

Abstract
High-throughput sequencing technology enables population-level surveys of human genomic variation. Here, we examine the joint allele frequency distributions across continental human populations and present an approach for combining complementary aspects of whole-genome, low-coverage data and targeted high-coverage data. We apply this approach to data generated by the pilot phase of the Thousand Genomes Project, including whole-genome 2–4× coverage data for 179 samples from HapMap European, Asian, and African panels as well as high-coverage target sequencing of the exons of 800 genes from 697 individuals in seven populations. We use the site frequency spectra obtained from these data to infer demographic parameters for an Out-of-Africa model for populations of African, European, and Asian descent and to predict, by a jackknife-based approach, the amount of genetic diversity that will be discovered as sample sizes are increased. We predict that the number of discovered nonsynonymous coding variants will reach 100,000 in each population after ∼1,000 sequenced chromosomes per population, whereas ∼2,500 chromosomes will be needed for the same number of synonymous variants. Beyond this point, the number of segregating sites in the European and Asian panel populations is expected to overcome that of the African panel because of faster recent population growth. Overall, we find that the majority of human genomic variable sites are rare and exhibit little sharing among diverged populations. Our results emphasize that replication of disease association for specific rare genetic variants across diverged populations must overcome both reduced statistical power because of rarity and higher population divergence.
Keywords: demographic inference, genetic drift, population genetics, human evolution
The Thousand Genomes Project (1000G) is the most extensive study to date of human genomic diversity (1). The pilot phase of the project consisted of whole-genome, low-coverage sequencing of 179 samples from four HapMap populations at 2–4× coverage, an exon pilot experiment that targeted exons from over 800 genes in 697 samples across seven HapMap populations with ∼50× coverage, and a trio pilot focusing on two mother–father–child trios (1). In this article, we present an approach for combining the low-coverage and the exon pilot data, and use it to estimate the joint allele frequency spectrum for individuals of European origin in Utah (CEU), Han Chinese individuals in Beijing (CHB), Japanese individuals in Tokyo (JPT), and Yoruba individuals in Ibadan, Nigeria (YRI). Our motivation for this analysis is that two pilot projects provide complementary information: the low-coverage pilot captures most of the common variation in the populations sequenced across the accessible human genome at the cost of missing some of the rarer variants, whereas the target capture data provide a more complete picture of rare variants on an interesting subset of the data. In this article, we are interested in leveraging the strengths of the exon and low-coverage pilots to obtain accurate estimates of population genetic parameters. We will focus in particular on the P-population site frequency spectrum Φ, a P-dimensional histogram that records the joint distribution of diallelic SNPs as displayed in Fig. 1.
Fig. 1.

The two-population joint SFS from panels of Chinese individuals from Beijing (CHB) and Yoruba individuals from Ibadan, Nigeria (YRI) for variants occurring in less than 15 of 100 sequenced chromosomes in both panels. Of the 3,366 variants in the overlap of the two panels, all but 194 sites are private to a single population.
More specifically, the value Φ(f1, f2, …, fP) of bin (f1, f2, …, fP) is the number of SNPs that occurs in f1 chromosomes from population 1, f2 chromosomes from population 2, etc. Because the allele frequency in diploid population i ranges from 0 to 2ni, where ni is the number of individuals sequenced in this population, Φ is a (2n1 + 1) × (2n2 + 1) × … × (2nP + 1) array. Because the number of individuals who are successfully sequenced at any given site may vary, ni is, in practice, chosen to be somewhat smaller than the total number of individual sequenced, and each site with n > ni sequenced individuals contributes to bin f in proportion to the probability that one finds f derived alleles in a random selection of ni of the n samples.
The one-population site frequency spectrum (SFS) is a staple of population genetics and is commonly used to reveal broad patterns of selection (2, 3) and demography (3–5). The multiple-population SFS has received increased attention recently (6–10), because it provides additional information about between-population structure. Many standard population genetic statistics, such as FST and Tajima's D, are summaries of the multiple-population SFS.
In this article, we study SFSs derived from the 1000G pilot project data. We develop methods to precisely estimate SFSs from high-throughput sequencing data and use this information to estimate demographic parameters for a detailed Out-of-Africa demographic model by using ∂a∂i (6), a software package that uses diffusion approximation to calculate expected SFSs across multiple populations (6, 10). We use these parameters to predict the number of variants to be discovered as the number of sequenced samples in the 1000G is increased. We also present a jackknife-based approach to the prediction of the number of undiscovered variants and compare the predictions of the two approaches.
Theory
Linear Error Model for SFSs from Low-Coverage Data.
Data of the kind collected by the 1000G low-coverage pilot (1) (2–4× coverage across 179 individuals) provide a large volume of data from which precise demographic inference can be drawn. However, the low coverage leads to biases that must be addressed to ensure accuracy of the inference (11, 12). We use an empirical approach to tune an error model for low-coverage sequencing based on a direct comparison of the SFSs generated by whole genome and capture experiments on the part of the genome sequenced by both experiments.
The usefulness of this approach relies on two observations. First, demographic inference does not require the knowledge of the particular sites that are variable, but rather requires statistical averages over all sites. Although it is impossible to infer which variable sites were missed, the average number of missed sites can be estimated directly. Second, the most significant bias caused by low coverage in the 1000G data is an elevated false-negative rate for rare variant genotype calls (1). Because the majority of genetic variants are rare, it is possible to infer error rates for such variants based on high-quality sequence data from a relatively small subset of the genome.
Because the SFS does not keep track of linkage information, we use an error model that acts independently on each genomic site. We suppose that the underlying true SFS 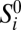 and the observed SFS Si for population p are related by a linear error model:
and the observed SFS Si for population p are related by a linear error model: 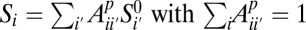 . In this model,
. In this model, 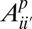 represents the proportion of sites with true frequency i′ that are assigned to frequency i. In the three-population case, which we will consider below, this model generalizes to
represents the proportion of sites with true frequency i′ that are assigned to frequency i. In the three-population case, which we will consider below, this model generalizes to
If we have Nc frequency bins per population, the number of parameters in this model is 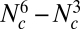 . We therefore need to introduce additional simplifying assumptions (justifications are provided below):
. We therefore need to introduce additional simplifying assumptions (justifications are provided below):
i) The errors occur independently in each population:
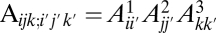 .
.ii) The probability
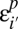 of missing a site decays exponentially with the number of variants present in the population:
of missing a site decays exponentially with the number of variants present in the population: 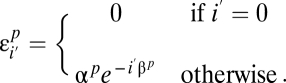
iii) If a site is found to be variable in one population, its frequency is estimated accurately:
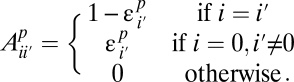
The resulting model has six parameters (amplitudes αp and error decay rates βp) and captures the bulk of the discrepancy between the high- and low-coverage data. It is motivated by the following observations:
i) The low-coverage SNP calls were made population by population. We assume, first, that the leading source of error is an insufficient number of variant reads to confidently call a variant and, second, that in a low-coverage experiment, the uncorrelated sampling fluctuations in read numbers play the largest role in the variation in read numbers.
ii) Variant calls in 1000G require multiple independent observations of a variant across a population to rule out read errors and call a variant genotype. This stringency strongly reduces the rate of false-positive calls, but it results in missing actual variants at a rate that depends on the expected number of nonreference reads observed at a given position across a population (1). The decay in the probability of detecting less than a fixed, small number c of reads for a variant present in i of N chromosome sequenced at depth d is dominated by
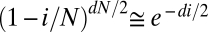 . We therefore fit a heuristic exponential error model αe−βi, where the effective read depth 2β accounts for fluctuations in read depth and read quality across the genome (Table 1).
. We therefore fit a heuristic exponential error model αe−βi, where the effective read depth 2β accounts for fluctuations in read depth and read quality across the genome (Table 1).iii) After enough variant reads are found in a 1000G population to justify a variant call, a single variant read is sufficient to make a genotype variant call, hence a reduced false-negative rate and low systematic bias in estimated frequencies when a variant has been identified.
Table 1.
Parameter values in the error model (Eq. 1)
| p | αp | βp | Mean mapped depth |
| CEU | 1.466 | 0.737 | 4.62× |
| CHB + JPT | 1.855 | 0.754 | 2.65× |
| YRI | 1.220 | 0.551 | 3.42× |
Note that the parameter values differ substantially between populations and that error rates decrease with increased coverage.
Estimating the Parameters of the Error Model.
Because errors are assumed to occur independently in each population, the error rates can be inferred directly from error rates in single-population SFSs. We compute the single-population SFSs at sites that are found to be variable in the exon pilot data and with at least 80 genotype calls in all three populations. The exon pilot and low-coverage pilot are then compared, and the optimal parameters αp and βp are obtained through a linear fit using the first three frequency bins of the compared spectra.
Note that this error model can be inverted to give a correction model for the SFS, which does not require the knowledge of the number of fixed variants (SI Appendix). However, the correction model may involve the subtraction of large numbers and has non-Poisson uncertainties. When inferring demographic parameters by maximum likelihood of a Poisson Random Field (6), we therefore incorporate the error model in our demographic model rather than attempt to correct the SFS.
Prediction of the Rate of Variant Discovery.
One practical use of inferring a demographic model is the ability to predict the number of variants that will be discovered in subsequent experiments. To study the impact of model choice on such predictions, we propose an alternate predictor of discovery rate based on sampling theory and inspired by an analogy with capture–recapture approaches to estimating animal population sizes (13–15) (let us consider rabbits for definiteness). In this analogy, a rabbit is akin to a SNP, a field trip is akin to an individual sequenced, and a rabbit capture is akin to the identification of a variant in a sequenced individual. In the absence of measurement errors, the probability of identifying a variant in a randomly chosen sequenced individual is proportional to the frequency of the variant in the population. This distribution of probabilities is akin to the variability in rabbit capture probability; a common SNP is akin to a trap-happy rabbit, and a rare SNP is akin to a trap-shy rabbit.
We propose a population genetics analog of the Burnham–Overton jackknife (16, 17) to estimate the total number V(N) of segregating sites in a sample of N chromosomes based on a subsample of n-sequenced chromosomes. This jackknife estimator uses the assumption that
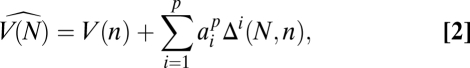 |
where 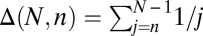 for a fixed jackknife order p. Explicit expressions for the
for a fixed jackknife order p. Explicit expressions for the 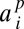 as well as performance benchmarking and additional discussion of this estimator are provided in SI Appendix.
as well as performance benchmarking and additional discussion of this estimator are provided in SI Appendix.
Results
After filtering away the exon pilot calls based on less than 15× coverage and individuals with substantial discrepancy with HapMap (SI Appendix), we compared the joint SFSs with the expected spectra obtained if each individual had been assigned to a population randomly in the independent sites model. That is, given an N × N spectrum φ(i, j), we have an expected spectrum of
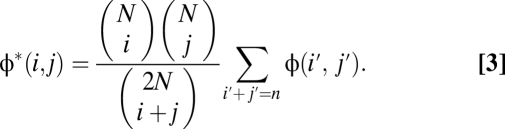 |
Figs. 2 and 3 indicate that, even for pairs of closely related populations, we find a substantial reduction in allele sharing for rare variants compared with a single randomly mixing population. In particular, Fig. 2, Right shows the Anscombe residuals between expectation and data. Blue strips along the axis correspond to a significant excess of variants private to one panel in the data, and they are accompanied by a reduction in shared variants (red). The residuals are larger (darker colors) for rare variants not only because of a larger number of sites but also because of reduced sharing. Indeed, we see in Fig. 3 that the amount of sharing, expressed as a proportion of the expectation in a panmictic population, is only a few percent between continental populations for variants present at 2% minor allele frequency (MAF) and about 60% for variants at 20% MAF. More closely related populations, such as CHB and JPT, still exhibit a 50% reduction in sharing at 2% MAF but barely any reduction for variants at 20% MAF. Interestingly, even closely related populations, such as CHB and CHD, exhibit a 20% reduction in sharing for 2% MAF. This finding is consistent with recent population structure, but although this analysis used only genotype calls with high-coverage data, such a reduction in sharing could also be partly explained by differences in the sequencing platform between the two populations.
Fig. 2.

Joint allele SFSs (all sites) for selected pairs of populations from the exome sequencing panel (Left) compared with expected spectra under site by site population label permutation (Center). Shown are sites occurring in, at most, 15 of 100 chromosomes. All population pairs, including two different panels of Chinese individuals sampled in Beijing, China and Denver, Colorado (CHB and CHD), as well as two groups of European origin (CEU and Tuscans from Italy, or TSI), show substantial residuals for rare variants (Right), consistent with reduced sharing. White bins contain less than one count.
Fig. 3.

The probability that two individuals carrying an allele of given minor frequency come from different populations, normalized by the expected frequency in a panmictic population, using the seven panels of the exome capture dataset. Sharing decreases dramatically as frequency approaches zero. The reduction in sharing at 50% frequency in some population pairs is caused by low overall numbers in that bin, and a single site (rs6662929) that exhibits inconsistent calls between different calling platforms and most likely has an incorrect homozygous reference call in some populations. Sites were binned by frequency: dots indicate the center of each bin, and solid lines are to guide the eye. Note that singletons are not shown, because there can be no sharing for such sites.
To increase the number of sites available for estimating joint SFS, we turned to the low-coverage pilot data. Direct comparison of low-coverage and exon capture genotype calls at sites called in the exon capture pilot shows a significant discrepancy for rare variants (SI Appendix, Figs. S1–S3) because of elevated rates of false-negative variant calls in low-coverage data. This finding results in biased estimates of the distribution of allele frequencies.
The bulk of the systematic discrepancy between high- and low-coverage SNP calls could be described using the simple false-negative model described above (Table 1 and SI Appendix, Figs. S1–S4). The most substantial discrepancy between this model and the data is in the CHB + JPT, possibly because this group was the metapopulation with the lowest coverage. In this case, the high-coverage singleton counts are 634% higher than the uncorrected low-coverage counts. After error correction, a discrepancy of 19% remains, with the corrected low-coverage site predicting more counts (SI Appendix, Fig. S3). Despite the high false-negative rate for singleton calls in the low-coverage data, its sheer volume provides an advantage in estimation precision over the much smaller exon pilot dataset. Similarly, the false-negative model for multiple-population SFSs (Eq. 1) was found to account for the bulk of the discrepancy between the multiple-population SFS derived from low- and high-coverage SNP calls (SI Appendix, Fig. S4).
We modeled the joint SFS for synonymous sites in African (YRI), Asian (CHB and JPT), and European (CEU) data sequenced in the low-coverage pilot using the 13-parameter demographic model used in ref. 6 (Fig. 4 and Table 2), taking into account the expected error model. The SFS was calculated using n = 40 samples per panel (80 chromosomes). We obtained maximum composite likelihood estimates for the 13 parameters using ∂a∂i, a diffusion-approximation-based package for estimating expected SFSs resulting from various demographic models (6) (Table 2). Our maximum likelihood parameters are broadly consistent with previously reported values using National Institute on Environmental Health Sciences (NIEHS) data (6). However, the resulting confidence intervals, determined by conventional bootstrap (likelihood profiles are provided in SI Appendix), are substantially narrower than those intervals resulting from NIEHS or high-coverage data alone. As an example, using a 25 y generation time, we find a time of split between African and Eurasian populations of TB = 51 thousand years ago (kya; 95% confidence interval = 45–69 kya). By contrast, the NIEHS data (6) resulted in a maximum likelihood estimate of TB = 140 kya (95% confidence interval = 40–270 kya). The inference based on the exon pilot alone yields TB = 98 kya (95% confidence interval = 43–210 kya). In general, the gain in precision was strongest for the parameters involved in more ancient events. Inference based on uncorrected low-coverage data yielded an unrealistic TB = 14 kya split.
Fig. 4.

An illustration of the inferred demographic model, with line width corresponding to population size and time flowing from left to right. The width of the red arrows is proportional to the migration intensity. Model details are provided in Table 2 and ref. 6.
Table 2.
Parameter estimates obtained using the NIEHS data (6), 1000G exon and low-coverage data, and 1000G exon pilot data only (this work)
| NIEHS | Low-coverage + exons | Exons | ||||
| Parameter | Estimate | 95% CI | Estimate | 95% CI | Estimate | 95% CI |
| NA | 7,300 | 4,400–10,100 | 7,310 | 6,984–7,739 | 7,310 | 3,647–9,208 |
| NAF | 12,300 | 11,500–13,900 | 14,474 | 13,419–16,184 | 15,388 | 14,240–17,542 |
| NB | 2,100 | 1,400–2,900 | 1,861 | 1,453–2,494 | 2,758 | 896–3,450 |
| NEUO | 1,000 | 500–1,900 | 1,032 | 677–1,290 | 1,620 | 991–2,759 |
| rEU (%) | 0.40 | 0.15–0.66 | 0.38 | 0.28–0.59 | 0.27 | 0.17–0.39 |
| NAS0 | 510 | 310–910 | 554 | 376–813 | 821 | 616–1,226 |
| rAS (%) | 0.55 | 0.23–0.88 | 0.48 | 0.30–0.75 | 0.31 | 0.18–0.42 |
| mAF−B (×10−5) | 25 | 15–34 | 15 | 12–19 | 20 | 5.5–29.6 |
| mAF−EU (×10−5) | 3.0 | 2.0–6.0 | 2.5 | 2.1–3.1 | 1.7 | 1.0–2.8 |
| mAF−AS (×10−5) | 1.9 | 0.3–10.4 | 0.78 | 0.4–1.2 | 0.58 | 0.23–1.24 |
| mEU−AS (×10−5) | 9.6 | 2.3–17.4 | 3.11 | 1.8–3.9 | 5.9 | 4.1–8.2 |
| TAF (kya) | 220 | 100–510 | 148 | 114–183 | 316 | 155–545 |
| TB (kya) | 140 | 40–270 | 51 | 45–69 | 98 | 43–210 |
| TEU − AS (kya) | 21.2 | 17.2–26.5 | 23 | 21–27 | 28 | 23–38 |
The 95% confidence intervals (CIs) in this work are obtained by bootstrap over coding regions. The equality of the maximum likelihood values for NA in the corrected low-coverage + exons data is a consequence of a normalization of the effective sequenced length (details in Methods). The resulting demographic model is illustrated in Fig. 4.
Beyond their fundamental interest as descriptors of human history, these parameters allow for a number of experimental predictions; given a demographic model, we can predict, for example, the number of synonymous variants to be discovered in samples of larger size that are currently in the process of being sequenced. We predicted the number of variants to be discovered in each of the three population considered (CEU, CHB + JPT, and YRI) as the sample size is increased using both the inferred demographic model and the jackknife estimator of the number of undiscovered variants presented in Methods (Fig. 5). Because the jackknife does not rely on assumptions about demography and selection, we also used it to predict the number of nonsynonymous sites to be discovered. The jackknife approach predicts that, as sample size is increased, the total number of segregating sites in CEU and CHB + JPT panels should overtake the number of segregating sites in the YRI population.
Fig. 5.

Observed and projected numbers of synonymous and nonsynonymous variants in CEU, CHB + JPT, and YRI as a function of the sample size (two times the number of individuals sequenced). Long and short dashes correspond to jackknife and model-based projections for synonymous sites, respectively. The dotted lines are jackknife projections for nonsynonymous sites. Discrepancies between projections are accounted for by the difference between the model prediction and observed number of singletons in the data.
Discussion
Our results illustrate that the vast majority of human variable sites are rare and that the majority of rare variants exhibit, at most, very little sharing among continental populations. We also find reduced sharing for rare variants compared with common variants among more closely related populations, such as CHB-JPT, CEU-TSI, and CHB-CHD. This lack of sharing can be explained by population divergence, and we expect that the fraction of newly discovered variable sites that are population-specific will keep increasing with sample size. This finding poses a formidable challenge for the reproduction of genome-wide association studies for rare functional variants across diverse populations, because the statistical difficulties caused by variant rarity within a population combine with increased between-population divergence.
We also show how sequencing a large number of individuals at low coverage is an efficient strategy not only for discovering the maximum number of variable sites but also for estimating demographic parameters, at least when error rates can be estimated. Different statistical methods have been proposed that include read depth information and models of sequencing errors to reduce biases in allele frequency estimation (11, 12). Because of the availability of high-coverage data for a subset of the genome in the populations studied here, we used direct comparison with high-coverage data to estimate and correct biases caused by low coverage. A significant advantage of the direct comparison approach is simplicity and computational efficiency; it can use existing curated genotype calls rather than require a full analysis of an error model at the individual read level. This advantage is particularly useful for data generated by 1000G, because multiple sequencing platforms and calling pipelines with different error modes have been used jointly. In general, the two approaches are not mutually exclusive, and when practical, a statistically corrected low-coverage SFS could be further corrected by comparison with targeted high-coverage data. Here, we used, as a reference, an exon capture dataset with >50× coverage and validation rates of 96.8% overall and 93.8% for singletons. We also restricted our analysis to a high-quality subset of the data (by selecting individuals with good coverage and HapMap concordance and selecting sites with sufficient coverage). The false-negative rate in the exon capture data was estimated to be below 5% for variants of at least 1% in frequency and 26% for variants below 1% in frequency. To avoid resulting biases in the frequency-dependent false-negative estimates for the low-coverage data, we restricted the comparison with sites where a high-coverage variant call had been made.
We found that the bulk of the discrepancy between high- and low-coverage data could be described by a simple model that uses only two parameters per population, and in which error rates decay exponentially with MAF, frequencies of detected variant sites are accurately determined, and errors occur independently in each population. The latter assumption is perhaps the most debatable: we expect at least some correlations in the coverage at a given site for different populations. An error model taking into account such correlations would, therefore, be desirable. However, given the limited data available to infer the parameters of the error model, the independence assumption is a reasonable tradeoff that allows for the capture of the bulk of the error patterns. Finally, the error rates likely differ between different genomic regions (such as coding vs. noncoding DNA), motivating our focus on exonic regions where high-coverage data were available. This finding emphasizes the importance of obtaining high-quality genotype data through sequencing or chip genotyping for representative noncoding regions.
The demographic model discussed in this paper was introduced in Gutenkunst et al. (6), where it was used to analyze the NIEHS intergenic data. Despite differences in putatively neutral sites (selected intergenics vs. synonymous), sequencing technology (Sanger vs. high throughput), and panel choice (CHB only vs. CHB + JPT), the inferred parameters are in broad agreement (Table 2). Inference based only on capture data provides overlapping 95% confidence intervals, with the single exception of Europe–Asia migration rate (1.8 − 3.9 × 10−5 vs. 4.1 − 8.2 × 10−5). The main difference between these three sets of parameter estimates is the width of the confidence intervals. The inference based on exon capture data provides reduced uncertainty compared with the NIEHS data, despite a comparable number of variable sites in the SFS; the additional number of samples per site results in more accurate frequency estimates that further constrain the demographic model. A much greater reduction in the confidence intervals is obtained by considering the low-coverage and exon capture data jointly (a 90% reduction of the confidence interval for the Out-of-Africa split time compared with a 27% reduction with the exon data only). Our estimate of the Out-of-Africa split time using the low-coverage data, 51 kya, is also in better agreement with both prior genetic and archaeological estimates of the modern human expansion out of Africa (18). It should be emphasized that, because we use a single Western African population as our African panel, the divergence described by our model might have occurred earlier than the actual Out-of-Africa event.
The narrow confidence intervals on some of the parameters should not obscure the fact that the parameter estimates are model-dependent. As a simple example, a model that does not allow for migration would require more recent split times to produce similar levels of population divergence. The demographic history of the four populations considered is much more eventful than what is accounted for by our model. Additional geographically intermediate populations from the Near East and Central Asia that were not included in our analysis might contribute significantly to the allele frequency distribution as ghost populations (19). Incorporating an appropriate number of source populations for estimates of migration has been a general limitation of two- and three-population models under isolation migration coalescent, approximate Bayesian computation, and diffusion-based approaches. This limitation might explain why our estimate of the divergence between East Asians and Europeans is more recent than estimates based on archaeological evidence (18), but is comparable with estimates of 23 kya (20) under an approximate Bayesian computation approach and 25 kya under an isolation migration approach with mtDNA X and Y sequence data (21).
Similarly, the current population sizes inferred from our model (15,500, 35,900, and 49,000 for YRI, CEU, and CHB, respectively) are still significantly lower than census sizes. Because our model accounts for some population size changes, these are expected to be in closer relationship to census sizes compared with the classical effective population size, but additional model refinement [such as structure within populations, generation overlap, and a recent increase in growth rate, which was observed in the work by Coventry et al. (22), in a sample of 10,422 European-Americans] will be needed to close the gap.
Predictions based on the demographic model and the jackknife approach differ as to the number of new variants to be discovered, particularly for CHB + JPT (Fig. 5). This difference is easily understood by considering the differences in the two approaches. The demographic model attempts to fit the complete SFS at the cost of model assumptions that might bias the results. By contrast, the jackknife approach focuses on the rare variants, and the model assumptions are weaker. The difference can be traced to the fact that the maximum likelihood demographic model predicts a number of singletons somewhat lower than the observed number (SI Appendix, Fig. S5). If this discrepancy is due to limitations in the model that fail to account for an excess of rare variants, we expect the jackknife estimator to be more accurate. By contrast, if the difference is because of inaccurate singleton frequency estimation (from sequencing errors leading to 6.2% of false-positive variants in the high-coverage data) or limitations of our correction model (SI Appendix, Figs. S1–S3), the demographic model is expected to provide more robust estimates.
Nonetheless, both methods predict at least 50,000 synonymous variants in the human genome when sequencing 1,000 individuals for the CEU and CHB populations, substantially more than would be predicted from population genetic models of constant size. The jackknife approach applied directly to the seven target capture populations shows similar patterns, with some variation within continents, in JPT samples showing less rare variants than the Chinese populations, and in TSI samples showing more rare variants than CEU (SI Appendix, Fig. S7). These results highlight the importance, for the planning of medical sequencing experiments, of accurate demographic models of human populations and the dramatic impact that recent human population growth has had on the structure of genetic variation. Specifically, our prediction that most genetic variants are rare and highly diverged suggests that genome-wide association studies aiming to correlate common disease susceptibility with rare variants may need extraordinarily large sample sizes and precise definitions of population samples to accurately compare frequencies in cases and controls. Eventually, a clear tradeoff will ensue between cataloging variants and genotyping vs. completely sequencing human genomes and comparing them among populations of cases and controls.
Methods
Numerics.
The unprecedented size of the 1,000 genomes data created challenges for the numerical solution of the diffusion equation. Namely, the number of grid points required to accurately estimate the three population SFS grows rapidly with the number of samples in each population. We optimized ∂a∂i and released version 1.5.0, in which the number of grid points necessary to achieve a given accuracy is reduced. As in ref. 6, we obtained SFSs with three different grid sizes (60, 70, and 80) and extrapolated to infinite grid size. Each likelihood evaluation took between 1 and 2 min on a 2.26-GHz processor. Optimization required hundreds to thousands of likelihood evaluations. Likelihoods were computed using the folded SFS to avoid biases caused by ancestral misidentification. Convergence of the maximum likelihood optimization process was ensured by restarting the search with modified initial conditions. The maximal likelihood parameters were chosen, but differences in parameter estimates from the different restarts were, on average, much smaller than the reported confidence intervals.
Conversion from Genetic to Physical Units.
The different parameters involved in the diffusion equation solved by ∂a∂i are normalized by the ancestral population size Na during the likelihood maximization. The optimal value of Na is calculated using the fact that the total number of segregating sites in a sample of n individuals is proportional to NaLμ, where μ is the mutation rate and L is the effective length sequenced. For this analysis, we used L = 5,007,837, the number of autosomal fourfold degenerate sites that passed quality control in all three populations, and μ = 2.36 × 10−8. For estimates based on exon data alone, we fixed the effective sequencing length to 68% of the target length by requesting equal values for NA in the corrected low-coverage and exon pilot estimates. The remaining 32% is composed of called sites that failed quality controls and sites for which no genotype call has been made. When performing bootstrap analysis, the total number of fourfold degenerate sites varied from bootstrap sample to bootstrap sample and was adjusted accordingly. Finally, to convert generation time to years, we used a generation time of 25 y. Estimated parameters are shown in Table 2.
Supplementary Material
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1019276108/-/DCSupplemental.
Contributor Information
David L. Altshuler, The 1000 Genomes Project.
Richard M. Durbin, The 1000 Genomes Project.
Gonçalo R. Abecasis, The 1000 Genomes Project
David R. Bentley, The 1000 Genomes Project
Aravinda Chakravarti, The 1000 Genomes Project.
Andrew G. Clark, The 1000 Genomes Project
Francis S. Collins, The 1000 Genomes Project
Francisco M. De La Vega, The 1000 Genomes Project
Peter Donnelly, The 1000 Genomes Project.
Michael Egholm, The 1000 Genomes Project.
Paul Flicek, The 1000 Genomes Project.
Stacey B. Gabriel, The 1000 Genomes Project
Richard A. Gibbs, The 1000 Genomes Project
Bartha M. Knoppers, The 1000 Genomes Project
Eric S. Lander, The 1000 Genomes Project
Hans Lehrach, The 1000 Genomes Project.
Elaine R. Mardis, The 1000 Genomes Project
Gil A. McVean, The 1000 Genomes Project
Debbie A. Nickerson, The 1000 Genomes Project
Leena Peltonen, The 1000 Genomes Project.
Alan J. Schafer, The 1000 Genomes Project
Stephen T. Sherry, The 1000 Genomes Project
Jun Wang, The 1000 Genomes Project.
Richard K. Wilson, The 1000 Genomes Project
Richard A. Gibbs, The 1000 Genomes Project.
David Deiros, The 1000 Genomes Project.
Mike Metzker, The 1000 Genomes Project.
Donna Muzny, The 1000 Genomes Project.
Jeff Reid, The 1000 Genomes Project.
David Wheeler, The 1000 Genomes Project.
Jun Wang, The 1000 Genomes Project.
Jingxiang Li, The 1000 Genomes Project.
Min Jian, The 1000 Genomes Project.
Guoqing Li, The 1000 Genomes Project.
Ruiqiang Li, The 1000 Genomes Project.
Huiqing Liang, The 1000 Genomes Project.
Geng Tian, The 1000 Genomes Project.
Bo Wang, The 1000 Genomes Project.
Jian Wang, The 1000 Genomes Project.
Wei Wang, The 1000 Genomes Project.
Huanming Yang, The 1000 Genomes Project.
Xiuqing Zhang, The 1000 Genomes Project.
Huisong Zheng, The 1000 Genomes Project.
Eric S. Lander, The 1000 Genomes Project.
David L. Altshuler, The 1000 Genomes Project
Lauren Ambrogio, The 1000 Genomes Project.
Toby Bloom, The 1000 Genomes Project.
Kristian Cibulskis, The 1000 Genomes Project.
Tim J. Fennell, The 1000 Genomes Project
Stacey B. Gabriel, The 1000 Genomes Project.
David B. Jaffe, The 1000 Genomes Project
Erica Shefler, The 1000 Genomes Project.
Carrie L. Sougnez, The 1000 Genomes Project
David R. Bentley, The 1000 Genomes Project.
Niall Gormley, The 1000 Genomes Project.
Sean Humphray, The 1000 Genomes Project.
Zoya Kingsbury, The 1000 Genomes Project.
Paula Koko-Gonzales, The 1000 Genomes Project.
Jennifer Stone, The 1000 Genomes Project.
Kevin J. McKernan, The 1000 Genomes Project.
Gina L. Costa, The 1000 Genomes Project
Jeffry K. Ichikawa, The 1000 Genomes Project
Clarence C. Lee, The 1000 Genomes Project
Ralf Sudbrak, The 1000 Genomes Project.
Hans Lehrach, The 1000 Genomes Project.
Tatiana A. Borodina, The 1000 Genomes Project
Andreas Dahl, The 1000 Genomes Project.
Alexey N. Davydov, The 1000 Genomes Project
Peter Marquardt, The 1000 Genomes Project.
Florian Mertes, The 1000 Genomes Project.
Wilfiried Nietfeld, The 1000 Genomes Project.
Philip Rosenstiel, The 1000 Genomes Project.
Stefan Schreiber, The 1000 Genomes Project.
Aleksey V. Soldatov, The 1000 Genomes Project
Bernd Timmermann, The 1000 Genomes Project.
Marius Tolzmann, The 1000 Genomes Project.
Michael Egholm, The 1000 Genomes Project.
Jason Affourtit, The 1000 Genomes Project.
Dana Ashworth, The 1000 Genomes Project.
Said Attiya, The 1000 Genomes Project.
Melissa Bachorski, The 1000 Genomes Project.
Eli Buglione, The 1000 Genomes Project.
Adam Burke, The 1000 Genomes Project.
Amanda Caprio, The 1000 Genomes Project.
Christopher Celone, The 1000 Genomes Project.
Shauna Clark, The 1000 Genomes Project.
David Conners, The 1000 Genomes Project.
Brian Desany, The 1000 Genomes Project.
Lisa Gu, The 1000 Genomes Project.
Lorri Guccione, The 1000 Genomes Project.
Kalvin Kao, The 1000 Genomes Project.
Andrew Kebbel, The 1000 Genomes Project.
Jennifer Knowlton, The 1000 Genomes Project.
Matthew Labrecque, The 1000 Genomes Project.
Louise McDade, The 1000 Genomes Project.
Craig Mealmaker, The 1000 Genomes Project.
Melissa Minderman, The 1000 Genomes Project.
Anne Nawrocki, The 1000 Genomes Project.
Faheem Niazi, The 1000 Genomes Project.
Kristen Pareja, The 1000 Genomes Project.
Ravi Ramenani, The 1000 Genomes Project.
David Riches, The 1000 Genomes Project.
Wanmin Song, The 1000 Genomes Project.
Cynthia Turcotte, The 1000 Genomes Project.
Shally Wang, The 1000 Genomes Project.
Elaine R. Mardis, The 1000 Genomes Project.
Richard K. Wilson, The 1000 Genomes Project.
David Dooling, The 1000 Genomes Project.
Lucinda Fulton, The 1000 Genomes Project.
Robert Fulton, The 1000 Genomes Project.
George Weinstock, The 1000 Genomes Project.
Richard M. Durbin, The 1000 Genomes Project.
John Burton, The 1000 Genomes Project.
David M. Carter, The 1000 Genomes Project
Carol Churcher, The 1000 Genomes Project.
Alison Coffey, The 1000 Genomes Project.
Anthony Cox, The 1000 Genomes Project.
Aarno Palotie, The 1000 Genomes Project.
Michael Quail, The 1000 Genomes Project.
Tom Skelly, The 1000 Genomes Project.
James Stalker, The 1000 Genomes Project.
Harold P. Swerdlow, The 1000 Genomes Project
Daniel Turner, The 1000 Genomes Project.
Anniek De Witte, The 1000 Genomes Project.
Shane Giles, The 1000 Genomes Project.
Richard A. Gibbs, The 1000 Genomes Project.
David Wheeler, The 1000 Genomes Project.
Matthew Bainbridge, The 1000 Genomes Project.
Danny Challis, The 1000 Genomes Project.
Aniko Sabo, The 1000 Genomes Project.
Fuli Yu, The 1000 Genomes Project.
Jin Yu, The 1000 Genomes Project.
Jun Wang, The 1000 Genomes Project.
Xiaodong Fang, The 1000 Genomes Project.
Xiaosen Guo, The 1000 Genomes Project.
Ruiqiang Li, The 1000 Genomes Project.
Yingrui Li, The 1000 Genomes Project.
Ruibang Luo, The 1000 Genomes Project.
Shuaishuai Tai, The 1000 Genomes Project.
Honglong Wu, The 1000 Genomes Project.
Hancheng Zheng, The 1000 Genomes Project.
Xiaole Zheng, The 1000 Genomes Project.
Yan Zhou, The 1000 Genomes Project.
Guoqing Li, The 1000 Genomes Project.
Jian Wang, The 1000 Genomes Project.
Huanming Yang, The 1000 Genomes Project.
Gabor T. Marth, The 1000 Genomes Project.
Erik P. Garrison, The 1000 Genomes Project
Weichun Huang, The 1000 Genomes Project.
Amit Indap, The 1000 Genomes Project.
Deniz Kural, The 1000 Genomes Project.
Wan-Ping Lee, The 1000 Genomes Project.
Wen Fung Leong, The 1000 Genomes Project.
Aaron R. Quinlan, The 1000 Genomes Project
Chip Stewart, The 1000 Genomes Project.
Michael P. Stromberg, The 1000 Genomes Project
Alistair N. Ward, The 1000 Genomes Project
Jiantao Wu, The 1000 Genomes Project.
Charles Lee, The 1000 Genomes Project.
Ryan E. Mills, The 1000 Genomes Project
Xinghua Shi, The 1000 Genomes Project.
Mark J. Daly, The 1000 Genomes Project.
Mark A. DePristo, The 1000 Genomes Project.
David L. Altshuler, The 1000 Genomes Project
Aaron D. Ball, The 1000 Genomes Project
Eric Banks, The 1000 Genomes Project.
Toby Bloom, The 1000 Genomes Project.
Brian L. Browning, The 1000 Genomes Project
Kristian Cibulskis, The 1000 Genomes Project.
Tim J. Fennell, The 1000 Genomes Project
Kiran V. Garimella, The 1000 Genomes Project
Sharon R. Grossman, The 1000 Genomes Project
Robert E. Handsaker, The 1000 Genomes Project
Matt Hanna, The 1000 Genomes Project.
Chris Hartl, The 1000 Genomes Project.
David B. Jaffe, The 1000 Genomes Project
Andrew M. Kernytsky, The 1000 Genomes Project
Joshua M. Korn, The 1000 Genomes Project
Heng Li, The 1000 Genomes Project.
Jared R. Maguire, The 1000 Genomes Project
Steven A. McCarroll, The 1000 Genomes Project
Aaron McKenna, The 1000 Genomes Project.
James C. Nemesh, The 1000 Genomes Project
Anthony A. Philippakis, The 1000 Genomes Project
Ryan E. Poplin, The 1000 Genomes Project
Alkes Price, The 1000 Genomes Project.
Manuel A. Rivas, The 1000 Genomes Project
Pardis C. Sabeti, The 1000 Genomes Project
Stephen F. Schaffner, The 1000 Genomes Project
Erica Shefler, The 1000 Genomes Project.
Ilya A. Shlyakhter, The 1000 Genomes Project
David N. Cooper, The 1000 Genomes Project.
Edward V. Ball, The 1000 Genomes Project
Matthew Mort, The 1000 Genomes Project.
Andrew D. Phillips, The 1000 Genomes Project
Peter D. Stenson, The 1000 Genomes Project
Jonathan Sebat, The 1000 Genomes Project.
Vladimir Makarov, The 1000 Genomes Project.
Kenny Ye, The 1000 Genomes Project.
Seungtai C. Yoon, The 1000 Genomes Project
Carlos D. Bustamante, The 1000 Genomes Project.
Andrew G. Clark, The 1000 Genomes Project.
Adam Boyko, The 1000 Genomes Project.
Jeremiah Degenhardt, The 1000 Genomes Project.
Simon Gravel, The 1000 Genomes Project.
Ryan N. Gutenkunst, The 1000 Genomes Project
Mark Kaganovich, The 1000 Genomes Project.
Alon Keinan, The 1000 Genomes Project.
Phil Lacroute, The 1000 Genomes Project.
Xin Ma, The 1000 Genomes Project.
Andy Reynolds, The 1000 Genomes Project.
Laura Clarke, The 1000 Genomes Project.
Paul Flicek, The 1000 Genomes Project.
Fiona Cunningham, The 1000 Genomes Project.
Javier Herrero, The 1000 Genomes Project.
Stephen Keenen, The 1000 Genomes Project.
Eugene Kulesha, The 1000 Genomes Project.
Rasko Leinonen, The 1000 Genomes Project.
William M. McLaren, The 1000 Genomes Project
Rajesh Radhakrishnan, The 1000 Genomes Project.
Richard E. Smith, The 1000 Genomes Project
Vadim Zalunin, The 1000 Genomes Project.
Xiangqun Zheng-Bradley, The 1000 Genomes Project.
Jan O. Korbel, The 1000 Genomes Project.
Adrian M. Stütz, The 1000 Genomes Project
Sean Humphray, The 1000 Genomes Project.
Markus Bauer, The 1000 Genomes Project.
R. Keira Cheetham, The 1000 Genomes Project.
Tony Cox, The 1000 Genomes Project.
Michael Eberle, The 1000 Genomes Project.
Terena James, The 1000 Genomes Project.
Scott Kahn, The 1000 Genomes Project.
Lisa Murray, The 1000 Genomes Project.
Aravinda Chakravarti, The 1000 Genomes Project.
Kai Ye, The 1000 Genomes Project.
Francisco M. De La Vega, The 1000 Genomes Project.
Yutao Fu, The 1000 Genomes Project.
Fiona C. L. Hyland, The 1000 Genomes Project
Jonathan M. Manning, The 1000 Genomes Project
Stephen F. McLaughlin, The 1000 Genomes Project
Heather E. Peckham, The 1000 Genomes Project
Onur Sakarya, The 1000 Genomes Project.
Yongming A. Sun, The 1000 Genomes Project
Eric F. Tsung, The 1000 Genomes Project
Mark A. Batzer, The 1000 Genomes Project.
Miriam K. Konkel, The 1000 Genomes Project
Jerilyn A. Walker, The 1000 Genomes Project
Ralf Sudbrak, The 1000 Genomes Project.
Marcus W. Albrecht, The 1000 Genomes Project
Vyacheslav S. Amstislavskiy, The 1000 Genomes Project
Ralf Herwig, The 1000 Genomes Project.
Dimitri V. Parkhomchuk, The 1000 Genomes Project
Stephen T. Sherry, The 1000 Genomes Project.
Richa Agarwala, The 1000 Genomes Project.
Hoda M. Khouri, The 1000 Genomes Project
Aleksandr O. Morgulis, The 1000 Genomes Project
Justin E. Paschall, The 1000 Genomes Project
Lon D. Phan, The 1000 Genomes Project
Kirill E. Rotmistrovsky, The 1000 Genomes Project
Robert D. Sanders, The 1000 Genomes Project
Martin F. Shumway, The 1000 Genomes Project
Chunlin Xiao, The 1000 Genomes Project.
Gil A. McVean, The 1000 Genomes Project.
Adam Auton, The 1000 Genomes Project.
Zamin Iqbal, The 1000 Genomes Project.
Gerton Lunter, The 1000 Genomes Project.
Jonathan L. Marchini, The 1000 Genomes Project
Loukas Moutsianas, The 1000 Genomes Project.
Simon Myers, The 1000 Genomes Project.
Afidalina Tumian, The 1000 Genomes Project.
Brian Desany, The 1000 Genomes Project.
James Knight, The 1000 Genomes Project.
Roger Winer, The 1000 Genomes Project.
David W. Craig, The 1000 Genomes Project.
Steve M. Beckstrom-Sternberg, The 1000 Genomes Project
Alexis Christoforides, The 1000 Genomes Project.
Ahmet A. Kurdoglu, The 1000 Genomes Project
John V. Pearson, The 1000 Genomes Project
Shripad A. Sinari, The 1000 Genomes Project
Waibhav D. Tembe, The 1000 Genomes Project
David Haussler, The 1000 Genomes Project.
Angie S. Hinrichs, The 1000 Genomes Project
Sol J. Katzman, The 1000 Genomes Project
Andrew Kern, The 1000 Genomes Project.
Robert M. Kuhn, The 1000 Genomes Project
Molly Przeworski, The 1000 Genomes Project.
Ryan D. Hernandez, The 1000 Genomes Project
Bryan Howie, The 1000 Genomes Project.
Joanna L. Kelley, The 1000 Genomes Project
S. Cord Melton, The 1000 Genomes Project.
Gonçalo R. Abecasis, The 1000 Genomes Project.
Yun Li, The 1000 Genomes Project.
Paul Anderson, The 1000 Genomes Project.
Tom Blackwell, The 1000 Genomes Project.
Wei Chen, The 1000 Genomes Project.
William O. Cookson, The 1000 Genomes Project
Jun Ding, The 1000 Genomes Project.
Hyun Min Kang, The 1000 Genomes Project.
Mark Lathrop, The 1000 Genomes Project.
Liming Liang, The 1000 Genomes Project.
Miriam F. Moffatt, The 1000 Genomes Project
Paul Scheet, The 1000 Genomes Project.
Carlo Sidore, The 1000 Genomes Project.
Matthew Snyder, The 1000 Genomes Project.
Xiaowei Zhan, The 1000 Genomes Project.
Sebastian Zöllner, The 1000 Genomes Project.
Philip Awadalla, The 1000 Genomes Project.
Ferran Casals, The 1000 Genomes Project.
Youssef Idaghdour, The 1000 Genomes Project.
John Keebler, The 1000 Genomes Project.
Eric A. Stone, The 1000 Genomes Project
Martine Zilversmit, The 1000 Genomes Project.
Lynn Jorde, The 1000 Genomes Project.
Jinchuan Xing, The 1000 Genomes Project.
Evan E. Eichler, The 1000 Genomes Project.
Gozde Aksay, The 1000 Genomes Project.
Can Alkan, The 1000 Genomes Project.
Iman Hajirasouliha, The 1000 Genomes Project.
Fereydoun Hormozdiari, The 1000 Genomes Project.
Jeffrey M. Kidd, The 1000 Genomes Project
S. Cenk Sahinalp, The 1000 Genomes Project.
Peter H. Sudmant, The 1000 Genomes Project
Elaine R. Mardis, The 1000 Genomes Project.
Ken Chen, The 1000 Genomes Project.
Asif Chinwalla, The 1000 Genomes Project.
Li Ding, The 1000 Genomes Project.
Daniel C. Koboldt, The 1000 Genomes Project
Mike D. McLellan, The 1000 Genomes Project
David Dooling, The 1000 Genomes Project.
George Weinstock, The 1000 Genomes Project.
John W. Wallis, The 1000 Genomes Project
Michael C. Wendl, The 1000 Genomes Project
Qunyuan Zhang, The 1000 Genomes Project.
Richard M. Durbin, The 1000 Genomes Project.
Cornelis A. Albers, The 1000 Genomes Project
Qasim Ayub, The 1000 Genomes Project.
Senduran Balasubramaniam, The 1000 Genomes Project.
Jeffrey C. Barrett, The 1000 Genomes Project
David M. Carter, The 1000 Genomes Project
Yuan Chen, The 1000 Genomes Project.
Donald F. Conrad, The 1000 Genomes Project
Petr Danecek, The 1000 Genomes Project.
Emmanouil T. Dermitzakis, The 1000 Genomes Project
Min Hu, The 1000 Genomes Project.
Ni Huang, The 1000 Genomes Project.
Matt E. Hurles, The 1000 Genomes Project
Hanjun Jin, The 1000 Genomes Project.
Luke Jostins, The 1000 Genomes Project.
Thomas M. Keane, The 1000 Genomes Project
Si Quang Le, The 1000 Genomes Project.
Sarah Lindsay, The 1000 Genomes Project.
Quan Long, The 1000 Genomes Project.
Daniel G. MacArthur, The 1000 Genomes Project
Stephen B. Montgomery, The 1000 Genomes Project
Leopold Parts, The 1000 Genomes Project.
James Stalker, The 1000 Genomes Project.
Chris Tyler-Smith, The 1000 Genomes Project.
Klaudia Walter, The 1000 Genomes Project.
Yujun Zhang, The 1000 Genomes Project.
Mark B. Gerstein, The 1000 Genomes Project.
Michael Snyder, The 1000 Genomes Project.
Alexej Abyzov, The 1000 Genomes Project.
Suganthi Balasubramanian, The 1000 Genomes Project.
Robert Bjornson, The 1000 Genomes Project.
Jiang Du, The 1000 Genomes Project.
Fabian Grubert, The 1000 Genomes Project.
Lukas Habegger, The 1000 Genomes Project.
Rajini Haraksingh, The 1000 Genomes Project.
Justin Jee, The 1000 Genomes Project.
Ekta Khurana, The 1000 Genomes Project.
Hugo Y. K. Lam, The 1000 Genomes Project
Jing Leng, The 1000 Genomes Project.
Xinmeng Jasmine Mu, The 1000 Genomes Project.
Alexander E. Urban, The 1000 Genomes Project
Zhengdong Zhang, The 1000 Genomes Project.
Yingrui Li, The 1000 Genomes Project.
Ruibang Luo, The 1000 Genomes Project.
Gabor T. Marth, The 1000 Genomes Project.
Erik P. Garrison, The 1000 Genomes Project
Deniz Kural, The 1000 Genomes Project.
Aaron R. Quinlan, The 1000 Genomes Project
Chip Stewart, The 1000 Genomes Project.
Michael P. Stromberg, The 1000 Genomes Project
Alistair N. Ward, The 1000 Genomes Project
Jiantao Wu, The 1000 Genomes Project.
Charles Lee, The 1000 Genomes Project.
Ryan E. Mills, The 1000 Genomes Project
Xinghua Shi, The 1000 Genomes Project.
Steven A. McCarroll, The 1000 Genomes Project.
Eric Banks, The 1000 Genomes Project.
Mark A. DePristo, The 1000 Genomes Project
Robert E. Handsaker, The 1000 Genomes Project
Chris Hartl, The 1000 Genomes Project.
Joshua M. Korn, The 1000 Genomes Project
Heng Li, The 1000 Genomes Project.
James C. Nemesh, The 1000 Genomes Project
Jonathan Sebat, The 1000 Genomes Project.
Vladimir Makarov, The 1000 Genomes Project.
Kenny Ye, The 1000 Genomes Project.
Seungtai C. Yoon, The 1000 Genomes Project
Jeremiah Degenhardt, The 1000 Genomes Project.
Mark Kaganovich, The 1000 Genomes Project.
Laura Clarke, The 1000 Genomes Project.
Richard E. Smith, The 1000 Genomes Project
Xiangqun Zheng-Bradley, The 1000 Genomes Project.
Jan O. Korbel, The 1000 Genomes Project
Sean Humphray, The 1000 Genomes Project.
R. Keira Cheetham, The 1000 Genomes Project.
Michael Eberle, The 1000 Genomes Project.
Scott Kahn, The 1000 Genomes Project.
Lisa Murray, The 1000 Genomes Project.
Kai Ye, The 1000 Genomes Project.
Francisco M. De La Vega, The 1000 Genomes Project.
Yutao Fu, The 1000 Genomes Project.
Heather E. Peckham, The 1000 Genomes Project
Yongming A. Sun, The 1000 Genomes Project
Mark A. Batzer, The 1000 Genomes Project.
Miriam K. Konkel, The 1000 Genomes Project
Jerilyn A. Walker, The 1000 Genomes Project
Chunlin Xiao, The 1000 Genomes Project.
Zamin Iqbal, The 1000 Genomes Project.
Brian Desany, The 1000 Genomes Project.
Tom Blackwell, The 1000 Genomes Project.
Matthew Snyder, The 1000 Genomes Project.
Jinchuan Xing, The 1000 Genomes Project.
Evan E. Eichler, The 1000 Genomes Project.
Gozde Aksay, The 1000 Genomes Project.
Can Alkan, The 1000 Genomes Project.
Iman Hajirasouliha, The 1000 Genomes Project.
Fereydoun Hormozdiari, The 1000 Genomes Project.
Jeffrey M. Kidd, The 1000 Genomes Project
Ken Chen, The 1000 Genomes Project.
Asif Chinwalla, The 1000 Genomes Project.
Li Ding, The 1000 Genomes Project.
Mike D. McLellan, The 1000 Genomes Project
John W. Wallis, The 1000 Genomes Project
Matt E. Hurles, The 1000 Genomes Project.
Donald F. Conrad, The 1000 Genomes Project
Klaudia Walter, The 1000 Genomes Project.
Yujun Zhang, The 1000 Genomes Project.
Mark B. Gerstein, The 1000 Genomes Project.
Michael Snyder, The 1000 Genomes Project.
Alexej Abyzov, The 1000 Genomes Project.
Jiang Du, The 1000 Genomes Project.
Fabian Grubert, The 1000 Genomes Project.
Rajini Haraksingh, The 1000 Genomes Project.
Justin Jee, The 1000 Genomes Project.
Ekta Khurana, The 1000 Genomes Project.
Hugo Y. K. Lam, The 1000 Genomes Project
Jing Leng, The 1000 Genomes Project.
Xinmeng Jasmine Mu, The 1000 Genomes Project.
Alexander E. Urban, The 1000 Genomes Project
Zhengdong Zhang, The 1000 Genomes Project.
Richard A. Gibbs, The 1000 Genomes Project.
Matthew Bainbridge, The 1000 Genomes Project.
Danny Challis, The 1000 Genomes Project.
Cristian Coafra, The 1000 Genomes Project.
Huyen Dinh, The 1000 Genomes Project.
Christie Kovar, The 1000 Genomes Project.
Sandy Lee, The 1000 Genomes Project.
Donna Muzny, The 1000 Genomes Project.
Lynne Nazareth, The 1000 Genomes Project.
Jeff Reid, The 1000 Genomes Project.
Aniko Sabo, The 1000 Genomes Project.
Fuli Yu, The 1000 Genomes Project.
Jin Yu, The 1000 Genomes Project.
Gabor T. Marth, The 1000 Genomes Project.
Erik P. Garrison, The 1000 Genomes Project
Amit Indap, The 1000 Genomes Project.
Wen Fung Leong, The 1000 Genomes Project.
Aaron R. Quinlan, The 1000 Genomes Project
Chip Stewart, The 1000 Genomes Project.
Alistair N. Ward, The 1000 Genomes Project
Jiantao Wu, The 1000 Genomes Project.
Kristian Cibulskis, The 1000 Genomes Project.
Tim J. Fennell, The 1000 Genomes Project
Stacey B. Gabriel, The 1000 Genomes Project
Kiran V. Garimella, The 1000 Genomes Project
Chris Hartl, The 1000 Genomes Project.
Erica Shefler, The 1000 Genomes Project.
Carrie L. Sougnez, The 1000 Genomes Project
Jane Wilkinson, The 1000 Genomes Project.
Andrew G. Clark, The 1000 Genomes Project.
Simon Gravel, The 1000 Genomes Project.
Fabian Grubert, The 1000 Genomes Project.
Laura Clarke, The 1000 Genomes Project.
Paul Flicek, The 1000 Genomes Project.
Richard E. Smith, The 1000 Genomes Project
Xiangqun Zheng-Bradley, The 1000 Genomes Project.
Stephen T. Sherry, The 1000 Genomes Project.
Hoda M. Khouri, The 1000 Genomes Project
Justin E. Paschall, The 1000 Genomes Project
Martin F. Shumway, The 1000 Genomes Project
Chunlin Xiao, The 1000 Genomes Project.
Gil A. McVean, The 1000 Genomes Project
Sol J. Katzman, The 1000 Genomes Project
Gonçalo R. Abecasis, The 1000 Genomes Project.
Tom Blackwell, The 1000 Genomes Project.
Elaine R. Mardis, The 1000 Genomes Project.
David Dooling, The 1000 Genomes Project.
Lucinda Fulton, The 1000 Genomes Project.
Robert Fulton, The 1000 Genomes Project.
Daniel C. Koboldt, The 1000 Genomes Project
Richard M. Durbin, The 1000 Genomes Project.
Senduran Balasubramaniam, The 1000 Genomes Project.
Allison Coffey, The 1000 Genomes Project.
Thomas M. Keane, The 1000 Genomes Project
Daniel G. MacArthur, The 1000 Genomes Project
Aarno Palotie, The 1000 Genomes Project.
Carol Scott, The 1000 Genomes Project.
James Stalker, The 1000 Genomes Project.
Chris Tyler-Smith, The 1000 Genomes Project.
Mark B. Gerstein, The 1000 Genomes Project.
Suganthi Balasubramanian, The 1000 Genomes Project.
Aravinda Chakravarti, The 1000 Genomes Project.
Bartha M. Knoppers, The 1000 Genomes Project.
Gonçalo R. Abecasis, The 1000 Genomes Project
Carlos D. Bustamante, The 1000 Genomes Project
Neda Gharani, The 1000 Genomes Project.
Richard A. Gibbs, The 1000 Genomes Project
Lynn Jorde, The 1000 Genomes Project.
Jane S. Kaye, The 1000 Genomes Project
Alastair Kent, The 1000 Genomes Project.
Taosha Li, The 1000 Genomes Project.
Amy L. McGuire, The 1000 Genomes Project
Gil A. McVean, The 1000 Genomes Project
Pilar N. Ossorio, The 1000 Genomes Project
Charles N. Rotimi, The 1000 Genomes Project
Yeyang Su, The 1000 Genomes Project.
Lorraine H. Toji, The 1000 Genomes Project
Chris TylerSmith, The 1000 Genomes Project.
Lisa D. Brooks, The 1000 Genomes Project
Adam L. Felsenfeld, The 1000 Genomes Project
Jean E. McEwen, The 1000 Genomes Project
Assya Abdallah, The 1000 Genomes Project.
Christopher R. Juenger, The 1000 Genomes Project
Nicholas C. Clemm, The 1000 Genomes Project
Francis S. Collins, The 1000 Genomes Project
Audrey Duncanson, The 1000 Genomes Project.
Eric D. Green, The 1000 Genomes Project
Mark S. Guyer, The 1000 Genomes Project
Jane L. Peterson, The 1000 Genomes Project
Alan J. Schafer, The 1000 Genomes Project
Gonçalo R. Abecasis, The 1000 Genomes Project
David L. Altshuler, The 1000 Genomes Project
Adam Auton, The 1000 Genomes Project.
Lisa D. Brooks, The 1000 Genomes Project
Richard M. Durbin, The 1000 Genomes Project
Richard A. Gibbs, The 1000 Genomes Project
Matt E. Hurles, The 1000 Genomes Project
Gil A. McVean, The 1000 Genomes Project
References
- 1.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyko AR, et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williamson SH, et al. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc Natl Acad Sci USA. 2005;102:7882–7887. doi: 10.1073/pnas.0502300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams AM, Hudson RR. Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single-nucleotide polymorphisms. Genetics. 2004;168:1699–1712. doi: 10.1534/genetics.104.030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marth GT, Czabarka E, Murvai J, Sherry ST. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–372. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen R, et al. Darwinian and demographic forces affecting human protein coding genes. Genome Res. 2009;19:838–849. doi: 10.1101/gr.088336.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawyer SA, Hartl DL. Population genetics of polymorphism and divergence. Genetics. 1992;132:1161–1176. doi: 10.1093/genetics/132.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bustamante CD, Wakeley J, Sawyer S, Hartl DL. Directional selection and the site-frequency spectrum. Genetics. 2001;159:1779–1788. doi: 10.1093/genetics/159.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi X, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch M. Estimation of allele frequencies from high-coverage genome-sequencing projects. Genetics. 2009;182:295–301. doi: 10.1534/genetics.109.100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 13.Ionita-Laza I, Lange C, M Laird N. Estimating the number of unseen variants in the human genome. Proc Natl Acad Sci USA. 2009;106:5008–5013. doi: 10.1073/pnas.0807815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ionita-Laza I, Laird NM. On the optimal design of genetic variant discovery studies. Stat Appl Genet Mol Biol. 2010;9:33. doi: 10.2202/1544-6115.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunge J, Fitzpatrick M. Estimating the number of species. A review. J Am Stat Assoc. 1993;88:364–373. [Google Scholar]
- 16.Burnham K, Overton W. Estimation of the size of a closed population when capture probabilities vary among animals. Biometrika. 1978;65:625–633. [Google Scholar]
- 17.Burnham K, Overton W. Robust estimation of population size when capture probabilities vary among animals. Ecology. 1979;60:927–936. [Google Scholar]
- 18.Klein RG, Hublin JJ. The Human Career. Human Biological and Cultural Origins. Chicago: University of Chicago Press; 1999. [Google Scholar]
- 19.Beerli P. Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Mol Ecol. 2004;13:827–836. doi: 10.1111/j.1365-294x.2004.02101.x. [DOI] [PubMed] [Google Scholar]
- 20.Laval G, Patin E, Barreiro LB, Quintana-Murci L. Formulating a historical and demographic model of recent human evolution based on resequencing data from noncoding regions. PLoS One. 2010;5:e10284. doi: 10.1371/journal.pone.0010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrigan D, et al. Inferring human population sizes, divergence times and rates of gene flow from mitochondrial, X and Y chromosome resequencing data. Genetics. 2007;177:2195–2207. doi: 10.1534/genetics.107.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coventry A, et al. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nat Commun. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.