll/IL-1 receptor pathway initiates profound opioid-induced proinflammatory signals that contribute to the pharmacodynamic actions of opioids.
Despite genetic knockout studies demonstrating the profound influence of Toll/IL-1 receptor signaling on opioid actions (Li et al., 2009, 2010; Hutchinson et al., 2010b,c; Zhang et al., 2010), such opioid-TLR responses are not always consistent (El Ghazi et al., 2010). Moreover, why has this TLR activity of opioids not been observed until now? The simple answer may be that perhaps the TLR activity of opioid was observed, even in the first studies using the stereoisomers of morphine, but that such observations have been overlooked. As previously discussed and reviewed here in detail, the nonclassic opioid sites described by others might be attributed in part to TLRs. It is very likely that other novel locations of opioid activity, beyond the TLRs, also exist.
Other explanations for this lack of discovery of TLR opioid activity may be found in the in silico docking data that implicates the soluble TLR accessory protein MD-2 as the computationally preferred docking site (Hutchinson et al., 2010c) (see Fig. 7). In vitro culture conditions may not consistently contain such a critical soluble component of the signaling system, resulting in substantial rightward shifts in TLR signaling potency for even classic TLR ligands such as LPS (Hutchinson et al., 2010c). Indeed, given that MD-2 is a soluble protein, rather than membrane-bound, the interaction of opioids with MD-2 may have been missed by the methods traditionally used for defining receptor binding. Moreover, the control parameters of all but a few studies were designed to eliminate the nonstereoselective binding and examine only the classic opioid sites, thus overlooking further characterization of these critical receptors.
Another explanation may lie in the heterologous desensitization literature, where opioid action at classic opioid receptors desensitizes chemokine receptor signaling, such as that of CXCR4. It is noteworthy that CXCR4 can colocate with TLR receptors, including TLR2 and TLR4 (Hajishengallis et al., 2008; Triantafilou et al., 2008) and has been attributed to profoundly modify TLR signaling. Therefore, opioid action at classic opioid receptors may also act to modify TLR signaling, whenever TLRs, CXCR4, and opioid receptors are expressed by the same cell. In addition, such an interaction may occur via common scaffolding proteins that are shared across the multiple receptor classes (Li et al., 2010) and perhaps may explain the actions of ultra-low-dose naloxone/naltrexone at filamin A (Wang et al., 2008), and the actions of a new compound, PTI-609 [discussed in section IV.C (Barbier et al., 2010; Burns and Wang, 2010)]. Future research on the complex receptor signaling requirements and cross-talk are needed to determine to what extent such interactions occur.
Given that both TLR4 and IL-1β, via Toll/IL-1 receptor signaling, have been implicated in enhanced nociception after opioid exposure and in neuropathic pain, this signaling pathway provides a common link between the two conditions. Moreover, this signaling pathway also provides a common initiating trigger to the central immune signaling associated with long-term opioid exposure and chronic pain, considering, respectively, the hypothesized opioid-TLR4 and endogenous danger signal-TLR4 relationships. This is not to say that all neuronal adaptations associated with long-term opioid exposure or neuropathic pain depend on Toll/IL-1 receptor signaling; rather, they are complemented and possibly facilitated by this system. Therefore, significant attention should be paid to the role of Toll/IL-1 receptor signaling when examining opioid pharmacodynamics.
C. What Are the Opportunities Provided by Pharmacological Targeting of Central Immune Signaling to Improve Opioid Analgesia?
Optimal patient analgesia with a few adverse or unwanted side effects should be the utmost goal. Therefore, if a physiological system is found to contribute to the unwanted adverse and/or side effects of a drug, then steps should be taken to minimize such actions. For opioids, based on preclinical evidence, central immune signaling is now a recognized contributor. Although not discussed here, other adverse actions of opioids, such as respiratory depression, addiction, and dependence are also associated with induction of central immune signaling (Hutchinson et al., 2007, 2008b, 2009a, 2010c; Bland et al., 2009; Zhang et al., 2010). Thus, to maintain the beneficial use of opioids, measures to minimize central immune signaling must be taken. For example, the clinical formulation of racemic methadone contains both the opioid-active and opioid-inactive isomers, for economic rather than clinical efficacy reasons. However, both isomers have demonstrated central immune signaling capacity. In this case, the long-term clinical consequence of apparently unnecessarily exposing the CNS to (+)-methadone needs to be considered.
Current preclinical data suggest that the analgesic efficacy of existing opioid formulations could be improved by coadministration with general glial attenuators, such as ibudilast, minocycline, propentofylline, or pentoxifylline; glutamate transporter enhancers such as ceftriaxone; treatments that block proinflammatory activity, such as IL-1ra; or treatments that favor anti-inflammatory conditions, such as amitriptyline or ultra-low-dose naltrexone/naloxone. Moreover, coadministration of opioids with such therapies may result in reduced response heterogeneity and a simpler way of dose finding, owing to the reduction of the central immune signaling action. Clinical evidence as proof of concept for such a treatment strategy is greatly needed. A recent preliminary report gives early indications that this approach may be worthy of further investigation, because ibudilast has been found to significantly decrease analgesic tolerance in a profoundly opioid-tolerant clinical population (http://investors.medicinova.com/phoenix.zhtml?c=183833&p=irol-newsArticle&ID=1507330&highlight=). The long-term systemic immunological consequences of such combined therapies clearly need to be examined in the future to appreciate increased risk associated with long-term immunomodulation.
A more specific approach to blocking opioid-induced central immune signaling may be via the blockade of TLRs known to trigger the proinflammatory cascade. The nonstereoselectivity of the opioid action at TLRs can theoretically be exploited in this fashion using (+)-isomer TLR inhibitors combined with traditional (−)-isomer opioid agonists. As discussed, such an approach leads to potentiated analgesia and reduced adverse events. Moreover, (+)-isomer TLR inhibitors by themselves serve in an antiallodynic role to block central immune signaling in neuropathic pain (Hutchinson et al., 2008c).
Finally, future drug development could combine the properties of a general glial attenuator and/or TLR inhibitor with potent opioid agonist activity in one small molecule. Such a hypothetical compound would have increased analgesic potency, maintain analgesia in neuropathic pain conditions, produce minimal analgesic tolerance (or cross-tolerance), and have reduced adverse effects such as addiction and dependence. Such a compound, PTI-609, seems to have been fortuitously made (Burns and Wang, 2010). PTI-609 was generated using a rational drug design approach based on the hypothesized filamin A site of action of ultra-low-dose naloxone/naltrexone. This compound maintains opioid receptor agonist activity as well as interaction with filamin A and has been shown to produce good analgesia and have little to no induction of conditioned place preference (reward). Moreover, the compound profoundly inhibits TLR4-dependent IL-1β, IL-6, and TNF-α production by human astrocytes. We wait with great interest as the use of such combined immune signaling-modifying and opioid analgesic therapies undergo clinical trials.
D. What Are the Broader Implications of Xenobiotic-Induced Central Immune Signaling?
The discoveries reviewed here of opioid-induced central immune signaling have implications for multiple research and clinical disciplines. The opioid-TLR4 activity reported here is in fact not “opioid” and rather can be viewed as “xenobiotic”-induced TLR signaling, owing to the activity of both opioid and nonopioid ligands. Given this, it has significant implications for the currently clinically used pharmacopeia and understanding of chemicals to which we are exposed in the environment. This collection of small molecules will probably include parent compounds and metabolites that also possess TLR activity, either as agonists or antagonists. If a clinically used compound or one of its metabolites possesses such TLR agonistic activity, side effects of the drug may be attributable to such undefined TLR agonist activity. In the case of opioids, such opioid-induced central immune signaling contributes to adverse recovery in preclinical models of spinal cord injury (Hook et al., 2009, 2011). Moreover, the shared central immune signaling properties of opioids with other drugs of abuse, such as methamphetamine, also raise the possibility that this drug-induced central immune signaling process may be a common phenomenon across drugs of abuse, resulting in amplification of drug-mediated activation of the mesolimbic dopamine reward pathway. Consequently, other xenobiotics that possess central immune signaling capacity may increase their abuse liability. Thus, care should be taken to identify potential TLR or central immune signaling properties of existing and new agents and to consider the impact this may have on their primary indication and/or on a specific patient population in which central immune signaling may have detrimental effects.
If compounds are found to block TLR or central immune signaling, then some of the clinical pharmacodynamic actions may be accounted for by this property. The newly discovered actions of amitriptyline (Hutchinson et al., 2010b) are of significant interest here, owing to its use as an antidepressant, perhaps lending additional support to the proinflammatory hypothesis of depression and illness responses. The identification of TLR inhibitory actions of several small molecules may provide new treatment options for a broad range of pathologic conditions for which blockade of central immune signaling may be beneficial.
V. Conclusions
This review has covered a wide-ranging preclinical literature (and, where available, clinical evidence) of classic and nonclassic opioid actions at neuronal and non-neuronal cells. It is apparent that an appreciation of all actions of opioids is required to fully understand the true neuronal and central immune signaling consequences of opioid exposure and how these combine to produce the behavioral response. Opioid actions at innate immune receptors, such as TLR4, and the ensuing central immune signaling cascade provide significant opportunities for new drug development. The excitement and promise provided by the wealth of preclinical opioid-induced central immune signaling literature reviewed here need to be capitalized upon in the short term by conducting quality clinical trials in patient populations. Fortuitously, such trials of opioid-induced central immune signaling and efficacy of central immune signaling-targeted therapies are emerging in the literature. Patient care and quality use of medicine must be at the heart of clinical pain management. Many patients do achieve adequate pain relief from existing opioid therapies, but many do not. Still, the long-term consequences of effective and/or ineffective long-term opioid treatment are of significant importance, now that we have a greater appreciation of the consequences of opioid-induced central immune signaling. Perhaps the emergence of new pharmacotherapies, using combined formulations or mixed function opioid therapies, that achieve profound analgesia while attenuating central immune signaling will bring about disease-modifying treatments rather than just symptomatic relief.
Acknowledgments
This work was supported in part by the Intramural Research Programs of the National Institutes of Health National Institute on Drug Abuse and the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism (to K.C.R.); the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grant K05-DA024044] (to L.R.W.); the National Health and Medical Research Council CJ Martin Fellowship [ID 465423, 2010] (to M.R.H.); the Australian Research Council Australian Research Fellowship [DP110100297, 2011] (to M.R.H.); a Faculty of Health Sciences Divisional Scholarship (to P.M.G.); the Israel Science Foundation [Grant 512/08] (to Y.S.); and the Leon and Clara Sznajderman Chair of Psychology (to Y.S.). Thanks to Tavik Morgenstern for assistance inpreparing the figures.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Hutchinson, Shavit, Grace, Rice, Maier, and Watkins.
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.004135.
- Akt
- protein kinase B
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CCL
- chemokine (C-C motif) ligand
- CCR
- chemokine (C-C motif) ligand receptor
- CD11b
- complement receptor 3
- CGRP
- calcitonin gene-related peptide
- CNS
- central nervous system
- DRG
- dorsal root ganglion
- ERK
- extracellular signal-regulated kinase
- GFAP
- glial fibrillary acidic protein
- GLAST
- glutamate/aspartate transporter
- GLT-1
- glutamate transporter-1
- H-290/51
- cis-7-methyl-9-methoxy-5,5a,6,10b-tetrahydroindeno[2,1-b]indole
- IL
- interleukin
- IL-1ra
- interleukin-1 receptor antagonist
- IP3
- inositol trisphosphate
- JNK
- c-Jun N-terminal kinase
- LPS
- lipopolysaccharide
- MAPK
- mitogen-activated protein kinase
- MD-2
- myeloid differentiation factor 2
- MEK
- mitogen activated protein kinase kinase
- MK801
- dizocilpine maleate
- MyD88
- myeloid differentiation primary response gene 88
- NF-κB
- nuclear factor κ-light-chain-enhancer of activated B cell
- NLR
- nucleotide-binding domain leucine-rich repeat-containing receptor
- NMDA
- N-methyl-d-aspartate
- TLR
- Toll-like receptor
- TNF
- Tumor necrosis factor
- U50488
- 2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide.
References
- Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. (2009) Chemokines and pain mechanisms. Brain Res Rev 60:125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. (2003) Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA 100:7947–7952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler MW, Rogers TJ. (2005) Are chemokines the third major system in the brain? J Leukoc Biol 78:1204–1209 [DOI] [PubMed] [Google Scholar]
- Agostini S, Eutamene H, Cartier C, Broccardo M, Improta G, Houdeau E, Petrella C, Ferrier L, Theodorou V, Bueno L. (2010) Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology 139:553–563, 563.e551–555 [DOI] [PubMed] [Google Scholar]
- Alonso E, Garrido E, Díez-Fernández C, Pérez-García C, Herradón G, Ezquerra L, Deuel TF, Alguacil LF. (2007) Yohimbine prevents morphine-induced changes of glial fibrillary acidic protein in brainstem and alpha2-adrenoceptor gene expression in hippocampus. Neurosci Lett 412:163–167 [DOI] [PubMed] [Google Scholar]
- Angst MS, Clark JD. (2006) Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology 104:570–587 [DOI] [PubMed] [Google Scholar]
- Araque A, Navarrete M. (2010) Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci 365:2375–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215 [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. (2010) Glial and neuronal control of brain blood flow. Nature 468:232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin PJ, Moalem-Taylor G. (2010) The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol 229:26–50 [DOI] [PubMed] [Google Scholar]
- Avdoshina V, Biggio F, Palchik G, Campbell LA, Mocchetti I. (2010) Morphine induces the release of CCL5 from astrocytes: potential neuroprotective mechanism against the HIV protein gp120. Glia 58:1630–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne JC, Shin NS. (2008) Efficacy of opioids for chronic pain: a review of the evidence. Clin J Pain 24:469–478 [DOI] [PubMed] [Google Scholar]
- Bandler R, Keay KA. (1996) Columnar organization in the midbrain periaqueductal gray and the integration of emotional expression. Prog Brain Res 107:285–300 [DOI] [PubMed] [Google Scholar]
- Bandler R, Shipley MT. (1994) Columnar organization in the midbrain periaqueductal gray: modules for emotional expression? Trends Neurosci 17:379–389 [DOI] [PubMed] [Google Scholar]
- Barbier L, Wang HY, Lin NH, Blasko A. (2010), inventors; Pain Therapeutics, Inc., assignee. Filamin A-binding anti-inflammatory analgesic. World patent WO2010051476 2010 June 5
- Batinić-Haberle I, Ndengele MM, Cuzzocrea S, Rebouças JS, Spasojević I, Salvemini D. (2009) Lipophilicity is a critical parameter that dominates the efficacy of metalloporphyrins in blocking the development of morphine antinociceptive tolerance through peroxynitrite-mediated pathways. Free Radic Biol Med 46:212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, Hornung V. (2011) Inflammasomes: current understanding and open questions. Cell Mol Life Sci 68:765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. (2002) Control of synaptic strength by glial TNFalpha. Science 295:2282–2285 [DOI] [PubMed] [Google Scholar]
- Bederson JB, Fields HL, Barbaro NM. (1990) Hyperalgesia during naloxone-precipitated withdrawal from morphine is associated with increased on-cell activity in the rostral ventromedial medulla. Somatosens Mot Res 7:185–203 [DOI] [PubMed] [Google Scholar]
- Beggs S, Liu XJ, Kwan C, Salter MW. (2010) Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol Pain 6:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behbehani MM, Fields HL. (1979) Evidence that an excitatory connection between the periaqueductal gray and nucleus raphe magnus mediates stimulation produced analgesia. Brain Res 170:85–93 [DOI] [PubMed] [Google Scholar]
- Beitner-Johnson D, Guitart X, Nestler EJ. (1993) Glial fibrillary acidic protein and the mesolimbic dopamine system: regulation by chronic morphine and lewis-fischer strain differences in the rat ventral tegmental area. J Neurochem 61:1766–1773 [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, Coscia CJ. (2005) Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem 280:27662–27669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ. (2003) Mu opioid transactivation and down-regulation of the epidermal growth factor receptor in astrocytes: implications for mitogen-activated protein kinase signaling. Mol Pharmacol 64:1391–1401 [DOI] [PubMed] [Google Scholar]
- Ben Achour S, Pascual O. (2010) Glia: the many ways to modulate synaptic plasticity. Neurochem Int 57:440–445 [DOI] [PubMed] [Google Scholar]
- Bennett J, Basivireddy J, Kollar A, Biron KE, Reickmann P, Jefferies WA, McQuaid S. (2010) Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J Neuroimmunol 229:180–191 [DOI] [PubMed] [Google Scholar]
- Berg-Johnsen J, Paulsen RE, Fonnum F, Langmoen IA. (1993) Changes in evoked potentials and amino acid content during fluorocitrate action studied in rat hippocampal cortex. Exp Brain Res 96:241–246 [DOI] [PubMed] [Google Scholar]
- Bessler H, Shavit Y, Mayburd E, Smirnov G, Beilin B. (2006) Postoperative pain, morphine consumption, and genetic polymorphism of IL-1beta and IL-1 receptor antagonist. Neurosci Lett 404:154–158 [DOI] [PubMed] [Google Scholar]
- Bettoni I, Comelli F, Rossini C, Granucci F, Giagnoni G, Peri F, Costa B. (2008) Glial TLR4 receptor as new target to treat neuropathic pain: efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia 56:1312–1319 [DOI] [PubMed] [Google Scholar]
- Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, Ter Meulen V, Schneider-Schaulies S. (2002) Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol 76:8729–8736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland ST, Hutchinson MR, Maier SF, Watkins LR, Johnson KW. (2009) The glial activation inhibitor AV411 reduces morphine-induced nucleus accumbens dopamine release. Brain Behav Immun 23:492–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SM, Yao H, Bethel-Brown C, Fuwang P, Williams R, Dhillon NK, Hegde R, Kumar A, Buch SJ. (2009) Morphine enhances Tat-induced activation in murine microglia. J Neurovirol 15:219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borish LC, Steinke JW. (2003) 2. Cytokines and chemokines. J Allergy Clin Immunol 111:S460–475 [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Turchan-Cholewo J, Smart EJ, Geurin T, Chauhan A, Reid R, Xu R, Nath A, Knapp PE, Hauser KF. (2008) Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia 56:1414–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. (2006) Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem 281:18081–18089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan MM, Hutchinson M, Watkins LR, Yin H. (2010) Toll-like receptor 4 in CNS pathologies. J Neurochem 114:13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn SJ, Hanley MR, Wilkin GP. (1985) Evidence for a kappa-opioid receptor on pituitary astrocytes: an autoradiographic study. Neurosci Lett 55:317–323 [DOI] [PubMed] [Google Scholar]
- Burbassi S, Sengupta R, Meucci O. (2010) Alterations of CXCR4 function in μ-opioid receptor-deficient glia. Eur J Neurosci 32:1278–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan TP, Jr, Vanderah TW, Lai J, Porreca F. (2002) Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci 22:5129–5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnard DM, Pittman QJ, Macvicar BA. (1991) Neurotransmitter-mediated changes in the electrophysiological properties of pituicytes. J Neuroendocrinol 3:433–439 [DOI] [PubMed] [Google Scholar]
- Burns LH, Wang HY. (2010) PTI-609: a novel analgesic that binds filamin A to control opioid signaling. Recent Pat CNS Drug Discov 5:210–220 [DOI] [PubMed] [Google Scholar]
- Burnstock G. (2006) Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev 58:58–86 [DOI] [PubMed] [Google Scholar]
- Cameron AA, Khan IA, Westlund KN, Willis WD. (1995) The efferent projections of the periaqueductal gray in the rat: a Phaseolus vulgaris-leucoagglutinin study. II. Descending projections. J Comp Neurol 351:585–601 [DOI] [PubMed] [Google Scholar]
- Cao F, Gao F, Xu AJ, Chen ZJ, Chen SS, Yang H, Yu HH, Mei W, Liu XJ, Xiao XP, et al. (2010) Regulation of spinal neuroimmune responses by prolonged morphine treatment in a rat model of cancer induced bone pain. Brain Res 1326:162–173 [DOI] [PubMed] [Google Scholar]
- Cao L, Tanga FY, Deleo JA. (2009) The contributing role of CD14 in toll-like receptor 4 dependent neuropathic pain. Neuroscience 158:896–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglioni AJ, Gallaway MC, Coulter JD. (1978) Spinal projections from the midbrain in monkey. J Comp Neurol 178:329–346 [DOI] [PubMed] [Google Scholar]
- Chacur M, Gutiérrez JM, Milligan ED, Wieseler-Frank J, Britto LR, Maier SF, Watkins LR, Cury Y. (2004) Snake venom components enhance pain upon subcutaneous injection: an initial examination of spinal cord mediators. Pain 111:65–76 [DOI] [PubMed] [Google Scholar]
- Chakfe Y, Seguin R, Antel JP, Morissette C, Malo D, Henderson D, Séguéla P. (2002) ADP and AMP induce interleukin-1beta release from microglial cells through activation of ATP-primed P2X7 receptor channels. J Neurosci 22:3061–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AC, Chao CC, Takemori AE, Gekker G, Hu S, Peterson PK, Portoghese PS. (1996) Arylacetamide-derived fluorescent probes: synthesis, biological evaluation, and direct fluorescent labeling of kappa opioid receptors in mouse microglial cells. J Med Chem 39:1729–1735 [DOI] [PubMed] [Google Scholar]
- Chang G, Chen L, Mao J. (2007) Opioid tolerance and hyperalgesia. Med Clin North Am 91:199–211 [DOI] [PubMed] [Google Scholar]
- Chang RC, Rota C, Glover RE, Mason RP, Hong JS. (2000) A novel effect of an opioid receptor antagonist, naloxone, on the production of reactive oxygen species by microglia: a study by electron paramagnetic resonance spectroscopy. Brain Res 854:224–229 [DOI] [PubMed] [Google Scholar]
- Chao CC, Gekker G, Hu S, Sheng WS, Shark KB, Bu DF, Archer S, Bidlack JM, Peterson PK. (1996) kappa opioid receptors in human microglia downregulate human immunodeficiency virus 1 expression. Proc Natl Acad Sci USA 93:8051–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Shark KB, Sheng WS, Gekker G, Peterson PK. (1997) Activation of mu opioid receptors inhibits microglial cell chemotaxis. J Pharmacol Exp Ther 281:998–1004 [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. (2000) Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 20:RC87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjie N, Alexander GJ, Sechzer JA, Lieberman KW. (1996) Prevention of cocaine-induced hyperactivity by a naloxone isomer with no opiate antagonist activity. Neurochem Res 21:691–693 [DOI] [PubMed] [Google Scholar]
- Chatterjie N, Sechzer JA, Lieberman KW, Alexander GJ. (1998) Dextro-naloxone counteracts amphetamine-induced hyperactivity. Pharmacol Biochem Behav 59:271–274 [DOI] [PubMed] [Google Scholar]
- Chen GY, Nuñez G. (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10:826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Geller EB, Rogers TJ, Adler MW. (2007a) The chemokine CX3CL1/fractalkine interferes with the antinociceptive effect induced by opioid agonists in the periaqueductal grey of rats. Brain Res 1153:52–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Geller EB, Rogers TJ, Adler MW. (2007b) Rapid heterologous desensitization of antinociceptive activity between mu or delta opioid receptors and chemokine receptors in rats. Drug Alcohol Depend 88:36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZR, Irvine RJ, Somogyi AA, Bochner F. (1991) Mu receptor binding of some commonly used opioids and their metabolites. Life Sci 48:2165–2171 [DOI] [PubMed] [Google Scholar]
- Cheng PY, Liu-Chen LY, Pickel VM. (1997) Dual ultrastructural immunocytochemical labeling of mu and delta opioid receptors in the superficial layers of the rat cervical spinal cord. Brain Res 778:367–380 [DOI] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, et al. (2005) Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 114:386–396 [DOI] [PubMed] [Google Scholar]
- Chien GL, Van Winkle DM. (1996) Naloxone blockade of myocardial ischemic preconditioning is stereoselective. J Mol Cell Cardiol 28:1895–1900 [DOI] [PubMed] [Google Scholar]
- Choi DW, Viseskul V. (1988) Opioids and non-opioid enantiomers selectively attenuate N-methyl-d-aspartate neurotoxicity on cortical neurons. Eur J Pharmacol 155:27–35 [DOI] [PubMed] [Google Scholar]
- Christie MJ. (2008) Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol 154:384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino PJ, Keene CD, Breyer RM, Montine KS, Montine TJ. (2008) Therapeutic targets in prostaglandin E2 signaling for neurologic disease. Curr Med Chem 15:1863–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Wodarski R, Guida F, Sasso O, Malcangio M. (2010) Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia 58:1710–1726 [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Malcangio M. (2009) The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci 29:6945–6954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S, Czyzyk T, Ansonoff M, Nitsche JF, Hsu MS, Nilsson L, Larsson K, Borsodi A, Toth G, Hill R, et al. (2002) Autoradiography of opioid and ORL1 ligands in opioid receptor triple knockout mice. Eur J Neurosci 16:1705–1712 [DOI] [PubMed] [Google Scholar]
- Collier HO. (1968) Supersensitivity and dependence. Nature 220:228–231 [DOI] [PubMed] [Google Scholar]
- Collo G, Neidhart S, Kawashima E, Kosco-Vilbois M, North RA, Buell G. (1997) Tissue distribution of the P2X7 receptor. Neuropharmacology 36:1277–1283 [DOI] [PubMed] [Google Scholar]
- Commons KG, Milner TA. (1996) Cellular and subcellular localization of delta opioid receptor immunoreactivity in the rat dentate gyrus. Brain Res 738:181–195 [DOI] [PubMed] [Google Scholar]
- Crain SM, Shen KF. (2000) Antagonists of excitatory opioid receptor functions enhance morphine's analgesic potency and attenuate opioid tolerance/dependence liability. Pain 84:121–131 [DOI] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. (2006) Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res 1069:235–243 [DOI] [PubMed] [Google Scholar]
- Cui Y, Liao XX, Liu W, Guo RX, Wu ZZ, Zhao CM, Chen PX, Feng JQ. (2008) A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain Behav Immun 22:114–123 [DOI] [PubMed] [Google Scholar]
- Dame JB, Juul SE. (2000) The distribution of receptors for the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum Dev 58:25–39 [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Pines G, Kanner BI. (1990) Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glycoprotein from rat brain. Biochemistry (Mosc) 29:6734–6740 [DOI] [PubMed] [Google Scholar]
- Dansereau MA, Gosselin RD, Pohl M, Pommier B, Mechighel P, Mauborgne A, Rostene W, Kitabgi P, Beaudet N, Sarret P, et al. (2008) Spinal CCL2 pronociceptive action is no longer effective in CCR2 receptor antagonist-treated rats. J Neurochem 106:757–769 [DOI] [PubMed] [Google Scholar]
- Darland T, Heinricher MM, Grandy DK. (1998) Orphanin FQ/nociceptin: a role in pain and analgesia, but so much more. Trends Neurosci 21:215–221 [DOI] [PubMed] [Google Scholar]
- Das KP, McMillian MK, Bing G, Hong JS. (1995) Modulatory effects of [Met5]-enkephalin on interleukin-1 beta secretion from microglia in mixed brain cell cultures. J Neuroimmunol 62:9–17 [DOI] [PubMed] [Google Scholar]
- Dave RS, Khalili K. (2010) Morphine treatment of human monocyte-derived macrophages induces differential miRNA and protein expression: impact on inflammation and oxidative stress in the central nervous system. J Cell Biochem 110:834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Buck DJ, Saffarian N, Mohan S, DeSilva U, Fernando SC, Stevens CW. (2008) Beta-funaltrexamine inhibits inducible nitric-oxide synthase expression in human astroglial cells. J Neuroimmune Pharmacol 3:150–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Buck DJ, Saffarian N, Stevens CW. (2007) The opioid antagonist, beta-funaltrexamine, inhibits chemokine expression in human astroglial cells. J Neuroimmunol 186:141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De A, Krueger JM, Simasko SM. (2003) Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res 981:133–142 [DOI] [PubMed] [Google Scholar]
- De Leo JA, Tawfik VL, LaCroix-Fralish ML. (2006) The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain 122:17–21 [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL. (2004) Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist 10:40–52 [DOI] [PubMed] [Google Scholar]
- Delfs JM, Kong H, Mestek A, Chen Y, Yu L, Reisine T, Chesselet MF. (1994) Expression of mu opioid receptor mRNA in rat brain: an in situ hybridization study at the single cell level. J Comp Neurol 345:46–68 [DOI] [PubMed] [Google Scholar]
- Dobrenis K, Makman MH, Stefano GB. (1995) Occurrence of the opiate alkaloid-selective mu3 receptor in mammalian microglia, astrocytes and Kupffer cells. Brain Res 686:239–248 [DOI] [PubMed] [Google Scholar]
- Dourish CT, Hawley D, Iversen SD. (1988) Enhancement of morphine analgesia and prevention of morphine tolerance in the rat by the cholecystokinin antagonist L-364,718. Eur J Pharmacol 147:469–472 [DOI] [PubMed] [Google Scholar]
- Doverty M, White JM, Somogyi AA, Bochner F, Ali R, Ling W. (2001) Hyperalgesic responses in methadone maintenance patients. Pain 90:91–96 [DOI] [PubMed] [Google Scholar]
- Drake CT, Chang PC, Harris JA, Milner TA. (2002) Neurons with mu opioid receptors interact indirectly with enkephalin-containing neurons in the rat dentate gyrus. Exp Neurol 176:254–261 [DOI] [PubMed] [Google Scholar]
- Düesberg U, von dem Bussche A, Kirschning C, Miyake K, Sauerbruch T, Spengler U. (2002) Cell activation by synthetic lipopeptides of the hepatitis C virus (HCV)–core protein is mediated by toll like receptors (TLRs) 2 and 4. Immunol Lett 84:89–95 [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Perez-Reyes E, Rice KC, Palmer MR. (1982) Stereoselectivity of opiate antagonists in rat hippocampus and neocortex: responses to (+) and (-) isomers of naloxone. Neuroscience 7:1691–1702 [DOI] [PubMed] [Google Scholar]
- Ebert B, Andersen S, Krogsgaard-Larsen P. (1995) Ketobemidone, methadone and pethidine are non-competitive N-methyl-D-aspartate (NMDA) antagonists in the rat cortex and spinal cord. Neurosci Lett 187:165–168 [DOI] [PubMed] [Google Scholar]
- El Ghazi I, Sheng WS, Hu S, Reilly BG, Lokensgard JR, Rock RB, Peterson PK, Wilcox GL, Armitage IM. (2010) Changes in the NMR metabolic profile of human microglial cells exposed to lipopolysaccharide or morphine. J Neuroimmune Pharmacol 5:574–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Knapp PE, Hauser KF. (2008a) CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat-exposed mice. J Neuroimmune Pharmacol 3:275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, Hauser KF. (2008b) Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca2+]i, NF-kappaB trafficking and transcription. PLoS ONE 3:e4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Ambati J, Bruce-Keller AJ, Knapp PE, Hauser KF. (2006) CCR2 mediates increases in glial activation caused by exposure to HIV-1 Tat and opiates. J Neuroimmunol 178:9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. (2001) TNF-alpha-induced c-Fos generation in the nucleus of the solitary tract is blocked by NBQX and MK-801. Am J Physiol Regul Integr Comp Physiol 281:R1394–R1400 [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Hansson E, Rönnbäck L. (1990) Delta and kappa opiate receptors in primary astroglial cultures from rat cerebral cortex. Neurochem Res 15:1123–1126 [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Hansson E, Rönnbäck L. (1991) Mu and delta opiate receptors in neuronal and astroglial primary cultures from various regions of the brain–coupling with adenylate cyclase, localisation on the same neurones and association with dopamine (D1) receptor adenylate cyclase. Neuropharmacology 30:1233–1239 [DOI] [PubMed] [Google Scholar]
- Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. (1992) Cloning of a delta opioid receptor by functional expression. Science 258:1952–1955 [DOI] [PubMed] [Google Scholar]
- Fairbanks CA, Wilcox GL. (2000) Spinal plasticity of acute opioid tolerance. J Biomed Sci 7:200–212 [DOI] [PubMed] [Google Scholar]
- Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F. (1996) Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol 156:1531–1539 [PubMed] [Google Scholar]
- Ferrer-Alcón M, García-Sevilla JA, Jaquet PE, La Harpe R, Riederer BM, Walzer C, Guimón J. (2000) Regulation of nonphosphorylated and phosphorylated forms of neurofilament proteins in the prefrontal cortex of human opioid addicts. J Neurosci Res 61:338–349 [DOI] [PubMed] [Google Scholar]
- Festa ED, Cecala C, Quinones-Jenab V, Jenab S. (2002) Cocaine modulates mu-opioid receptor mRNA but not c-fos mRNA levels in primary cortical astrocytes. Brain Res Bull 58:285–288 [DOI] [PubMed] [Google Scholar]
- Fields HL. (1988) Sources of variability in the sensation of pain. Pain 33:195–200 [DOI] [PubMed] [Google Scholar]
- Fields HL. (1992) Is there a facilitating component to central pain modulation? Am Pain Soc J 1:71–78 [Google Scholar]
- Fields HL, Basbaum AI. (1978) Brainstem control of spinal pain-transmission neurons. Annu Rev Physiol 40:217–248 [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM. (1985) Anatomy and physiology of a nociceptive modulatory system. Philos Trans R Soc Lond B Biol Sci 308:361–374 [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P. (1991) Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci 14:219–245 [DOI] [PubMed] [Google Scholar]
- Fishbain DA, Cole B, Lewis JE, Gao J, Rosomoff RS. (2009) Do opioids induce hyperalgesia in humans? An evidence-based structured review. Pain Med 10:829–839 [DOI] [PubMed] [Google Scholar]
- Foo H, Mason P. (2003) Discharge of raphe magnus ON and OFF cells is predictive of the motor facilitation evoked by repeated laser stimulation. J Neurosci 23:1933–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. (2007) Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 21:47–59 [DOI] [PubMed] [Google Scholar]
- Freeman SE, Patil VV, Durham PL. (2008) Nitric oxide-proton stimulation of trigeminal ganglion neurons increases mitogen-activated protein kinase and phosphatase expression in neurons and satellite glial cells. Neuroscience 157:542–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. (2010) Targeting astrocyte signaling for chronic pain. Neurotherapeutics 7:482–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, Lai J, Porreca F. (2002) Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci 22:6747–6755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido E, Pérez-García C, Alguacil LF, Díez-Fernández C. (2005) The alpha2-adrenoceptor antagonist yohimbine reduces glial fibrillary acidic protein upregulation induced by chronic morphine administration. Neurosci Lett 383:141–144 [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Pickel VM. (2002) Alpha(2A)-adrenergic receptors are present in mu-opioid receptor containing neurons in rat medial nucleus tractus solitarius. Synapse 43:208–218 [DOI] [PubMed] [Google Scholar]
- Glaum SR, Miller RJ, Hammond DL. (1994) Inhibitory actions of delta 1-, delta 2-, and mu-opioid receptor agonists on excitatory transmission in lamina II neurons of adult rat spinal cord. J Neurosci 14:4965–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A, Lowney LI, Pal BK. (1971) Stereospecific and nonspecific interactions of the morphine congener levorphanol in subcellular fractions of mouse brain. Proc Natl Acad Sci USA 68:1742–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordh T, Chu H, Sharma HS. (2006) Spinal nerve lesion alters blood-spinal cord barrier function and activates astrocytes in the rat. Pain 124:211–221 [DOI] [PubMed] [Google Scholar]
- Gorodinsky A, Barg J, Belcheva MM, Levy R, McHale RJ, Vogel Z, Coscia CJ. (1995) Dynorphins modulate DNA synthesis in fetal brain cell aggregates. J Neurochem 65:1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goucke CR. (2003) The management of persistent pain. Med J Aust 178:444–447 [DOI] [PubMed] [Google Scholar]
- Grace PM, Hutchinson MR, Bishop A, Somogyi AA, Mayrhofer G, Rolan PE. (2011) Adoptive transfer of peripheral immune cells potentiates allodynia in a graded chronic constriction injury model of neuropathic pain. Brain Behav Immun 25:503–513 [DOI] [PubMed] [Google Scholar]
- Graeber MB. (2010) Changing face of microglia. Science 330:783–788 [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ. (2010) Microglia: biology and pathology. Acta Neuropathol 119:89–105 [DOI] [PubMed] [Google Scholar]
- Grimm MC, Ben-Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, Ali H, Richardson R, Snyderman R, Oppenheim JJ. (1998) Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med 188:317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudt TJ, Williams JT. (1994) mu-Opioid agonists inhibit spinal trigeminal substantia gelatinosa neurons in guinea pig and rat. J Neurosci 14:1646–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gul H, Yildiz O, Dogrul A, Yesilyurt O, Isimer A. (2000) The interaction between IL-1beta and morphine: possible mechanism of the deficiency of morphine-induced analgesia in diabetic mice. Pain 89:39–45 [DOI] [PubMed] [Google Scholar]
- Guo RX, Zhang M, Liu W, Zhao CM, Cui Y, Wang CH, Feng JQ, Chen PX. (2009) NMDA receptors are involved in upstream of the spinal JNK activation in morphine antinociceptive tolerance. Neurosci Lett 467:95–99 [DOI] [PubMed] [Google Scholar]
- Gurwell JA, Duncan MJ, Maderspach K, Stiene-Martin A, Elde RP, Hauser KF. (1996) Kappa-opioid receptor expression defines a phenotypically distinct subpopulation of astroglia: relationship to Ca2+ mobilization, development, and the antiproliferative effect of opioids. Brain Res 737:175–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagino Y, Kariura Y, Manago Y, Amano T, Wang B, Sekiguchi M, Nishikawa K, Aoki S, Wada K, Noda M. (2004) Heterogeneity and potentiation of AMPA type of glutamate receptors in rat cultured microglia. Glia 47:68–77 [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. (2008) Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci USA 105:13532–13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson E, Rönnbäck L. (1983) Incorporation of 3H-valine into soluble protein of cultivated astroglial cells after morphine treatment. J Neurosci Res 10:279–288 [DOI] [PubMed] [Google Scholar]
- Hansson E, Rönnbäck L. (1985) Amino acid incorporation during morphine intoxication. II: Electrophoretic separation of extracellular proteins from cerebral hemisphere slices and astroglia-enriched primary cultures. J Neurosci Res 14:479–490 [DOI] [PubMed] [Google Scholar]
- Hansson E, Westerlund A, Björklund U, Olsson T. (2008) mu-Opioid agonists inhibit the enhanced intracellular Ca2+ responses in inflammatory activated astrocytes co-cultured with brain endothelial cells. Neuroscience 155:1237–1249 [DOI] [PubMed] [Google Scholar]
- Harris JA, Chang PC, Drake CT. (2004) Kappa opioid receptors in rat spinal cord: sex-linked distribution differences. Neuroscience 124:879–890 [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, et al. (1998) Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA 95:10896–10901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Paulsen RE, Johnsen A, Fonnum F. (1992) Selective inhibition of glial cell metabolism in vivo by fluorocitrate. Brain Res 576:120–124 [DOI] [PubMed] [Google Scholar]
- Hauser KF, Harris-White ME, Jackson JA, Opanashuk LA, Carney JM. (1998) Opioids disrupt Ca2+ homeostasis and induce carbonyl oxyradical production in mouse astrocytes in vitro: transient increases and adaptation to sustained exposure. Exp Neurol 151:70–76 [DOI] [PubMed] [Google Scholar]
- Hauser KF, Stiene-Martin A. (1991) Characterization of opioid-dependent glial development in dissociated and organotypic cultures of mouse central nervous system: critical periods and target specificity. Brain Res Dev Brain Res 62:245–255 [DOI] [PubMed] [Google Scholar]
- Hauser KF, Stiene-Martin A, Mattson MP, Elde RP, Ryan SE, Godleske CC. (1996) mu-Opioid receptor-induced Ca2+ mobilization and astroglial development: morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca2+-dependent mechanism. Brain Res 720:191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay JL, Kaboutari J, White JM, Salem A, Irvine R. (2009) Model of methadone-induced hyperalgesia in rats and effect of memantine. Eur J Pharmacol 626:229–233 [DOI] [PubMed] [Google Scholar]
- Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. (2010) Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain 10:316–322 [DOI] [PubMed] [Google Scholar]
- Heijnen CJ, Kavelaars A, Ballieux RE. (1991) Beta-endorphin: cytokine and neuropeptide. Immunol Rev 119:41–63 [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Barbaro NM, Fields HL. (1989) Putative nociceptive modulating neurons in the rostral ventromedial medulla of the rat: firing of on- and off-cells is related to nociceptive responsiveness. Somatosens Mot Res 6:427–439 [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Tortorici V, Fields HL. (1994) Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience 63:279–288 [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ, Tershner SA, Poore LH, Bellgowan PS. (1998) Antinociception following opioid stimulation of the basolateral amygdala is expressed through the periaqueductal gray and rostral ventromedial medulla. Brain Res 779:104–118 [DOI] [PubMed] [Google Scholar]
- Hickey WF. (1999) Leukocyte traffic in the central nervous system: the participants and their roles. Semin Immunol 11:125–137 [DOI] [PubMed] [Google Scholar]
- Højsted J, Sjøgren P. (2007) An update on the role of opioids in the management of chronic pain of nonmalignant origin. Curr Opin Anaesthesiol 20:451–455 [DOI] [PubMed] [Google Scholar]
- Holdridge SV, Armstrong SA, Taylor AM, Cahill CM. (2007) Behavioural and morphological evidence for the involvement of glial cell activation in delta opioid receptor function: implications for the development of opioid tolerance. Mol Pain 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GM, Hebert SL, Rogers RC, Hermann GE. (2004) Immunocytochemical localization of TNF type 1 and type 2 receptors in the rat spinal cord. Brain Res 1025:210–219 [DOI] [PubMed] [Google Scholar]
- Hong J, Cho IH, Kwak KI, Suh EC, Seo J, Min HJ, Choi SY, Kim CH, Park SH, Jo EK, et al. (2010) Microglial Toll-like receptor 2 contributes to kainic acid-induced glial activation and hippocampal neuronal cell death. J Biol Chem 285:39447–39457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook MA, Moreno G, Woller S, Puga D, Hoy K, Jr, Balden R, Grau JW. (2009) Intrathecal morphine attenuates recovery of function after a spinal cord injury. J Neurotrauma 26:741–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook MA, Washburn SN, Moreno G, Woller SA, Puga D, Lee KH, Grau JW. (2011) An IL-1 receptor antagonist blocks a morphine-induced attenuation of locomotor recovery after spinal cord injury. Brain Behav Immun 25:349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath RJ, DeLeo JA. (2009) Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci 29:998–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath RJ, Landry RP, Romero-Sandoval EA, DeLeo JA. (2010a) Morphine tolerance attenuates the resolution of postoperative pain and enhances spinal microglial p38 and extracellular receptor kinase phosphorylation. Neuroscience 169:843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath RJ, Romero-Sandoval EA, De Leo JA. (2010b) Inhibition of microglial P2X4 receptors attenuates morphine tolerance, Iba1, GFAP and mu opioid receptor protein expression while enhancing perivascular microglial ED2. Pain 150:401–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:3749–3752 [PubMed] [Google Scholar]
- Hu S, Chao CC, Hegg CC, Thayer S, Peterson PK. (2000) Morphine inhibits human microglial cell production of, and migration towards, RANTES. J Psychopharmacol 14:238–243 [DOI] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Lokensgard JR, Peterson PK. (2002) Morphine induces apoptosis of human microglia and neurons. Neuropharmacology 42:829–836 [DOI] [PubMed] [Google Scholar]
- Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. (1975) Identification of two related penatpeptides from the brain with potent opiate agonist activity. Nature 258:577–580 [DOI] [PubMed] [Google Scholar]
- Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH. (2002) Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia 37:314–327 [PubMed] [Google Scholar]
- Hurley RW, Hammond DL. (2000) The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J Neurosci 20:1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. (2007) Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. ScientificWorldJournal 7:98–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL, Somogyi AA, et al. (2008a) Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun 22:1178–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, Somogyi AA, Yin H, Maier SF, Rice KC, et al. (2010a) Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience 167:880–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL, Brzeski A, Northcutt A, Vietz CM, Judd CM, et al. (2009a) Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast). Brain Behav Immun 23:240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Loram LC, Zhang Y, Shridhar M, Rezvani N, Berkelhammer D, Phipps S, Foster PS, Landgraf K, Falke JJ, et al. (2010b) Evidence that tricyclic small molecules may possess toll-like receptor and myeloid differentiation protein 2 activity. Neuroscience 168:551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL, Loram LC, Rozeske RR, Bland ST, Maier SF, et al. (2008b) Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun 22:1248–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Ramos KM, Loram LC, Wieseler J, Sholar PW, Kearney JJ, Lewis MT, Crysdale NY, Zhang Y, Harrison JA, et al. (2009b) Evidence for a role of heat shock protein-90 in toll like receptor 4 mediated pain enhancement in rats. Neuroscience 164:1821–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, et al. (2008c) Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur J Neurosci 28:20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, et al. (2010c) Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun 24:83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima I, Minamikawa J, Jacobson AE, Brossi A, Rice KC. (1978) Studies in the (+)-morphinan series. 5. Synthesis and biological properties of (+)-naloxone. J Med Chem 21:398–400 [DOI] [PubMed] [Google Scholar]
- Inoue K. (2009) [The mechanism and control of neuropathic pain]. Rinsho Shinkeigaku 49:779–782 [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. (2004) Toll-like receptor control of the adaptive immune responses. Nat Immunol 5:987–995 [DOI] [PubMed] [Google Scholar]
- Jacquet YF, Klee WA, Rice KC, Iijima I, Minamikawa J. (1977) Stereospecific and nonstereospecific effects of (+)- and (-)-morphine: evidence for a new class of receptors? Science 198:842–845 [DOI] [PubMed] [Google Scholar]
- Ji RR. (2010) Targeting microglial purinergic signaling to improve morphine analgesia. Pain 150:377–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, et al. (2004) A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci 24:7353–7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Westbrook RF. (2005) Inhibition of morphine analgesia by LPS: role of opioid and NMDA receptors and spinal glia. Behav Brain Res 156:75–83 [DOI] [PubMed] [Google Scholar]
- Juni A, Klein G, Pintar JE, Kest B. (2007) Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 147:439–444 [DOI] [PubMed] [Google Scholar]
- Kamei J, Kawashima N, Kasuya Y. (1992) Role of spleen or spleen products in the deficiency in morphine-induced analgesia in diabetic mice. Brain Res 576:139–142 [DOI] [PubMed] [Google Scholar]
- Kamei J, Kawashima N, Suzuki T, Misawa M, Kasuya Y. (1993) The effects of cyclosporine on morphine-induced antinociception in diabetic mice. Neurosci Lett 158:213–216 [DOI] [PubMed] [Google Scholar]
- Kehl LJ, Kovács KJ, Larson AA. (2004) Tolerance develops to the effect of lipopolysaccharides on movement-evoked hyperalgesia when administered chronically by a systemic but not an intrathecal route. Pain 111:104–115 [DOI] [PubMed] [Google Scholar]
- Khurdayan VK, Buch S, El-Hage N, Lutz SE, Goebel SM, Singh IN, Knapp PE, Turchan-Cholewo J, Nath A, Hauser KF. (2004) Preferential vulnerability of astroglia and glial precursors to combined opioid and HIV-1 Tat exposure in vitro. Eur J Neurosci 19:3171–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL, Gavériaux-Ruff C. (2002) Exploring the opioid system by gene knockout. Prog Neurobiol 66:285–306 [DOI] [PubMed] [Google Scholar]
- Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, Choi SY, Park K, Kim JS, Akira S, et al. (2007a) A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem 282:14975–14983 [DOI] [PubMed] [Google Scholar]
- Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, et al. (2007b) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell 130:906–917 [DOI] [PubMed] [Google Scholar]
- Kim JP, Goldberg MP, Choi DW. (1987) High concentrations of naloxone attenuate N-methyl-d-aspartate receptor-mediated neurotoxicity. Eur J Pharmacol 138:133–136 [DOI] [PubMed] [Google Scholar]
- Kimball ES, Raffa RB. (1989) Obligatory role of B cells and adherent accessory cells in the transfer of a defect in morphine-mediated antinociception in C57BL/6J-bg/bg (beige-J) mice. J Neuroimmunol 22:185–192 [DOI] [PubMed] [Google Scholar]
- King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. (2005) Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals 14:194–205 [DOI] [PubMed] [Google Scholar]
- Kleibeuker W, Ledeboer A, Eijkelkamp N, Watkins LR, Maier SF, Zijlstra J, Heijnen CJ, Kavelaars A. (2007) A role for G protein-coupled receptor kinase 2 in mechanical allodynia. Eur J Neurosci 25:1696–1704 [DOI] [PubMed] [Google Scholar]
- Knapp PE, Hauser KF. (1996) mu-Opioid receptor activation enhances DNA synthesis in immature oligodendrocytes. Brain Res 743:341–345 [DOI] [PubMed] [Google Scholar]
- Knapp PE, Itkis OS, Zhang L, Spruce BA, Bakalkin G, Hauser KF. (2001) Endogenous opioids and oligodendroglial function: possible autocrine/paracrine effects on cell survival and development. Glia 35:156–165 [DOI] [PubMed] [Google Scholar]
- Knapp PE, Maderspach K, Hauser KF. (1998) Endogenous opioid system in developing normal and jimpy oligodendrocytes: mu and kappa opioid receptors mediate differential mitogenic and growth responses. Glia 22:189–201 [DOI] [PubMed] [Google Scholar]
- Komatsu T, Sakurada S, Katsuyama S, Sanai K, Sakurada T. (2009) Mechanism of allodynia evoked by intrathecal morphine-3-glucuronide in mice. Int Rev Neurobiol 85:207–219 [DOI] [PubMed] [Google Scholar]
- Kong LY, Jeohn G, Hudson PM, Du L, Liu B, Hong JS. (2000) Reduction of lipopolysaccharide-induced neurotoxicity in mouse mixed cortical neuron/glia cultures by ultralow concentrations of dynorphins. J Biomed Sci 7:241–247 [DOI] [PubMed] [Google Scholar]
- Kong LY, McMillian MK, Hudson PM, Jin L, Hong JS. (1997) Inhibition of lipopolysaccharide-induced nitric oxide and cytokine production by ultralow concentrations of dynorphins in mixed glia cultures. J Pharmacol Exp Ther 280: 61–66 [PubMed] [Google Scholar]
- Kovelowski CJ, Ossipov MH, Sun H, Lai J, Malan TP, Porreca F. (2000) Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain 87:265–273 [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. (2009) Toll-like receptors and innate immunity. Biochem Biophys Res Commun 388:621–625 [DOI] [PubMed] [Google Scholar]
- Kuypers HG, Maisky VA. (1975) Retrograde axonal transport of horseradish peroxidase from spinal cord to brainstem cell groups in the cat. Neurosci Lett 1:9–14 [DOI] [PubMed] [Google Scholar]
- Lai JP, Zhan GX, Campbell DE, Douglas SD, Ho WZ. (2000) Detection of substance P and its receptor in human fetal microglia. Neuroscience 101:1137–1144 [DOI] [PubMed] [Google Scholar]
- Lan LS, Ping YJ, Na WL, Miao J, Cheng QQ, Ni MZ, Lei L, Fang LC, Guang RC, Jin Z, et al. (2010) Down-regulation of Toll-like receptor 4 gene expression by short interfering RNA attenuates bone cancer pain in a rat model. Mol Pain 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang DW, Darrah HK, Hedley-Whyte J, Laasberg LH. (1975) Uptake into brain proteins of 35S-methionine during morphine tolerance. J Pharmacol Exp Ther 192:521–530 [PubMed] [Google Scholar]
- Laughlin TM, Bethea JR, Yezierski RP, Wilcox GL. (2000) Cytokine involvement in dynorphin-induced allodynia. Pain 84:159–167 [DOI] [PubMed] [Google Scholar]
- Laughlin TM, Vanderah TW, Lashbrook J, Nichols ML, Ossipov M, Porreca F, Wilcox GL. (1997) Spinally administered dynorphin A produces long-lasting allodynia: involvement of NMDA but not opioid receptors. Pain 72:253–260 [DOI] [PubMed] [Google Scholar]
- Lazriev IL, Kiknadze GI, Kutateladze II, Nebieridze MI. (2001) Effect of morphine on the number and branching of astrocytes in various regions of rat brain. Bull Exp Biol Med 131:248–250 [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Liu T, Shumilla JA, Mahoney JH, Vijay S, Gross MI, Vargas JA, Sultzbaugh L, Claypool MD, Sanftner LM, et al. (2006a) The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biol 2:279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Mahoney JH, Milligan ED, Martin D, Maier SF, Watkins LR. (2006b) Spinal cord glia and interleukin-1 do not appear to mediate persistent allodynia induced by intramuscular acidic saline in rats. J Pain 7:757–767 [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. (2005) Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 115: 71–83 [DOI] [PubMed] [Google Scholar]
- Lee KM, Jeon SM, Cho HJ. (2010) Interleukin-6 induces microglial CX3CR1 expression in the spinal cord after peripheral nerve injury through the activation of p38 MAPK. Eur J Pain 14:682.e1–12 [DOI] [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, Rice KC, Watkins LR. (2010) Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience 165:569–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li H, Zhang Y, Sun X, Hanley GA, LeSage G, Zhang Y, Sun S, Peng Y, Yin D. (2010) Toll-like receptor 2 is required for opioids-induced neuronal apoptosis. Biochem Biophys Res Commun 391:426–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun X, Zhang Y, Huang J, Hanley G, Ferslew KE, Peng Y, Yin D. (2009) Morphine promotes apoptosis via TLR2, and this is negatively regulated by beta-arrestin 2. Biochem Biophys Res Commun 378:857–861 [DOI] [PubMed] [Google Scholar]
- Li Y, Wang X, Tian S, Guo CJ, Douglas SD, Ho WZ. (2002) Methadone enhances human immunodeficiency virus infection of human immune cells. J Infect Dis 185:118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao SL, Chen WY, Raung SL, Chen CJ. (2003) Neuroprotection of naloxone against ischemic injury in rats: role of mu receptor antagonism. Neurosci Lett 345:169–172 [DOI] [PubMed] [Google Scholar]
- Lilius TO, Rauhala PV, Kambur O, Kalso EA. (2009) Modulation of morphine-induced antinociception in acute and chronic opioid treatment by ibudilast. Anesthesiology 111:1356–1364 [DOI] [PubMed] [Google Scholar]
- Lin SL, Tsai RY, Shen CH, Lin FH, Wang JJ, Hsin ST, Wong CS. (2010a) Co-administration of ultra-low dose naloxone attenuates morphine tolerance in rats via attenuation of NMDA receptor neurotransmission and suppression of neuroinflammation in the spinal cords. Pharmacol Biochem Behav 96:236–245 [DOI] [PubMed] [Google Scholar]
- Lin SL, Tsai RY, Tai YH, Cherng CH, Wu CT, Yeh CC, Wong CS. (2010b) Ultra-low dose naloxone upregulates interleukin-10 expression and suppresses neuroinflammation in morphine-tolerant rat spinal cords. Behav Brain Res 207:30–36 [DOI] [PubMed] [Google Scholar]
- Lipovsky MM, Gekker G, Hu S, Hoepelman AI, Peterson PK. (1998) Morphine enhances complement receptor-mediated phagocytosis of Cryptococcus neoformans by human microglia. Clin Immunol Immunopathol 87:163–167 [DOI] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. (2000a) Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther 293:607–617 [PubMed] [Google Scholar]
- Liu B, Du L, Kong LY, Hudson PM, Wilson BC, Chang RC, Abel HH, Hong JS. (2000b) Reduction by naloxone of lipopolysaccharide-induced neurotoxicity in mouse cortical neuron-glia co-cultures. Neuroscience 97:749–756 [DOI] [PubMed] [Google Scholar]
- Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. (2002a) Role of nitric oxide in inflammation-mediated neurodegeneration. Ann NY Acad Sci 962:318–331 [DOI] [PubMed] [Google Scholar]
- Liu B, Jiang JW, Wilson BC, Du L, Yang SN, Wang JY, Wu GC, Cao XD, Hong JS. (2000c) Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J Pharmacol Exp Ther 295:125–132 [PubMed] [Google Scholar]
- Liu B, Qin L, Yang SN, Wilson BC, Liu Y, Hong JS. (2001) Femtomolar concentrations of dynorphins protect rat mesencephalic dopaminergic neurons against inflammatory damage. J Pharmacol Exp Ther 298:1133–1141 [PubMed] [Google Scholar]
- Liu S, Yang J, Wang L, Jiang M, Qiu Q, Ma Z, Liu L, Li C, Ren C, Zhou J, et al. (2010a) Tibia tumor-induced cancer pain involves spinal p38 mitogen-activated protein kinase activation via TLR4-dependent mechanisms. Brain Res 1346:213–223 [DOI] [PubMed] [Google Scholar]
- Liu W, Wang CH, Cui Y, Mo LQ, Zhi JL, Sun SN, Wang YL, Yu HM, Zhao CM, Feng JQ, et al. (2006) Inhibition of neuronal nitric oxide synthase antagonizes morphine antinociceptive tolerance by decreasing activation of p38 MAPK in the spinal microglia. Neurosci Lett 410:174–177 [DOI] [PubMed] [Google Scholar]
- Liu WT, Han Y, Liu YP, Song AA, Barnes B, Song XJ. (2010b) Spinal matrix metalloproteinase-9 contributes to physical dependence on morphine in mice. J Neurosci 30:7613–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XJ, Salter MW. (2005) Purines and pain mechanisms: recent developments. Curr Opin Investig Drugs 6:65–75 [PubMed] [Google Scholar]
- Liu Y, Qin L, Wilson BC, An L, Hong JS, Liu B. (2002b) Inhibition by naloxone stereoisomers of beta-amyloid peptide (1–42)-induced superoxide production in microglia and degeneration of cortical and mesencephalic neurons. J Pharmacol Exp Ther 302:1212–1219 [DOI] [PubMed] [Google Scholar]
- Loh HH, Hitzemann RJ. (1974) Effect of morphine on the turnover and synthesis of (leu-3H)-protein and (ch-14C)-phosphatidylcholine in discrete regions of the rat brain. Biochem Pharmacol 23:1753–1765 [DOI] [PubMed] [Google Scholar]
- Lowney LI, Schulz K, Lowery PJ, Goldstein A. (1974) Partial purification of an opiate receptor from mouse brain. Science 183:749–753 [DOI] [PubMed] [Google Scholar]
- Lu X, Bing G, Hagg T. (2000) Naloxone prevents microglia-induced degeneration of dopaminergic substantia nigra neurons in adult rats. Neuroscience 97:285–291 [DOI] [PubMed] [Google Scholar]
- Maderspach K, Takács J, Niewiadomska G, Csillag A. (1995) Postsynaptic and extrasynaptic localization of kappa-opioid receptor in selected brain areas of young rat and chick using an anti-receptor monoclonal antibody. J Neurocytol 24:478–486 [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Fernandez SF, Chawda R, Shanahan TC, Schwartz SA. (2008) Tight junction regulation by morphine and HIV-1 tat modulates blood-brain barrier permeability. J Clin Immunol 28:528–541 [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Aalinkeel R, Chawda RP, Sykes DE, Nair MP. (2005) Morphine modulates chemokine gene regulation in normal human astrocytes. Clin Immunol 115:323–332 [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MP. (2002) Morphine regulates gene expression of alpha- and beta-chemokines and their receptors on astroglial cells via the opioid mu receptor. J Immunol 169:3589–3599 [DOI] [PubMed] [Google Scholar]
- Mallard C, Wang X, Hagberg H. (2009) The role of Toll-like receptors in perinatal brain injury. Clin Perinatol 36:763–772, v–vi [DOI] [PubMed] [Google Scholar]
- Maneckjee R, Minna JD. (1992) Nonconventional opioid binding sites mediate growth inhibitory effects of methadone on human lung cancer cells. Proc Natl Acad Sci USA 89:1169–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneckjee R, Minna JD. (1997) Characterization of methadone receptor subtypes present in human brain and lung tissues. Life Sci 61:PL 333–PL 338 [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ. (1995) Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci 18:22–29 [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Peschanski M. (1982) Spinal projections from the periaqueductal grey and dorsal raphe in the rat, cat and monkey. Neuroscience 7:2769–2776 [DOI] [PubMed] [Google Scholar]
- Mantz J, Cordier J, Giaume C. (1993) Effects of general anesthetics on intercellular communications mediated by gap junctions between astrocytes in primary culture. Anesthesiology 78:892–901 [DOI] [PubMed] [Google Scholar]
- Mao J. (2006) Opioid-induced abnormal pain sensitivity. Curr Pain Headache Rep 10:67–70 [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. (1995a) Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain 61:353–364 [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. (1995b) Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain 62:259–274 [DOI] [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G. (2002) Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22:8312–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranto J, Rappaport J, Datta PK. (2008) Regulation of complement component C3 in astrocytes by IL-1beta and morphine. J Neuroimmune Pharmacol 3:43–51 [DOI] [PubMed] [Google Scholar]
- Marie-Claire C, Courtin C, Roques BP, Noble F. (2004) Cytoskeletal genes regulation by chronic morphine treatment in rat striatum. Neuropsychopharmacology 29:2208–2215 [DOI] [PubMed] [Google Scholar]
- Mason P. (1997) Physiological identification of pontomedullary serotonergic neurons in the rat. J Neurophysiol 77:1087–1098 [DOI] [PubMed] [Google Scholar]
- Mason P. (1999) Central mechanisms of pain modulation. Curr Opin Neurobiol 9:436–441 [DOI] [PubMed] [Google Scholar]
- Mattioli TA, Milne B, Cahill CM. (2010) Ultra-low dose naltrexone attenuates chronic morphine-induced gliosis in rats. Mol Pain 6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer DJ, Price DD. (1976) Central nervous system mechanisms of analgesia. Pain 2:379–404 [DOI] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, Price DD. (1999) Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci USA 96:7731–7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Namovic MT, Donnelly-Roberts DL, Harris RR, Zhang XF, Shieh CC, Wismer CT, Zhu CZ, Gauvin DM, et al. (2007) P2X7-related modulation of pathological nociception in rats. Neuroscience 146:1817–1828 [DOI] [PubMed] [Google Scholar]
- McGreal EP. (2009) Structural basis of pattern recognition by innate immune molecules. Adv Exp Med Biol 653:139–161 [DOI] [PubMed] [Google Scholar]
- McLennan GP, Kiss A, Miyatake M, Belcheva MM, Chambers KT, Pozek JJ, Mohabbat Y, Moyer RA, Bohn LM, Coscia CJ. (2008) Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J Neurochem 107:1753–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. (1994) The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology 33:1471–1478 [DOI] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. (1993) Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain 52:127–136 [DOI] [PubMed] [Google Scholar]
- Mika J. (2008) Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol Rep 60:297–307 [PubMed] [Google Scholar]
- Mika J, Osikowicz M, Makuch W, Przewlocka B. (2007) Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol 560:142–149 [DOI] [PubMed] [Google Scholar]
- Mika J, Rojewska E, Makuch W, Przewlocka B. (2010) Minocycline reduces the injury-induced expression of prodynorphin and pronociceptin in the dorsal root ganglion in a rat model of neuropathic pain. Neuroscience 165:1420–1428 [DOI] [PubMed] [Google Scholar]
- Mika J, Wawrzczak-Bargiela A, Osikowicz M, Makuch W, Przewlocka B. (2009) Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain Behav Immun 23:75–84 [DOI] [PubMed] [Google Scholar]
- Milligan ED, Mehmert KK, Hinde JL, Harvey LO, Martin D, Tracey KJ, Maier SF, Watkins LR. (2000) Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res 861:105–116 [DOI] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Watkins LR. (2008) Glia in pathological pain: a role for fractalkine. J Neuroimmunol 198:113–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. (2003) Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci 23:1026–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10:23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O'Connor KA, Verge GM, Chapman G, Green P, Foster AC, et al. (2004) Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci 20:2294–2302 [DOI] [PubMed] [Google Scholar]
- Mitra S, Sinatra RS. (2004) Perioperative management of acute pain in the opioid-dependent patient. Anesthesiology 101:212–227 [DOI] [PubMed] [Google Scholar]
- Miyake K. (2007) Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol 19:3–10 [DOI] [PubMed] [Google Scholar]
- Miyatake M, Rubinstein TJ, McLennan GP, Belcheva MM, Coscia CJ. (2009) Inhibition of EGF-induced ERK/MAP kinase-mediated astrocyte proliferation by mu opioids: integration of G protein and beta-arrestin 2-dependent pathways. J Neurochem 110:662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller T, Kann O, Verkhratsky A, Kettenmann H. (2000) Activation of mouse microglial cells affects P2 receptor signaling. Brain Res 853:49–59 [DOI] [PubMed] [Google Scholar]
- Morioka N, Inoue A, Hanada T, Kumagai K, Takeda K, Ikoma K, Hide I, Tamura Y, Shiomi H, Dohi T, et al. (2002) Nitric oxide synergistically potentiates interleukin-1 beta-induced increase of cyclooxygenase-2 mRNA levels, resulting in the facilitation of substance P release from primary afferent neurons: involvement of cGMP-independent mechanisms. Neuropharmacology 43:868–876 [DOI] [PubMed] [Google Scholar]
- Moulédous L, Diaz MF, Gutstein HB. (2004) Modulation of extracellular signal-regulated kinase (ERK) activity by acute and chronic opioid treatment in neuronal and glial cell lines. J Neurochem 90:1371–1377 [DOI] [PubMed] [Google Scholar]
- Muller WA. (2009) Mechanisms of transendothelial migration of leukocytes. Circ Res 105:223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. (2007) Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest 117:3530–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscoli C, Doyle T, Dagostino C, Bryant L, Chen Z, Watkins LR, Ryerse J, Bieberich E, Neumman W, Salvemini D. (2010) Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J Neurosci 30:15400–15408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Mantovani A. (2001) Toll-like receptors (TLRs) signalling and expression pattern. J Endotoxin Res 7:297–300 [PubMed] [Google Scholar]
- Nakagawa T, Satoh M. (2004) Involvement of glial glutamate transporters in morphine dependence. Ann NY Acad Sci 1025:383–388 [DOI] [PubMed] [Google Scholar]
- Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A, Kuzumaki N, Nagumo Y, Suzuki T. (2006a) Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology 31:2476–2488 [DOI] [PubMed] [Google Scholar]
- Narita M, Miyatake M, Shibasaki M, Tsuda M, Koizumi S, Narita M, Yajima Y, Inoue K, Suzuki T. (2005) Long-lasting change in brain dynamics induced by methamphetamine: enhancement of protein kinase C-dependent astrocytic response and behavioral sensitization. J Neurochem 93:1383–1392 [DOI] [PubMed] [Google Scholar]
- Narita M, Suzuki M, Imai S, Narita M, Ozaki S, Kishimoto Y, Oe K, Yajima Y, Yamazaki M, Suzuki T. (2004a) Molecular mechanism of changes in the morphine-induced pharmacological actions under chronic pain-like state: suppression of dopaminergic transmission in the brain. Life Sci 74:2655–2673 [DOI] [PubMed] [Google Scholar]
- Narita M, Suzuki M, Narita M, Niikura K, Nakamura A, Miyatake M, Yajima Y, Suzuki T. (2006b) mu-Opioid receptor internalization-dependent and -independent mechanisms of the development of tolerance to mu-opioid receptor agonists: comparison between etorphine and morphine. Neuroscience 138:609–619 [DOI] [PubMed] [Google Scholar]
- Narita M, Suzuki M, Narita M, Yajima Y, Suzuki R, Shioda S, Suzuki T. (2004b) Neuronal protein kinase C gamma-dependent proliferation and hypertrophy of spinal cord astrocytes following repeated in vivo administration of morphine. Eur J Neurosci 19:479–484 [DOI] [PubMed] [Google Scholar]
- Ndengele MM, Cuzzocrea S, Masini E, Vinci MC, Esposito E, Muscoli C, Petrusca DN, Mollace V, Mazzon E, Li D, et al. (2009) Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther 329: 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert MJ, Kincaid W, Heinricher MM. (2004) Nociceptive facilitating neurons in the rostral ventromedial medulla. Pain 110:158–165 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318 [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB. (2010) Spinal glia and chronic pain. Metabolism 59 (Suppl 1):S21–S26 [DOI] [PubMed] [Google Scholar]
- O'Neill LA. (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev 226:10–18 [DOI] [PubMed] [Google Scholar]
- Obata K, Katsura H, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Akira S, Noguchi K. (2008) Toll-like receptor 3 contributes to spinal glial activation and tactile allodynia after nerve injury. J Neurochem 105:2249–2259 [DOI] [PubMed] [Google Scholar]
- Ochshorn M, Novick DM, Kreek MJ. (1990) In vitro studies of the effect of methadone on natural killer cell activity. Isr J Med Sci 26:421–425 [PubMed] [Google Scholar]
- Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH. (2005) Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol 193:384–393 [DOI] [PubMed] [Google Scholar]
- Ohtori S, Takahashi K, Moriya H, Myers RR. (2004) TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine (Phila Pa 1976) 29:1082–1088 [DOI] [PubMed] [Google Scholar]
- Oka T, Aou S, Hori T. (1994) Intracerebroventricular injection of interleukin-1 beta enhances nociceptive neuronal responses of the trigeminal nucleus caudalis in rats. Brain Res 656:236–244 [DOI] [PubMed] [Google Scholar]
- Olsson Y. (1990) Microenvironment of the peripheral nervous system under normal and pathological conditions. Crit Rev Neurobiol 5:265–311 [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Malan TP, Jr, Hruby VJ, Porreca F. (2004) Antinociceptive and nociceptive actions of opioids. J Neurobiol 61:126–148 [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. (2005) Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers 80:319–324 [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Vanderah TW, Porreca F. (2003) Induction of pain facilitation by sustained opioid exposure: relationship to opioid antinociceptive tolerance. Life Sci 73:783–800 [DOI] [PubMed] [Google Scholar]
- Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M. (2001) Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res 905:254–258 [DOI] [PubMed] [Google Scholar]
- Palma C, Minghetti L, Astolfi M, Ambrosini E, Silberstein FC, Manzini S, Levi G, Aloisi F. (1997) Functional characterization of substance P receptors on cultured human spinal cord astrocytes: synergism of substance P with cytokines in inducing interleukin-6 and prostaglandin E2 production. Glia 21:183–193 [DOI] [PubMed] [Google Scholar]
- Pannese E. (1981) The satellite cells of the sensory ganglia. Adv Anat Embryol Cell Biol 65:1–111 [DOI] [PubMed] [Google Scholar]
- Pavlovic ZW, Cooper ML, Bodnar RJ. (1996) Opioid antagonists in the periaqueductal gray inhibit morphine and beta-endorphin analgesia elicited from the amygdala of rats. Brain Res 741:13–26 [DOI] [PubMed] [Google Scholar]
- Perry VH, Hume DA, Gordon S. (1985) Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 15:313–326 [DOI] [PubMed] [Google Scholar]
- Persson AI, Thorlin T, Bull C, Zarnegar P, Ekman R, Terenius L, Eriksson PS. (2003) Mu- and delta-opioid receptor antagonists decrease proliferation and increase neurogenesis in cultures of rat adult hippocampal progenitors. Eur J Neurosci 17:1159–1172 [DOI] [PubMed] [Google Scholar]
- Persson PA, Thorlin T, Rönnbäck L, Hansson E, Eriksson PS. (2000) Differential expression of delta opioid receptors and mRNA in proliferating astrocytes during the cell cycle. J Neurosci Res 61:371–375 [DOI] [PubMed] [Google Scholar]
- Pert CB, Snowman AM, Snyder SH. (1974) Localization of opiate receptor binding in synaptic membranes of rat brain. Brain Res 70:184–188 [DOI] [PubMed] [Google Scholar]
- Pert CB, Snyder SH. (1973) Opiate receptor: demonstration in nervous tissue. Science 179:1011–1014 [DOI] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Hu S, Sheng WS, Molitor TW, Chao CC. (1995) Morphine stimulates phagocytosis of Mycobacterium tuberculosis by human microglial cells: involvement of a G protein-coupled opiate receptor. Adv Neuroimmunol 5:299–309 [DOI] [PubMed] [Google Scholar]
- Peterson PK, Molitor TW, Chao CC. (1998) The opioid-cytokine connection. J Neuroimmunol 83:63–69 [DOI] [PubMed] [Google Scholar]
- Piaton G, Gould RM, Lubetzki C. (2010) Axon-oligodendrocyte interactions during developmental myelination, demyelination and repair. J Neurochem 114:1243–1260 [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088 [DOI] [PubMed] [Google Scholar]
- Pomeroy SL, Behbehani MM. (1979) Physiologic evidence for a projection from the periaqueductal gray to nucleus raphe magnus in the rat. Brain Res 176:143–147 [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. (1997) Astrocytic neurotransmitter receptors in situ and in vivo. Prog Neurobiol 51:439–455 [DOI] [PubMed] [Google Scholar]
- Potrebic SB, Fields HL, Mason P. (1994) Serotonin immunoreactivity is contained in one physiological cell class in the rat rostral ventromedial medulla. J Neurosci 14:1655–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pud D, Cohen D, Lawental E, Eisenberg E. (2006) Opioids and abnormal pain perception: new evidence from a study of chronic opioid addicts and healthy subjects. Drug Alcohol Depend 82:218–223 [DOI] [PubMed] [Google Scholar]
- Qian L, Tan KS, Wei SJ, Wu HM, Xu Z, Wilson B, Lu RB, Hong JS, Flood PM. (2007) Microglia-mediated neurotoxicity is inhibited by morphine through an opioid receptor-independent reduction of NADPH oxidase activity. J Immunol 179:1198–1209 [DOI] [PubMed] [Google Scholar]
- Qin L, Block ML, Liu Y, Bienstock RJ, Pei Z, Zhang W, Wu X, Wilson B, Burka T, Hong JS. (2005) Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. FASEB J 19:550–557 [DOI] [PubMed] [Google Scholar]
- Quan N, He L, Lai W. (2003) Endothelial activation is an intermediate step for peripheral lipopolysaccharide induced activation of paraventricular nucleus. Brain Res Bull 59:447–452 [DOI] [PubMed] [Google Scholar]
- Raffa RB, Kimball ES, Mathiasen JR. (1988) The analgesic defect of C57BL/6J-bgJ/bgJ (beige-J: Chediak-Higashi syndrome) mice transmitted by adoptive transfer of spleen cells to normal littermates. Life Sci 42:1231–1236 [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. (2002) The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci 22:9980–9989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. (2003a) Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 306:624–630 [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, Rutkowski MD, DeLeo JA. (2003b) Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain 104:655–664 [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. (2004a) Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology 29:327–334 [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. (2004b) Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci 20:467–473 [DOI] [PubMed] [Google Scholar]
- Ramírez F, Vanegas H. (1989) Tooth pulp stimulation advances both medullary off-cell pause and tail flick. Neurosci Lett 100:153–156 [DOI] [PubMed] [Google Scholar]
- Ramos KM, Jiang Y, Svensson CI, Calcutt NA. (2007) Pathogenesis of spinally mediated hyperalgesia in diabetes. Diabetes 56:1569–1576 [DOI] [PubMed] [Google Scholar]
- Ramos KM, Lewis MT, Morgan KN, Crysdale NY, Kroll JL, Taylor FR, Harrison JA, Sloane EM, Maier SF, Watkins LR. (2010) Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience 169:1888–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp SE, Ready LB, Nessly ML. (1995) Acute pain management in patients with prior opioid consumption: a case-controlled retrospective review. Pain 61:195–201 [DOI] [PubMed] [Google Scholar]
- Rawls SM, Zielinski M, Patel H, Sacavage S, Baron DA, Patel D. (2010) Beta-lactam antibiotic reduces morphine analgesic tolerance in rats through GLT-1 transporter activation. Drug Alcohol Depend 107:261–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. (2009) CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 29:11982–11992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AJ, Patel S, Fox A, Walker K, Urban L. (2000) Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain 4:247–257 [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. (1996) Enhanced descending modulation of nociception in rats with persistent hindpaw inflammation. J Neurophysiol 76:3025–3037 [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. (2008) Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anaesthesiol 21:570–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Gibby CC, El Osta B, Spitz MR, Parsons H, Kurzrock R, Wu X, Shete S, Bruera E. (2008) The influence of tumor necrosis factor-alpha -308 G/A and IL-6-174 G/C on pain and analgesia response in lung cancer patients receiving supportive care. Cancer Epidemiol Biomarkers Prev 17:3262–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S. (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9:429–439 [DOI] [PubMed] [Google Scholar]
- Rock RB, Hu S, Sheng WS, Peterson PK. (2006) Morphine stimulates CCL2 production by human neurons. J Neuroinflammation 3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Mackie K, Pickel VM. (2001) Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat caudate putamen nucleus. J Neurosci 21:823–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TJ, Steele AD, Howard OM, Oppenheim JJ. (2000) Bidirectional heterologous desensitization of opioid and chemokine receptors. Ann NY Acad Sci 917:19–28 [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Horvath RJ, DeLeo JA. (2008) Neuroimmune interactions and pain: focus on glial-modulating targets. Curr Opin Investig Drugs 9:726–734 [PMC free article] [PubMed] [Google Scholar]
- Rönnbäck L, Hansson E. (1985) Amino acid incorporation during morphine intoxication. I: Dose and time effects of morphine on protein synthesis in specific regions of the rat brain and in astroglia-enriched primary cultures. J Neurosci Res 14:461–477 [DOI] [PubMed] [Google Scholar]
- Rönnbäck L, Hansson E. (1988) Are astroglial cells involved in morphine tolerance? Neurochem Res 13:87–103 [DOI] [PubMed] [Google Scholar]
- Roth-Schechter B, Mandel P. (1976) A study on the development of barbiturate tolerance and dependence in hamster glial cells in culture. Biochem Pharmacol 25:563–571 [DOI] [PubMed] [Google Scholar]
- Roy S, Ge BL, Loh HH, Lee NM. (1992) Characterization of [3H]morphine binding to interleukin-1-activated thymocytes. J Pharmacol Exp Ther 263:451–456 [PubMed] [Google Scholar]
- Roy S, Ge BL, Ramakrishnan S, Lee NM, Loh HH. (1991) [3H]morphine binding is enhanced by IL-1-stimulated thymocyte proliferation. FEBS Lett 287:93–96 [DOI] [PubMed] [Google Scholar]
- Roy S, Sedqi M, Ramakrishnan S, Barke RA, Loh HH. (1996) Differential effects of opioids on the proliferation of a macrophage cell line, Bac 1.2F5. Cell Immunol 169:271–277 [DOI] [PubMed] [Google Scholar]
- Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. (1995) Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptor mRNA. Brain Res Mol Brain Res 34:209–220 [DOI] [PubMed] [Google Scholar]
- Ruzicka BB, Thompson RC, Watson SJ, Akil H. (1996) Interleukin-1 beta-mediated regulation of mu-opioid receptor mRNA in primary astrocyte-enriched cultures. J Neurochem 66:425–428 [DOI] [PubMed] [Google Scholar]
- Saito O, Svensson CI, Buczynski MW, Wegner K, Hua XY, Codeluppi S, Schaloske RH, Deems RA, Dennis EA, Yaksh TL. (2010) Spinal glial TLR4-mediated nociception and production of prostaglandin E(2) and TNF. Br J Pharmacol 160:1754–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, De Koninck Y, Henry JL. (1993) Physiological roles for adenosine and ATP in synaptic transmission in the spinal dorsal horn. Prog Neurobiol 41:125–156 [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. (2001) Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410:471–475 [DOI] [PubMed] [Google Scholar]
- Satoh M, Minami M. (1995) Molecular pharmacology of the opioid receptors. Pharmacol Ther 68:343–364 [DOI] [PubMed] [Google Scholar]
- Sawaya BE, Deshmane SL, Mukerjee R, Fan S, Khalili K. (2009) TNF alpha production in morphine-treated human neural cells is NF-kappaB-dependent. J Neuroimmune Pharmacol 4:140–149 [DOI] [PubMed] [Google Scholar]
- Schaible HG, Neugebauer V, Cervero F, Schmidt RF. (1991) Changes in tonic descending inhibition of spinal neurons with articular input during the development of acute arthritis in the cat. J Neurophysiol 66:1021–1032 [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. (2007) The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 10:1361–1368 [DOI] [PubMed] [Google Scholar]
- Schug SA, Zech D, Grond S. (1992) Adverse effects of systemic opioid analgesics. Drug Saf 7:200–213 [DOI] [PubMed] [Google Scholar]
- Schwartz JP, Nishiyama N, Wilson D, Taniwaki T. (1994) Receptor-mediated regulation of neuropeptide gene expression in astrocytes. Glia 11:185–190 [DOI] [PubMed] [Google Scholar]
- Sharma HS, Ali SF. (2006) Alterations in blood-brain barrier function by morphine and methamphetamine. Ann NY Acad Sci 1074:198–224 [DOI] [PubMed] [Google Scholar]
- Sharma HS, Sjöquist PO, Ali SF. (2010) Alterations in blood-brain barrier function and brain pathology by morphine in the rat. Neuroprotective effects of antioxidant H-290/51. Acta Neurochir Suppl 106:61–66 [DOI] [PubMed] [Google Scholar]
- Shavit Y, Fish G, Wolf G, Mayburd E, Meerson Y, Yirmiya R, Beilin B. (2005a) The effects of perioperative pain management techniques on food consumption and body weight after laparotomy in rats. Anesth Analg 101:1112–1116, table of contents [DOI] [PubMed] [Google Scholar]
- Shavit Y, Wolf G, Goshen I, Livshits D, Yirmiya R. (2005b) Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain 115:50–59 [DOI] [PubMed] [Google Scholar]
- Shen CH, Tsai RY, Shih MS, Lin SL, Tai YH, Chien CC, Wong CS. (2011) Etanercept restores the antinociceptive effect of morphine and suppresses spinal neuroinflammation in morphine-tolerant rats. Anesth Analg 112:454–459 [DOI] [PubMed] [Google Scholar]
- Sheng WS, Hu S, Lokensgard JR, Peterson PK. (2003) U50,488 inhibits HIV-1 Tat-induced monocyte chemoattractant protein-1 (CCL2) production by human astrocytes. Biochem Pharmacol 65:9–14 [DOI] [PubMed] [Google Scholar]
- Shi XQ, Zekki H, Zhang J. (2011) The role of TLR2 in nerve injury-induced neuropathic pain is essentially mediated through macrophages in peripheral inflammatory response. Glia 59:231–241 [DOI] [PubMed] [Google Scholar]
- Shinder V, Amir R, Devor M. (1998) Cross-excitation in dorsal root ganglia does not depend on close cell-to-cell apposition. Neuroreport 9:3997–4000 [DOI] [PubMed] [Google Scholar]
- Sibinga NE, Goldstein A. (1988) Opioid peptides and opioid receptors in cells of the immune system. Annu Rev Immunol 6:219–249 [DOI] [PubMed] [Google Scholar]
- Simka M. (2009) Blood brain barrier compromise with endothelial inflammation may lead to autoimmune loss of myelin during multiple sclerosis. Curr Neurovasc Res 6:132–139 [DOI] [PubMed] [Google Scholar]
- Simon EJ, Hiller JM, Edelman I. (1973) Stereospecific binding of the potent analgesic (3H) etorphine to rat-brain homogenate. Proc Nat Acad Sci USA 70:1947–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin F, Slowe S, Becker JA, Matthes HW, Filliol D, Chluba J, Kitchen I, Kieffer BL. (2001) Analysis of [3H]bremazocine binding in single and combinatorial opioid receptor knockout mice. Eur J Pharmacol 414:189–195 [DOI] [PubMed] [Google Scholar]
- Simpkins CO, Ives N, Tate E, Johnson M. (1985) Naloxone inhibits superoxide release from human neutrophils. Life Sci 37:1381–1386 [DOI] [PubMed] [Google Scholar]
- Singh VK. (1980) Suppressive effects of methadone on human blood lymphocytes. Immunol Lett 2:177 [Google Scholar]
- Singla A, Stojanovic MP, Chen L, Mao J. (2007) A differential diagnosis of hyperalgesia, toxicity, and withdrawal from intrathecal morphine infusion. Anesth Analg 105:1816–1819 [DOI] [PubMed] [Google Scholar]
- Slatkin NE, Rhiner M. (2003) Treatment of opiate-related sedation: utility of the cholinesterase inhibitors. J Support Oncol 1:53–63 [PubMed] [Google Scholar]
- Smith K. (2010) Neuroscience: settling the great glia debate. Nature 468:160–162 [DOI] [PubMed] [Google Scholar]
- Song L, Pachter JS. (2004) Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc Res 67:78–89 [DOI] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. (2001) The involvement of glial cells in the development of morphine tolerance. Neurosci Res 39:281–286 [DOI] [PubMed] [Google Scholar]
- Sowa G, Gekker G, Lipovsky MM, Hu S, Chao CC, Molitor TW, Peterson PK. (1997) Inhibition of swine microglial cell phagocytosis of Cryptococcus neoformans by femtomolar concentrations of morphine. Biochem Pharmacol 53:823–828 [DOI] [PubMed] [Google Scholar]
- Sperlágh B, Baranyi M, Haskó G, Vizi ES. (2004) Potent effect of interleukin-1 beta to evoke ATP and adenosine release from rat hippocampal slices. J Neuroimmunol 151:33–39 [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. (2003) Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci 116:4615–4628 [DOI] [PubMed] [Google Scholar]
- Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D'Acquisto F, Malcangio M. (2010) Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem 114:1143–1157 [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25:3219–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Gurwell JA, Hauser KF. (1991) Morphine alters astrocyte growth in primary cultures of mouse glial cells: evidence for a direct effect of opiates on neural maturation. Brain Res Dev Brain Res 60:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Hauser KF. (1990) Opioid-dependent growth of glial cultures: suppression of astrocyte DNA synthesis by met-enkephalin. Life Sci 46:91–98 [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Hauser KF. (1991) Glial growth is regulated by agonists selective for multiple opioid receptor types in vitro. J Neurosci Res 29:538–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Hauser KF. (1993) Morphine suppresses DNA synthesis in cultured murine astrocytes from cortex, hippocampus and striatum. Neurosci Lett 157:1–3 [DOI] [PubMed] [Google Scholar]
- Stokes KB, Lee NM. (1976) Morphine effects of RNA synthesis in purified oligodendroglial nuclei. Proc West Pharmacol Soc 19:48–54 [PubMed] [Google Scholar]
- Suarez-Roca H, Abdullah L, Zuniga J, Madison S, Maixner W. (1992) Multiphasic effect of morphine on the release of substance P from rat trigeminal nucleus slices. Brain Res 579:187–194 [DOI] [PubMed] [Google Scholar]
- Suder P, Bodzon-Kulakowska A, Mak P, Bierczynska-Krzysik A, Daszykowski M, Walczak B, Lubec G, Kotlinska JH, Silberring J. (2009) The proteomic analysis of primary cortical astrocyte cell culture after morphine administration. J Proteome Res 8:4633–4640 [DOI] [PubMed] [Google Scholar]
- Svensson CI, Hua XY, Powell HC, Lai J, Porreca F, Yaksh TL. (2005a) Prostaglandin E2 release evoked by intrathecal dynorphin is dependent on spinal p38 mitogen activated protein kinase. Neuropeptides 39:485–494 [DOI] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, et al. (2003) Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem 86:1534–1544 [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schäfers M, Jones TL, Powell H, Sorkin LS. (2005b) Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett 379:209–213 [DOI] [PubMed] [Google Scholar]
- Svingos AL, Colago EE. (2002) Kappa-opioid and NMDA glutamate receptors are differentially targeted within rat medial prefrontal cortex. Brain Res 946:262–271 [DOI] [PubMed] [Google Scholar]
- Svingos AL, Moriwaki A, Wang JB, Uhl GR, Pickel VM. (1996) Ultrastructural immunocytochemical localization of mu-opioid receptors in rat nucleus accumbens: extrasynaptic plasmalemmal distribution and association with Leu5-enkephalin. J Neurosci 16:4162–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer S, De Leo J. (2011) Propentofylline: glial modulation, neuroprotection, and alleviation of chronic pain. Handb Exp Pharmacol (200):235–250 [DOI] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA. (2001) Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience 103:529–539 [DOI] [PubMed] [Google Scholar]
- Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ. (2002) Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci USA 99:10276–10281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YH, Tsai RY, Lin SL, Yeh CC, Wang JJ, Tao PL, Wong CS. (2009) Amitriptyline suppresses neuroinflammation-dependent interleukin-10-p38 mitogen-activated protein kinase-heme oxygenase-1 signaling pathway in chronic morphine-infused rats. Anesthesiology 110:1379–1389 [DOI] [PubMed] [Google Scholar]
- Tai YH, Wang YH, Tsai RY, Wang JJ, Tao PL, Liu TM, Wang YC, Wong CS. (2007) Amitriptyline preserves morphine's antinociceptive effect by regulating the glutamate transporter GLAST and GLT-1 trafficking and excitatory amino acids concentration in morphine-tolerant rats. Pain 129:343–354 [DOI] [PubMed] [Google Scholar]
- Tai YH, Wang YH, Wang JJ, Tao PL, Tung CS, Wong CS. (2006) Amitriptyline suppresses neuroinflammation and up-regulates glutamate transporters in morphine-tolerant rats. Pain 124:77–86 [DOI] [PubMed] [Google Scholar]
- Takagi K, Fukuda H, Watanabe M, Sato M. (1960) Studies on antitussives. III. (+)-Morphine and its derivatives. Yakugaku Zasshi 80:1506–1509 [Google Scholar]
- Takayama N, Ueda H. (2005a) Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. J Neurosci 25:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama N, Ueda H. (2005b) Morphine-induced overexpression of prepro-nociceptin/orphanin FQ in cultured astrocytes. Peptides 26:2513–2517 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. (2004) Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res 48:463–469 [DOI] [PubMed] [Google Scholar]
- Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, Cheng A, Siler DA, Markesbery WR, Arumugam TV, Mattson MP. (2008) Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol 213:114–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. (2005) The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA 102:5856–5861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, DeLeo JA. (2004) Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int 45:397–407 [DOI] [PubMed] [Google Scholar]
- Tawfik VL, LaCroix-Fralish ML, Nutile-McMenemy N, DeLeo JA. (2005) Transcriptional and translational regulation of glial activation by morphine in a rodent model of neuropathic pain. J Pharmacol Exp Ther 313:1239–1247 [DOI] [PubMed] [Google Scholar]
- Telleria-Diaz A, Schmidt M, Kreusch S, Neubert AK, Schache F, Vazquez E, Vanegas H, Schaible HG, Ebersberger A. (2010) Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: involvement of prostaglandins and endocannabinoids. Pain 148:26–35 [DOI] [PubMed] [Google Scholar]
- Terayama R, Guan Y, Dubner R, Ren K. (2000) Activity-induced plasticity in brain stem pain modulatory circuitry after inflammation. Neuroreport 11:1915–1919 [DOI] [PubMed] [Google Scholar]
- Terenius L. (1973) Stereospecific interaction between narcotic analgesics and a synaptic plasma membrane fraction of rat cerebral cortex. Acta Pharmacol Toxicol (Copenh) 32:317–320 [DOI] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB. (2009) CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain 13:263–272 [DOI] [PubMed] [Google Scholar]
- Thomas PT, House RV, Bhargava HN. (1995) Direct cellular immunomodulation produced by diacetylmorphine (heroin) or methadone. Gen Pharmacol 26:123–130 [DOI] [PubMed] [Google Scholar]
- Thorlin T, Eriksson PS, Persson PA, Aberg ND, Hansson E, Rönnbäck L. (1998a) Delta-opioid receptors on astroglial cells in primary culture: mobilization of intracellular free calcium via a pertussis sensitive G protein. Neuropharmacology 37:299–311 [DOI] [PubMed] [Google Scholar]
- Thorlin T, Eriksson PS, Rönnbäck L, Hansson E. (1998b) Receptor-activated Ca2+ increases in vibrodissociated cortical astrocytes: a nonenzymatic method for acute isolation of astrocytes. J Neurosci Res 54:390–401 [DOI] [PubMed] [Google Scholar]
- Thorlin T, Persson PA, Eriksson PS, Hansson E, Rönnbäck L. (1999) Delta-opioid receptor immunoreactivity on astrocytes is upregulated during mitosis. Glia 25:370–378 [DOI] [PubMed] [Google Scholar]
- Tilleux S, Hermans E. (2007) Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85:2059–2070 [DOI] [PubMed] [Google Scholar]
- Toda N, Kishioka S, Hatano Y, Toda H. (2009) Modulation of opioid actions by nitric oxide signaling. Anesthesiology 110:166–181 [DOI] [PubMed] [Google Scholar]
- Trang T, Beggs S, Salter MW. (2006) Purinoceptors in microglia and neuropathic pain. Pflugers Arch 452:645–652 [DOI] [PubMed] [Google Scholar]
- Trescot AM, Datta S, Lee M, Hansen H. (2008) Opioid pharmacology. Pain Physician 11:S133–153 [PubMed] [Google Scholar]
- Triantafilou M, Lepper PM, Briault CD, Ahmed MA, Dmochowski JM, Schumann C, Triantafilou K. (2008) Chemokine receptor 4 (CXCR4) is part of the lipopolysaccharide “sensing apparatus”. Eur J Immunol 38:192–203 [DOI] [PubMed] [Google Scholar]
- Tryoen-Toth P, Gavériaux-Ruff C, Labourdette G. (2000) Down-regulation of mu-opioid receptor expression in rat oligodendrocytes during their development in vitro. J Neurosci Res 60:10–20 [DOI] [PubMed] [Google Scholar]
- Tryoen-Toth P, Gavériaux-Ruff C, Maderspach K, Labourdette G. (1998) Regulation of kappa-opioid receptor mRNA level by cyclic AMP and growth factors in cultured rat glial cells. Brain Res Mol Brain Res 55:141–150 [DOI] [PubMed] [Google Scholar]
- Tsan MF, Gao B. (2004) Cytokine function of heat shock proteins. Am J Physiol Cell Physiol 286:C739–C744 [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. (2003) P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424:778–783 [DOI] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Ding Q, Keller JN, Hauser KF, Knapp PE, Bruce-Keller AJ. (2008) Cell-specific actions of HIV-Tat and morphine on opioid receptor expression in glia. J Neurosci Res 86:2100–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, Hauser KF, Bruce-Keller AJ. (2009) Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem 108:202–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Ueda M. (2009) Mechanisms underlying morphine analgesic tolerance and dependence. Front Biosci 14:5260–5272 [DOI] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. (2008) Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci 28:11263–11268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MO, Smith DJ. (1994) Nuclei within the rostral ventromedial medulla mediating morphine antinociception from the periaqueductal gray. Brain Res 652:9–16 [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. (2002) HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem 277:15107–15112 [DOI] [PubMed] [Google Scholar]
- van Dorp EL, Kest B, Kowalczyk WJ, Morariu AM, Waxman AR, Arout CA, Dahan A, Sarton EY. (2009) Morphine-6beta-glucuronide rapidly increases pain sensitivity independently of opioid receptor activity in mice and humans. Anesthesiology 110:1356–1363 [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Zhang ET, Malan TP, Jr, Ossipov MH, Lai J, et al. (2000) Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci 20:7074–7079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Laughlin T, Lashbrook JM, Nichols ML, Wilcox GL, Ossipov MH, Malan TP, Jr, Porreca F. (1996) Single intrathecal injections of dynorphin A or des-Tyr-dynorphins produce long-lasting allodynia in rats: blockade by MK-801 but not naloxone. Pain 68:275–281 [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. (2001) Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci 21:279–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Xie JY, Yie JX, Kowal J, Ossipov MH, King T, Porreca F. (2006) Descending facilitation from the rostral ventromedial medulla maintains visceral pain in rats with experimental pancreatitis. Gastroenterology 130:2155–2164 [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. (2004) Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci 20:1150–1160 [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, et al. (2003) Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23:8692–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmar P, Kullmann JS, Thilo B, Claussen MC, Rothhammer V, Jacobi H, Sellner J, Nessler S, Korn T, Hemmer B. (2010) Active immunization with amyloid-beta 1–42 impairs memory performance through TLR2/4-dependent activation of the innate immune system. J Immunol 185:6338–6347 [DOI] [PubMed] [Google Scholar]
- Wadachi R, Hargreaves KM. (2006) Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J Dent Res 85:49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29:3974–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Frankfurt M, Burns LH. (2008) High-affinity naloxone binding to filamin a prevents mu opioid receptor-Gs coupling underlying opioid tolerance and dependence. PLoS ONE 3:e1554. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang Z, Ma W, Chabot JG, Quirion R. (2009) Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB J 23:2576–2586 [DOI] [PubMed] [Google Scholar]
- Wang Z, Ma W, Chabot JG, Quirion R. (2010a) Calcitonin gene-related peptide as a regulator of neuronal CaMKII-CREB, microglial p38-NFκB and astroglial ERK-Stat1/3 cascades mediating the development of tolerance to morphine-induced analgesia. Pain 151:194–205 [DOI] [PubMed] [Google Scholar]
- Wang Z, Ma W, Chabot JG, Quirion R. (2010b) Morphological evidence for the involvement of microglial p38 activation in CGRP-associated development of morphine antinociceptive tolerance. Peptides 31:2179–2184 [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hansen MK, Nguyen KT, Lee JE, Maier SF. (1999) Dynamic regulation of the proinflammatory cytokine, interleukin-1beta: molecular biology for non-molecular biologists. Life Sci 65:449–481 [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. (2003) Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov 2:973–985 [DOI] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. (1997) Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain 71:225–235 [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. (2001) Glial activation: a driving force for pathological pain. Trends Neurosci 24:450–455 [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. (2003) Immune and glial involvement in physiological and pathological exaggerated pain states, in Proceedings of the 10th World Congress on Pain (Dostrovsky JO, Carr DB, Koltzenburg M. eds) pp 369–385, IASP Press, Seattle, WA [Google Scholar]
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF. (2005) Glia: novel counter-regulators of opioid analgesia. Trends Neurosci 28:661–669 [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. (2007a) Norman Cousins Lecture. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun 21:131–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Milligan ED, Maier SF. (2007b) “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev 56:148–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. (2009) The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30:581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman AR, Arout C, Caldwell M, Dahan A, Kest B. (2009) Acute and chronic fentanyl administration causes hyperalgesia independently of opioid receptor activity in mice. Neurosci Lett 462:68–72 [DOI] [PubMed] [Google Scholar]
- Weber M, Modemann S, Schipper P, Trauer H, Franke H, Illes P, Geiger KD, Hengstler JG, Kleemann WJ. (2006) Increased polysialic acid neural cell adhesion molecule expression in human hippocampus of heroin addicts. Neuroscience 138:1215–1223 [DOI] [PubMed] [Google Scholar]
- Wei F, Guo W, Zou S, Ren K, Dubner R. (2008) Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci 28:10482–10495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen ZH, Wu GJ, Chang YC, Wang JJ, Wong CS. (2005) Dexamethasone modulates the development of morphine tolerance and expression of glutamate transporters in rats. Neuroscience 133:807–817 [DOI] [PubMed] [Google Scholar]
- Wen ZH, Wu GJ, Hsu LC, Chen WF, Chen JY, Shui HA, Chou AK, Wong CS. (2008) N-Methyl-d-aspartate receptor antagonist MK-801 attenuates morphine tolerance and associated glial fibrillary acid protein up-regulation: a proteomic approach. Acta Anaesthesiol Scand 52:499–508 [DOI] [PubMed] [Google Scholar]
- White F, Wilson N. (2010) Opiate-induced hypernociception and chemokine receptors. Neuropharmacology 58:35–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Wilson NM. (2008) Chemokines as pain mediators and modulators. Curr Opin Anaesthesiol 21:580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EH, Weninger W, Hunter CA. (2010) Trafficking of immune cells in the central nervous system. J Clin Invest 120:1368–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y. (2006) Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 120:315–324 [DOI] [PubMed] [Google Scholar]
- Wolf G, Yirmiya R, Goshen I, Iverfeldt K, Holmlund L, Takeda K, Shavit Y. (2003) Impairment of interleukin-1 (IL-1) signaling reduces basal pain sensitivity in mice: genetic, pharmacological and developmental aspects. Pain 104:471–480 [DOI] [PubMed] [Google Scholar]
- Wu FX, Bian JJ, Miao XR, Huang SD, Xu XW, Gong DJ, Sun YM, Lu ZJ, Yu WF. (2010) Intrathecal siRNA against Toll-like receptor 4 reduces nociception in a rat model of neuropathic pain. Int J Med Sci 7:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Schwasinger ET, Terashvili M, Tseng LF. (2007) Dextro-morphine attenuates the morphine-produced conditioned place preference via the sigma(1) receptor activation in the rat. Eur J Pharmacol 562:221–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Sun HS, Cheng CW, Terashvili M, Tseng LF. (2006a) Dextro-naloxone or levo-naloxone reverses the attenuation of morphine antinociception induced by lipopolysaccharide in the mouse spinal cord via a non-opioid mechanism. Eur J Neurosci 24:2575–2580 [DOI] [PubMed] [Google Scholar]
- Wu HE, Sun HS, Cheng CW, Tseng LF. (2006b) p38 mitogen-activated protein kinase inhibitor SB203580 reverses the antianalgesia induced by dextro-morphine or morphine in the mouse spinal cord. Eur J Pharmacol 550:91–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Sun HS, Terashivili M, Schwasinger E, Sora I, Hall FS, Uhl GR, Tseng LF. (2006c) dextro- and levo-morphine attenuate opioid delta and kappa receptor agonist produced analgesia in mu-opioid receptor knockout mice. Eur J Pharmacol 531:103–107 [DOI] [PubMed] [Google Scholar]
- Wu HE, Thompson J, Sun HS, Terashvili M, Tseng LF. (2005) Antianalgesia: stereoselective action of dextro-morphine over levo-morphine on glia in the mouse spinal cord. J Pharmacol Exp Ther 314:1101–1108 [DOI] [PubMed] [Google Scholar]
- Wu N, Lu XQ, Yan HT, Su RB, Wang JF, Liu Y, Hu G, Li J. (2008) Aquaporin 4 deficiency modulates morphine pharmacological actions. Neurosci Lett 448:221–225 [DOI] [PubMed] [Google Scholar]
- Xie N, Li H, Wei D, LeSage G, Chen L, Wang S, Zhang Y, Chi L, Ferslew K, He L, et al. (2010) Glycogen synthase kinase-3 and p38 MAPK are required for opioid-induced microglia apoptosis. Neuropharmacology 59:444–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. (2007) Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by kappa opioid activation of p38 mitogen-activated protein kinase. J Neurosci 27:2570–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Wiesenfeld-Hallin Z, Hughes J, Horwell DC, Hökfelt T. (1992) CI988, a selective antagonist of cholecystokininB receptors, prevents morphine tolerance in the rat. Br J Pharmacol 105:591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Harty GJ, Onofrio BM. (1986) High dose of spinal morphine produce a nonopiate receptor-mediated hyperesthesia: clinical and theoretic implications. Anesthesiology 64:590–597 [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. (1978) Narcotic analgestics: CNS sites and mechanisms of action as revealed by intracerebral injection techniques. Pain 4:299–359 [DOI] [PubMed] [Google Scholar]
- Zagon IS, McLaughlin PJ. (1991) Identification of opioid peptides regulating proliferation of neurons and glia in the developing nervous system. Brain Res 542:318–323 [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. (2006) Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem 97:772–783 [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. (2007) Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci 27:12396–12406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Rogers TJ, Caterina M, Oppenheim JJ. (2004) Proinflammatory chemokines, such as C-C chemokine ligand 3, desensitize mu-opioid receptors on dorsal root ganglia neurons. J Immunol 173:594–599 [DOI] [PubMed] [Google Scholar]
- Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, Berman BM, Lao L. (2008) IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain 135:232–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Barres BA. (2010) Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol 20:588–594 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li H, Li Y, Sun X, Zhu M, Hanley G, Lesage G, Yin D. (2010) Essential role of toll-like receptor 2 in morphine-induced microglia activation in mice. Neurosci Lett 489:43–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. (2007) Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci 27:2357–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Chen ML, Zhang YQ, Zhao ZQ. (2010) Involvement of spinal microglial P2X7 receptor in generation of tolerance to morphine analgesia in rats. J Neurosci 30:8042–8047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Vincler MA, Parker R, Eisenach JC. (2006) Spinal cord dynorphin expression increases, but does not drive microglial prostaglandin production or mechanical hypersensitivity after incisional surgery in rats. Pain 125:43–52 [DOI] [PubMed] [Google Scholar]
- Zhuo M, Gebhart GF. (1992) Characterization of descending facilitation and inhibition of spinal nociceptive transmission from the nuclei reticularis gigantocellularis and gigantocellularis pars alpha in the rat. J Neurophysiol 67:1599–1614 [DOI] [PubMed] [Google Scholar]
- Zhuo M, Gebhart GF. (1997) Biphasic modulation of spinal nociceptive transmission from the medullary raphe nuclei in the rat. J Neurosci 97:746–758 [DOI] [PubMed] [Google Scholar]