

Abstract
Although agonists bind directly in the heptahelical domain (HD) of most class-I rhodopsin-like G protein coupled receptors (GPCRs), class-III agonists bind in the extracellular domain of their receptors. Indeed, the latter possess a large extracellular domain composed of a cysteine-rich domain and a Venus flytrap module. Both the low sequence homology and the structural organization of class-III GPCRs raised the question of whether or not the HD of these receptors functions the same way as rhodopsin-like GPCRs. Here, we show that the HD of metabotropic glutamate receptor 5 (mGlu5) displays the same agonist-independent constitutive activity as the wild-type receptor. Moreover, we show that the noncompetitive antagonist MPEP [2-methyl-6-(phenylethynyl)-pyridine hydrochloride] and the positive allosteric modulator DFB (3,3′-difluorobenzaldazine) act as inverse agonist and full agonist, respectively, on the mGlu5 HD in the absence of the extracellular domain. This finding illustrates that, like rhodopsin-like receptors, the HD of mGluRs can constitutively couple to G proteins and be negatively and positively regulated by ligands. These data show that the HD of mGluRs behave like any other class-I GPCRs in terms of G protein coupling and regulation by various types of ligands.
Keywords: G protein-coupled receptor, allostery, modulator, activation mechanism, inverse agonism
G protein-coupled receptors (GPCRs) represent >1% of the total mammalian genes. They have been successful during animal evolution in recognizing a wide range of stimuli from photon to large glycoproteins (1–3). These receptors transduce the extracellular signals in cellular responses via heterotrimeric G proteins. On the basis of sequence similarity, mammalian GPCRs have been classified into five major classes (3), but all share a common central domain composed of seven transmembrane helices, the heptahelical domain (HD). This domain is assumed to adopt various active and inactive conformations, the former being stabilized by agonists whereas the latter are stabilized by inverse agonists (4, 5). In most cases, these ligands directly interact in the HD, but agonists can act within an additional extracellular domain (1, 6). This is the case of class-III GPCRs (7–10).
Class-III GPCRs include receptors for the main neurotransmitters glutamate and γ-aminobutyric acid (GABA) as well as receptors for Ca2+, sweet taste compounds, and pheromones (10). Their agonist-binding domain is homologous to bacterial periplasmic binding proteins involved in the trafficking of ions, amino acids, and sugars in the periplasm of Gram-negative bacteria (7, 10, 11). This finding was confirmed by the determination of the crystal structure of the metabotropic glutamate receptor 1 (mGlu1) extracellular domain. This domain has a bilobate structure that adopts a closed conformation on agonist binding in the cleft that separates both lobes (12, 13) and is often called a “Venus flytrap” module (VFTM).
How agonist binding in the VFTM leads to the activation of the HD remains unknown. However, the determination of the crystal structure of the mGlu1 VFTM with and without bound glutamate together with the demonstration that these receptors are constitutive dimers (14, 15) lead to a model for activation of class-III GPCRs (10, 12, 16). Accordingly, agonist binding in at least one VFTM of these dimeric receptors leads to a large conformational change of the dimer of VFTMs, possibly forcing the two HDs to interact with each other differently. This result is expected to stabilize their active state. Such a peculiar structural organization of the receptor protein, associated with a very low sequence identity of their HD compared with that of other GPCRs, raised the question of whether or not the HDs of all GPCRs function the same way.
We previously reported that mGlu1a and mGlu5 metabotropic glutamate receptors (mGluRs) display constitutive activity (17, 18). A precise analysis of the constitutive activity of these receptors led us to propose that it resulted from a spontaneous activity of their HD rather than from a spontaneous closure of their VFTM (19). In agreement with this proposal, the noncompetitive antagonists MPEP [2-methyl-6-(phenylethynyl)pyridine hydrochloride] and BAY36-7620, which bind in the HD (20, 21), are the only antagonists that display inverse agonist activity.
In the present study, we examined whether the HD of such receptors could activate G proteins in the absence of the VFTM. We found that mGlu5 HD was able to spontaneously activate Gq-type G proteins, and that this constitutive activity could be inhibited by the known mGlu5 inverse agonist. Of interest, the positive modulator of mGlu5, although devoid of agonist activity on the wild-type receptor, acted as a full agonist on mGlu5 HD in the absence of the large extracellular domain. In summary, we provide insight on the mechanism of action of negative and positive allosteric modulators of class-III GPCRs. Moreover, our data show that the HD of class-III GPCRs displays constitutive activity and can be positively or negatively regulated by ligands, like any other class I GPCRs.
Materials and Methods
Materials. Glutamic acid was purchased from Sigma. l-Quisqualic acid and MPEP were purchased from Tocris Cookson (Bristol, U.K.). Glutamate-pyruvate transaminase was purchased from Roche (Basel, Switzerland). Culture medium, FCS, and other products used for cell culture were purchased from GIBCO/BRL/Life Technologies (Cergy Pontoise, France). [3H]Myo-inositol (23.4 Ci/mol) (1 Ci = 37 GBq) was purchased from Amersham Pharmacia (Saclay, France).
Contruction of mGlu5 Mutants. The construction of the N-terminal hemagglutinin (HA)-tagged rat mGlu5a pRKG5a-NHA has been described (22). The plasmid expressing the Δ5 mutant was obtained by inserting between the MluI and XbaI sites of pRK5-NHA, the mGlu5a cDNA coding for the HD and the C-terminal tail between the residues P568 and the C-terminal end obtained by PCR. The final plasmid encodes for a protein possessing the signal peptide of mGlu5 followed by a HA tag and then by the HD and the intracellular C terminus of mGlu5a. The same strategy was used to generate Δ5Δ, which corresponds to the mGlu5a HD segment between P568 and L864 (Fig. 1A).
Fig. 1.

Cell surface expression of mGlu5, Δ5, and Δ5Δ. (A) Schematic representation of mGlu5, Δ5, and Δ5Δ and location of the sites of truncation. The open box represents the HA tag, and the gray box corresponds to the signal peptide of mGlu5.(B) Surface expression of mGlu5, Δ5, and Δ5Δ in HEK 293 cells was detected by immunofluorescence on nonpermeabilized cells. (C) Quantification of cell surface expression of mGlu5, Δ5, and Δ5Δ by ELISA on intact cells. Cells were transfected with 0.6, 5, and 5 μg of plasmids expressing mGlu5, Δ5, and Δ5Δ, respectively.
Cell Culture and Transfection. HEK293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS and transfected by electroporation as described (23). Ten million cells were transfected with plasmid DNA encoding mGlu5 (0.6 μg), Δ5 (5 μg), Δ5Δ (5 μg) and completed to a total amount of 10 μg of plasmid DNA with pRK6. To avoid any influence of glutamate in the assay medium released by the cells, the high-affinity glutamate transporter EAAC1 was also cotransfected with the receptor.
Synthesis of 3,3′-Difluorobenzaldazine (DFB). DFB was synthesized according to Buu-Hoi and Saint-Ruf (24).
Immunofluorescence. Immunofluorescence assay was carried out as described (22). Briefly, 24 h after transfection, cells plated on coverslips were washed with PBS and incubated for 90 min at 37°C with an anti-HA mouse monoclonal antibody (clone 12CA5, Roche) at 1.33 μg/ml in PBS and 0.2% gelatin. The primary antibody was then detected with a Cy3 secondary antibody (1 μg/ml) (Jackson ImmunoResearch). Coverslips were mounted and observed by using an Axiophot2 microscope (Zeiss, LePecq, France).
Cell Surface Quantification by ELISA. Twenty-four hours after transfection, cells were washed twice with PBS, fixed with PBS and 4% paraformaldehyde, and incubated for 30 min with an anti-HA rat monoclonal antibody (0.5 μg/ml, clone 3F10, Roche) in PBS containing 5% FCS. Cells were then incubated with a secondary goat antibody conjugated to peroxydase (1 μg/ml, Jackson ImmunoResearch). Secondary antibody was detected and quantified by chemiluminescence using Supersignal West Femto (Pierce) and a Wallac Victor2 luminescence counter (Molecular Devices).
Inositol Phosphate (IP) Determination. IP accumulation in transfected cells was performed in 96-well microplates after cell labeling overnight with [3H]myo-inositol. The IP formation determination was performed after a 30-min incubation in the presence of 10 mM LiCl and in the presence or absence of the indicated compounds. For basal determination of IP production, glutamate-pyruvate transaminase (1 unit/ml) and 2 mM pyruvate were added to the reaction. The reaction was stopped with 0.1 M formic acid. Supernatants were recovered, and IP produced were purified in 96-well plates by ion exchange chromatography using DOWEX resin. Radioactivity was measured by using a Wallac 1450 MicroBeta microplate liquid scintillation counter (Molecular Devices). Results are expressed as the amount of IP produced over the radioactivity present in the 10% Triton X-100 and 0.1 M NaOH-solubilized membrane fraction, plus the produced IP. The dose–response curves were fitted by using the GraphPad (San Diego) PRISM program and the following equation:
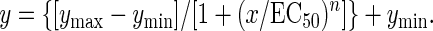 |
[1] |
Intracellular Calcium Measurements. Cells were seeded after transfection in polyornithine-coated, black-walled, clear bottom 96-well plates and cultured for 24 h. Cells were washed with freshly prepared buffer and loaded with 1 μM Ca2+-sensitive fluorescent dye Fluo-4 a.m. (Molecular Probes) for 1 h at 37°C. Cells were washed and incubated with 50 μl of buffer. A drug plate was prepared with the various concentrations of agonist to be tested, and 50 μl of 2×-drug solution was added in each well after 20 s of recording. Fluorescence signals (excitation 485 nm, emission 525 nm) were measured by using the fluorescence microplate reader Flexstation (Molecular Devices) at sampling intervals of 1.5 s for 60 s.
All data represented correspond to means ± SEM from representative experiments performed in triplicate.
Results
Expression of Wild-Type and Truncated mGlu5. To study the functional properties of mGlu5 HD, two truncated constructs were created. The first one, Δ5, was obtained by removing the VFTM and the cysteine-rich domain (CRD), and the second, Δ5Δ, was generated by truncating most of the C-terminal tail of Δ5 (Fig. 1 A). Both N-terminal HA-tagged truncated mutants were expressed at the cell surface as shown by immunostaining of nonpermeabilized cells (Fig. 1B). Under the same conditions, no fluorescence signal was detected at the surface of control cells transfected with the N-terminal HA-tagged GABAB1 subunit of the GABAB receptor, which cannot reach the cell surface alone (25, 26) or with an empty pRK6 plasmid (not shown).
Surface expression of the HA-tagged mGlu5, Δ5, and Δ5Δ was then quantified by using an ELISA assay. These experiments revealed that, for an equal quantity of plasmid DNA transfected in HEK293 cells, the two truncated mutants were 5 to 10 times less expressed at the cell surface than wild-type receptor (data not shown). To compare the functional properties of these constructs, conditions were set up to achieve a similar surface expression level as depicted in Fig. 1C.
mGlu5 Truncated Mutants Are Constitutively Active. The G protein-coupling activity of mGlu5, Δ5, and Δ5Δ was examined by measuring IP accumulation. We found a higher IP formation in cells expressing any of these three constructs compared with mock-transfected cells (Fig. 2A). This basal activity represented 291%, 211%, and 284% of the control IP production, for mGlu5, Δ5, and Δ5Δ, respectively. Among these three constructs, only the wild-type mGlu5 could be stimulated by glutamate (Fig. 2 A). Even when applied at high concentration (up to 10 mM, data not shown), glutamate had no effect on the truncated mutants (Fig. 2 A). These data show that the removal of the large extracellular domain suppresses the ability of glutamate to activate the receptor but does not prevent the truncated constructs to activate G proteins spontaneously.
Fig. 2.

Like mGlu5, Δ5 and Δ5Δ are constitutively active. (A) IP production measured in mGlu5, Δ5, and Δ5Δ or mock-transfected HEK 293 cells under basal conditions (open bars) or in the presence of 1 mM Glu (filled bars). Basal IP formation in mock-transfected cell is highlighted by a dotted line. Data correspond to the ratio between total IP produced by the cells and the total radioactivity remaining in the membranes plus the produced IPs. (B) MPEP decreased the basal IP production in HEK293 cells expressing mGlu5 (open circles) and Δ5Δ receptors (filled circles). Results are expressed as the percentage of the basal IP production measured in the absence of MPEP.
As mentioned above, the truncated receptors did not reach efficiently the cell surface and accumulated in the endoplasmic reticulum (ER). Therefore, it is possible that the observed high basal IP formation was due to the intracellular accumulation of mGlu5 rather than to a constitutive activity of the receptor at the cell surface. However, we found that a longer version of Δ5Δ with six additional residues at the C-terminal end did not reach the cell surface and accumulated in the ER as shown by immunofluorescence studies and ELISA assays performed on intact and permeabilized cells (data not shown). No high basal IP formation could be measured in cells expressing this construct (data not shown). The same was true for a mGlu5 chimeric construct bearing the ER retention signal of the GABAB1 receptor subunit (data not shown). This result confirmed that the basal activity measured was mostly generated by receptors at the cell surface.
Constitutive Activity of Δ5 and Δ5Δ Is Inhibited by a Negative Allosteric Modulator. The noncompetitive antagonist MPEP binds within the HD of mGlu5 (20, 27). This compound, often called a negative allosteric modulator, also inhibits the constitutive activity of mGlu5. As observed on the wild-type receptor (48.2 ± 15.1% inhibition, n = 8), MPEP also inhibited 42.8 ± 1.5% (n = 3) and 46.7 ± 15.3% (n = 7) of the basal IP formation measured in cells expressing Δ5 and Δ5Δ, respectively (Fig. 2B). These effects of MPEP were dose-dependent with IC50 values determined on Δ5 (7.6 ± 6.6 nM, n = 3, not shown) or Δ5Δ (10.4 ± 6.6 nM, n = 7) similar to that determined on mGlu5 (8.1 ± 4.3 nM, n = 8) (Fig. 2B). These data confirm that basal IP formation measured in cells expressing Δ5 or Δ5Δ originates from a constitutive phospholipase C activation by these constructs, and that MPEP does not require the extracellular domain of mGlu5 to interact with the receptor and acts as an inverse agonist. Furthermore, these data confirm the correct folding of the mGlu5 HD in the absence of both the large extracellular domain and the long C-terminal tail.
In Contrast to the Wild-Type Receptor, the Truncated Mutants Are Activated by a Positive Allosteric Modulator. Recently, positive allosteric modulators of various mGlu receptors have been identified (10, 28, 29). Such compounds display no or very low agonist activity but largely potentiate the action of agonists interacting in the VFTM. Analysis of the putative binding site of the mGlu1 and mGlu2 modulators suggested that they bind in the HD of these receptors (29, 30). Very recently, DFB has been reported as a positive allosteric modulator of mGlu5 (31). In agreement with this study, we found that DFB did not activate mGlu5 when care was taken to remove as much as possible glutamate from the medium (Fig. 3). However, DFB potentiated the action of glutamate, decreasing its EC50 value 2-fold [32.2 ± 9.2 (n = 9) to 17.4 ± 4.4 μM(n = 4) in the absence and presence of 100 μM DFB, respectively] (Fig. 3A). The same effect was observed with the agonist quisqualate [EC50 23.1 ± 2.3 (n = 5) and 13.1 ± 5.5 nM (n = 3) in the absence and presence of DFB, respectively, data not illustrated]. The maximal effect of glutamate was, however, not modified by DFB. As shown in Fig. 3B, DFB potentiated quisqualate-induced activation of mGlu5 with an EC50 value of 11.3 ± 1.4 μM. A similar effect of DFB was observed when the Ca2+ signal was measured in cells expressing mGlu5 (Fig. 3A).
Fig. 3.

DFB potentiates agonist-induced activity of wild-type mGlu5. (A) Effect of increasing concentrations of glutamate in the absence (CTR, open circles) or presence (DFB, filled circles) of 100 μM DFB on IP production in cells expressing mGlu5.(B) Effect of increasing concentration of DFB was measured on cells expressing mGlu5 in the absence (open circles) or presence of 10 nM quisqualate (filled circles). (Inset) The effect of 10 nM quisqualate on intracellular Ca2+ concentration was measured under control condition (C) or in the presence of 100 μM DFB (DFB). Vertical bar represents a change in the fluorescence signal of 1,000 units. Data are expressed as the percentage of the maximal effect measured with quisqualate plus DFB.
The effect of DFB was then examined on the truncated receptors Δ5 and Δ5Δ. In contrast to what was observed with the wild-type receptor, DFB activated these constructs directly (Fig. 4A). This effect was dose-dependent, with an EC50 value of 12.9 ± 6.3 μM (n = 6)(Fig. 4A), identical to that measured for the potentiating effect on the wild-type receptor (Fig. 3A). A similar effect was obtained with Δ5 (data not shown). DFB was also able to induce a Ca2+ signal in cells expressing Δ5 or Δ5Δ (Fig. 4B), with EC50 values of 20.2 ± 1.7 μM and 19.6 ± 1.2 μM, respectively. These data indicate that, whereas DFB is a clear positive allosteric modulator devoid of agonist activity on mGlu5, it acts as an agonist on the truncated receptors.
Fig. 4.

Direct activation of Δ5Δ by the positive allosteric regulator DFB. (A) Effect of increasing doses of DFB on Δ5Δ. DFB dose-dependently activates the truncated receptor Δ5Δ. The curve has been normalized such that the basal response is zero and the maximum is 100%. (Inset) IP formation (% above the basal) was induced by DFB only in cells expressing Δ5Δ and not in cells expressing mGlu5. (B) Direct stimulation of Δ5 and Δ5Δ by 100 μM DFB as revealed by intracellular Ca2+ measurement with Fluo-4. (C) Activity of mGlu5 (squares) and Δ5Δ (circles) as a function of their membrane expression. Cells were transfected with increasing amounts of cDNA coding for these receptors, and surface expression of mGlu5 and Δ5Δ was measured by ELISA on intact cells. Basal (open symbols) and glutamate-(1 mM) or DFB-(1 mM) (filled symbols) induced IP formation was measured in parallel.
DFB-Induced Activity of Δ5Δ Is Close to the Agonist-Induced Activity of mGlu5. To compare the activities of mGlu5 and Δ5Δ, cell surface expression and basal and agonist-induced IP production were measured in cells transfected with various amounts of plasmid DNA. Both basal and glutamate-induced IP productions were directly proportional to the number of receptors at the cell surface, the slope of the correlation lines being indicative of the specific (amount of IP produced per receptor) basal and glutamate-induced activity of the receptor (Fig. 4C). When the same analysis was performed with Δ5Δ in the absence and presence of DFB, the same correlation lines were obtained, showing that the specific constitutive activity of Δ5Δ is similar to that of mGlu5, and that DFB-induced activity of Δ5Δ is similar to the glutamate-induced activity of the wild-type receptor (Fig. 4C).
Inhibition of the DFB-Induced Response of Δ5Δ by MPEP. Because both DFB and MPEP were found to act on Δ5Δ, we examined whether MPEP could inhibit the action of DFB. As shown in Fig. 5, MPEP inhibited the effect of DFB on IP production or increase in intracellular Ca2+ in cells expressing Δ5Δ. However, even at high concentration, MPEP only partly inhibited the effect of DFB on Δ5Δ. This finding indicates a complex interaction between MPEP and DFB binding sites, in agreement with the partial inhibition of [3H]methoxyPEPy (an analog of MPEP) binding by DFB on the wild-type receptor (31).
Fig. 5.

MPEP inhibits partially DFB-induced activity on Δ5Δ. (A) Effect of 10, 30, and 100 nM MPEP on IP production induced by 300 μM DFB in cells expressing Δ5Δ. (B) Effect of 10 and 100 nM MPEP on intracellular Ca2+ release induced by 300 μM DFB on cells expressing Δ5Δ.
Discussion
The present study demonstrates that mGlu5 HD can activate PLC in the absence of both the large extracellular domain (the VFTM and the CRD) and the long C-terminal intracellular tail. Indeed, this domain of mGlu5 displays a constitutive activity that is similar to that of the wild-type receptor. Moreover, we show that the inverse agonist MPEP conserved its activity on this truncated mutant. Finally, our data demonstrate that the positive allosteric modulator DFB is acting as a full agonist on this domain. These data shed light on the possible mechanism of action of such positive allosteric modulators of class-III GPCRs.
Our data clearly indicate that both the inverse agonist MPEP and the positive allosteric regulator DFB do not need the large extracellular domain of mGlu5 to exert their action. Indeed, our data indicate that they directly interact within the HD of this receptor and therefore at a site distinct from the orthosteric ligand-binding site located in the VFTM (10, 12). This finding is in agreement with the proposed binding site of the noncompetitive antagonists of mGlu5 (MPEP) (20, 27), and mGlu1 (21, 30, 32). Of interest, when a detailed analysis of these antagonists' binding sites have been performed by site-directed mutagenesis and molecular modeling, the binding pocket was found to be equivalent to that of retinal in rhodopsin (20, 27, 30). Although positive modulators of mGlu receptors have been identified only recently (28, 29, 31), one study analyzed the binding site of the mGlu2 potentiator LY487379 (29). In that case again, the binding pocket was found to be located within the HD, with important residues located in transmembrane domains IV and V. In agreement with the latter study, our data also indicate that the positive modulator DFB binds in the HD of mGlu5. However, the effect of DFB was not fully inhibited by MPEP, in agreement with the reported partial inhibition of MPEP binding by DFB (31). This finding indicates a complex interaction between these two ligands, suggesting that they act at different sites. This result reinforces the need for a detailed analysis of the DFB site.
Group-I mGluRs, mGlu1a, mGlu5a, and mGlu5b display constitutive activity in heterologous expression systems (17, 18). The absence of inverse agonist activity of competitive antagonists known to prevent VFTM closure led us to propose that the constitutive activity originates from the HD able to reach an active state even if the VFTM stays open (19, 21). In agreement with this possibility, noncompetitive antagonists interacting in the HD have inverse agonist activity (20, 21). Our present data showing that the HD of mGlu5 displays the same constitutive activity as the wild-type receptor strongly support this idea. A kinetic model recently developed also confirmed this idea and, of interest, showed that the allosteric coupling between the VFTM and the HD is not strict (19). In other words, the change in conformation of one domain influences only the equilibrium constant between the two states of the other domain but does not force it to adopt a specific conformation. Such a finding is likely of importance because we recently reported that the constitutive activity of group I mGluRs is tightly regulated by the intracellular Homer proteins in neurons (33). As such, group I mGluRs can be activated either by extracellular glutamate or by the intracellular protein Homer1a. The low allosteric coupling between the HD and the VFTM allows the intracellular protein Homer1a not to increase dramatically glutamate affinity. Thus, the receptor can retain its ability to be further activated by extracellular glutamate in a physiological range of concentration (19).
Our data confirm that the positive allosteric modulator of mGlu5 DFB is devoid of agonist activity on the wild-type receptor (it is not able to activate the full-length receptor by itself) (31). We previously proposed that such a ligand is not able to directly stabilize the active state of the HD but instead facilitates the active closed state of the VFTM to activate it (19). This proposal is clearly not consistent with our present data showing that DFB activates the HD expressed alone to a similar extent as glutamate on the full-length receptor. Accordingly, it seems that the presence of the VFTM prevents DFB from activating the HD, a conclusion not consistent with a weak allosteric coupling between the VFTM and the HD. How can one reconcile these observations? Recent findings revealed that GPCRs likely exist in at least three states: an Rg (ground) state that corresponds to the totally inactive state stabilized by inverse agonists, an R state that is able to activate G proteins although with a low efficacy, and an R* state that corresponds to the active state of the receptor stabilized by full agonists (34, 35). By analogy, we propose that the HD of mGlu5 also exists in these three states: HDg, HD, and HD*. The equilibrium between HDg and HD may not be controlled by the VFTM. This equilibrium would be at the origin of the observed constitutive activity not inhibited by competitive antagonists, but by noncompetitive ones directly acting in the HD (Fig. 6). In contrast, the HD* state would require the VFTM to be in the active state, such that DFB would not be able to activate the receptor without agonists. In contrast, in the absence of VFTM, the HD would be able to reach more freely the fully active state HD* in the presence of DFB (Fig. 6). Because mGluRs are constitutive dimers, it is possible that the HD can oscillate between HDg and HD states when the dimer of VFTMs is in the resting orientation (R in Fig. 6). On the other hand, when the dimer of VFTMs is in the active orientation (A in Fig. 6), the HD can reach the HD* state, which is stabilized by DFB. Accordingly, it is possible that the formation of a specific form of the HD dimer is stabilized by DFB. However, further experiments are required to confirm this proposal.
Fig. 6.

Schematic representation of the possible action of inverse agonists and positive modulators of mGlu5. (Upper) The constitutive dimer of mGlu5 is shown to be composed of a VFTM (top), a CRD (middle), and a HD. The HD is proposed to oscillate between a slightly active state (HD) and a totally inactive ground state (HDg), the latter being stabilized by inverse agonist. This equilibrium can occur even though the dimer of VFTM stays in the resting state (R). The dimer of VFTMs is assumed to reach an active orientation (A) in the presence of agonist, leading to the stabilization of a fully active state of the dimer of HDs (HD*). The positive allosteric modulator, DFB, is proposed to bind with a higher affinity on HD*, stabilizing the fully active state of the receptor, leading to an increased affinity of the receptor for agonists (19). (Lower)Inthe absence of the large extracellular domain, the HD can reach more freely the fully active state HD*, allowing the positive modulators to act as full agonists.
The periplasmic binding proteins are known to bind their ligand in the periplasmic space and to deliver it to a transmembrane complex responsible for the transport of the molecule inside the bacteria (11, 36). Among the hypotheses for the activation mechanism of class-III GPCRs, it was proposed that the VFTM would bind the ligand and deliver it to another site within the HD leading to its activation (37). Our data show that the mGlu5 HD cannot be activated by glutamate up to a concentration of 10 mM. This result is not due to the inability of this domain to be activated because DFB can fully activate this domain. Such an observation favors therefore the second proposal, which originates from the crystal structure of the mGlu1 VFTM (10, 12, 16). Indeed, this domain forms dimers, and a large change in conformation of the dimer is observed on agonist binding (Fig. 6). This result is assumed to stabilize a specific conformation of the dimer of associated HDs, leading to their activation (Fig. 6). As mentioned above, class-III VFTMs are involved not only in ligand binding but also in the dimerization process of these receptors (14, 15, 38). Such a dimer formation is assumed to be crucial for the intramolecular transduction between the VFTM and the HD (10). Whether dimerization of GPCRs is required for G protein activation is still a matter of intense debate (39–42). At least our data show that the stabilization (and even disulfide cross-linking between the subunits) of class-III dimers by the VFTM is not required for the HD to activate G proteins. Accordingly, either the HD can dimerize by itself, or dimerization is not required for G protein activation.
There is actually a lot of pharmaceutical interest in identifying new allosteric modulators of mGluRs as potential new therapeutic agents. Our data show that the use of the HD of mGluRs may be a good tool to identify such new ligands because an agonist rather than a positive modulator has to be identified. The group of Conklin also highlighted the potential use of RASSL (receptor activated solely by synthetic ligands; ref. 43). HDs of mGluRs may constitute new possibilities to develop such tools that can be targeted in a specific neuronal compartment not attainable with other mutant GPCRs.
Acknowledgments
We thank Dr. P. Rondard for critical reading of the manuscript and all other members of J.P.P.'s laboratory for constant support. This work was supported by Centre National de la Recherche Scientifique, the Action Concertée Incitative “Molécules et Cibles Thérapeutiques” from the French government, the “Comité Parkinson” from the Fondation de France, the Fondation pour la Recherche Médicale and the Region Languedoc-Roussillon. C.G., J.K., and J.L. were supported by fellowships from the Fondation pour la Recherche Médicale (to C.G.), Centre National de la Recherche Scientifique (to J.K.), and Aventis Pharma (to J.L.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: DFB, 3,3′-difluorobenzaldazine; GPCR, G protein-coupled receptor; HA, hemagglutinin; HD, heptahelical domain; CRD, cysteine-rich domain; IP, inositol phosphate; MPEP, 2-methyl-6-(phenylethynyl)-pyridine hydrochloride; VFTM, Venus flytrap module; GABA, γ-aminobutyric acid; mGluR, metabotropic glutamate receptor.
References
- 1.Bockaert, J. & Pin, J.-P. (1999) EMBO J. 18, 1723-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bockaert, J., Claeysen, S., Becamel, C., Pinloche, S. & Dumuis, A. (2002) Int. Rev. Cytol. 212, 63-132. [DOI] [PubMed] [Google Scholar]
- 3.Fredriksson, R., Lagerstrom, M. C., Lundin, L. G. & Schioth, H. B. (2003) Mol. Pharmacol. 63, 1256-1272. [DOI] [PubMed] [Google Scholar]
- 4.Lefkowitz, R. J., Cotecchia, S., Samama, P. & Costa, T. (1993) Trends Pharmacol. Sci. 14, 303-307. [DOI] [PubMed] [Google Scholar]
- 5.Leff, P. (1995) Trends Pharmacol. Sci. 16, 89-97. [DOI] [PubMed] [Google Scholar]
- 6.Ji, T. H., Grossmann, M. & Ji, I. (1998) J. Biol. Chem. 273, 17299-17302. [DOI] [PubMed] [Google Scholar]
- 7.O'Hara, P. J., Sheppard, P. O., Thøgersen, H., Venezia, D., Haldeman, B. A., McGrane, V., Houamed, K. M., Thomsen, C., Gilbert, T. L. & Mulvihill, E. R. (1993) Neuron 11, 41-52. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi, K., Tsuchida, K., Tanabe, Y., Masu, M. & Nakanishi, S. (1993) J. Biol. Chem. 268, 19341-19345. [PubMed] [Google Scholar]
- 9.Okamoto, N., Hori, S., Akazawa, C., Hayashi, Y., Shigemoto, R., Mizuno, N. & Nakanishi, S. (1994) J. Biol. Chem. 269, 1231-1236. [PubMed] [Google Scholar]
- 10.Pin, J.-P., Galvez, T. & Prezeau, L. (2003) Pharmacol. Ther. 98, 325-354. [DOI] [PubMed] [Google Scholar]
- 11.Felder, C., Graul, R., Lee, A., Merkle, H. & Sadee, W. (1999) AAPS Pharmsci. 1, 10.1208/ps010202. [DOI] [PMC free article] [PubMed]
- 12.Kunishima, N., Shimada, Y., Tsuji, Y., Sato, T., Yamamoto, M., Kumasaka, T., Nakanishi, S., Jingami, H. & Morikawa, K. (2000) Nature 407, 971-977. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchiya, D., Kunishima, N., Kamiya, N., Jingami, H. & Morikawa, K. (2002) Proc. Natl. Acad. Sci. USA 99, 2660-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsuji, Y., Shimada, Y., Takeshita, T., Kajimura, N., Nomura, S., Sekiyama, N., Otomo, J., Usukura, J., Nakanishi, S. & Jingami, H. (2000) J. Biol. Chem. 275, 28144-28151. [DOI] [PubMed] [Google Scholar]
- 15.Romano, C., Yang, W.-L. & O'Malley, K. L. (1996) J. Biol. Chem. 271, 28612-28616. [DOI] [PubMed] [Google Scholar]
- 16.Jensen, A. A., Greenwood, J. R. & Bräuner-Osborne, H. (2002) Trends Pharmacol. Sci. 23, 491-493. [DOI] [PubMed] [Google Scholar]
- 17.Joly, C., Gomeza, J., Brabet, I., Curry, K., Bockaert, J. & Pin, J.-P. (1995) J. Neurosci. 15, 3970-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prézeau, L., Gomeza, J., Ahern, S., Mary, S., Galvez, T., Bockaert, J. & Pin, J.-P. (1996) Mol. Pharmacol. 49, 422-429. [PubMed] [Google Scholar]
- 19.Parmentier, M.-L., Prézeau, L., Bockaert, J. & Pin, J.-P. (2002) Trends Pharmacol. Sci. 23, 268-274. [DOI] [PubMed] [Google Scholar]
- 20.Pagano, A., Rüegg, D., Litschig, S., Stoehr, N., Stierlin, C., Heinrich, M., Floersheim, P., Prézeau, L., Carroll, F., Pin, J.-P., et al. (2000) J. Biol. Chem. 275, 33750-33758. [DOI] [PubMed] [Google Scholar]
- 21.Carroll, F. Y., Stolle, A., Beart, P. M., Voerste, A., Brabet, I., Mauler, F., Joly, C., Antonicek, H., Bockaert, J., Müller, T., et al. (2001) Mol. Pharmacol. 59, 965-973. [PMC free article] [PubMed] [Google Scholar]
- 22.Ango, F., Albani-Torregrossa, S., Joly, C., Robbe, D., Michel, J.-M., Pin, J.-P., Bockaert, J. & Fagni, L. (1999) Neuropharmacology 38, 793-803. [DOI] [PubMed] [Google Scholar]
- 23.Brabet, I., Parmentier, M.-L., De Colle, C., Bockaert, J., Acher, F. & Pin, J.-P. (1998) Neuropharmacology 37, 1043-1051. [DOI] [PubMed] [Google Scholar]
- 24.Buu-Hoi, N. P. & Saint-Ruf, G. (1967) Bull. Soc. Chim. Fr., 955-960. [PubMed]
- 25.Margeta-Mitrovic, M., Jan, Y. N. & Jan, L. Y. (2000) Neuron 27, 97-106. [DOI] [PubMed] [Google Scholar]
- 26.Pagano, A., Rovelli, G., Mosbacher, J., Lohmann, T., Duthey, B., Stauffer, D., Ristig, D., Schuler, V., Meigel, I., Lampert, C., et al. (2001) J. Neurosci. 21, 1189-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malherbe, P., Kratochwil, N., Zenner, M. T., Piussi, J., Diener, C., Kratzeisen, C., Fischer, C. & Porter, R. H. P. (2003) Mol. Pharmacol. 64, 823-832. [DOI] [PubMed] [Google Scholar]
- 28.Knoflach, F., Mutel, V., Jolidon, S., Kew, J. N., Malherbe, P., Vieira, E., Wichmann, J. & Kemp, J. A. (2001) Proc. Natl. Acad. Sci. USA 98, 13402-13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaffhauser, H. J., Rowe, B. A., Morales, S., Chavez-Noriega, L. E., Yin, R., Jachec, C., Rao, S. P., Bain, G., Pinkerton, A. B., Vernier, J.-M., et al. (2003) Mol. Pharmacol. 64, 798-810. [DOI] [PubMed] [Google Scholar]
- 30.Malherbe, P., Kratochwil, N., Knoflach, F., Zenner, M. T., Kew, J. N., Kratzeisen, C., Maerki, H. P., Adam, G. & Mutel, V. (2003) J. Biol. Chem. 278, 8340-8347. [DOI] [PubMed] [Google Scholar]
- 31.O'Brien, J. A., Lemaire, W., Chen, T.-B., Chang, R. S. L., Jacobson, M. A., Ha, S. N., Lindsley, C. W., Schaffhauser, H. J., Sur, C., Pettibone, D. J., et al. (2003) Mol. Pharmacol. 64, 731-740. [DOI] [PubMed] [Google Scholar]
- 32.Litschig, S., Gasparini, F., Rueegg, D., Munier, N., Flor, P. J., Vranesic, I.-T., Prézeau, L., Pin, J.-P., Thomsen, C. & Kuhn, R. (1999) Mol. Pharmacol. 55, 453-461. [PubMed] [Google Scholar]
- 33.Ango, F., Prézeau, L., Muller, T., Worley, P. F., Pin, J. P., Bockaert, J. & Fagni, L. (2001) Nature 411, 962-965. [DOI] [PubMed] [Google Scholar]
- 34.Joubert, L., Claeysen, S., Sebben, M., Bessis, A. S., Clark, R. D., Martin, R. S., Bockaert, J. & Dumuis, A. (2002) J. Biol. Chem. 277, 25502-25511. [DOI] [PubMed] [Google Scholar]
- 35.Okada, T., Ernst, O. P., Palczewski, K. & Hofmann, K. P. (2001) Trends Biochem. Sci. 26, 318-324. [DOI] [PubMed] [Google Scholar]
- 36.Quiocho, F. A. (1990) Phil. Trans. R. Soc. London B 326, 341-351. [DOI] [PubMed] [Google Scholar]
- 37.Pin, J.-P. & Bockaert, J. (1995) Curr. Opin. Neurobiol. 5, 342-349. [DOI] [PubMed] [Google Scholar]
- 38.Ray, K. & Hauschild, B. C. (2000) J. Biol. Chem. 275, 34245-34251. [DOI] [PubMed] [Google Scholar]
- 39.Bouvier, M. (2001) Nat. Rev. Neurosci. 2, 274-286. [DOI] [PubMed] [Google Scholar]
- 40.Baneres, J. L., Martin, A., Hullot, P., Girard, J. P., Rossi, J. C. & Parello, J. (2003) J. Mol. Biol. 329, 801-814. [DOI] [PubMed] [Google Scholar]
- 41.Liang, Y., Fotiadis, D., Filipek, S., Saperstein, D. A., Palczewski, K. & Engel, A. (2003) J. Biol. Chem. 278, 21655-21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamm, H. E. (2001) Proc. Natl. Acad. Sci. USA 98, 4819-4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scearce-Levie, K., Coward, P., Redfern, C. H. & Conklin, B. R. (2001) Trends Pharmacol. Sci. 22, 414-420. [DOI] [PubMed] [Google Scholar]