

Abstract
Induction of NF-κB-dependent transcription requires phosphorylation and subsequent degradation of I-κB, an inhibitor of NF-κB, followed by nuclear translocation and DNA binding of NF-κB. Tumor necrosis factor receptor-associated factor 2 (TRAF2) plays a role in NF-κB activation in response to cytokines such as tumor necrosis factor α (TNFα). In this study, we purified and characterized a novel kinase (T2K, also known as TBK1 or NAK), which associates with TRAF2 and exhibits kinase activity towards I-κBα in vitro. The physiological function of T2K was investigated using T2K-deficient mice. Heterozygotes appear normal, but t2k–/– animals die at ∼E14.5 of massive liver degeneration and apoptosis. Never theless, hematopoietic progenitors from T2K-deficient fetal liver support normal lymphocyte development. Furthermore, t2k–/– embryonic fibroblasts and thymocytes do not display increased sensitivity to TNFα-induced apoptosis. In response to either TNFα or IL-1 induction, t2k–/– embryonic fibroblasts exhibit normal degradation of I-κB and κB-binding activity. However, NF-κB-directed transcription is dramatically reduced. These results demonstrate that, like I-κB kinase β and the RelA subunit of NF-κB, T2K is critical in protecting embryonic liver from apoptosis. However, T2K has a unique role in the activation of NF-κB-directed transcription, apparently independent of I-κB degradation and NF-κB DNA binding.
Keywords: apoptosis/I-κB kinase/NF-κB/T2K/TNF signaling
Introduction
Rel-family NF-κB transcription factors, including the prototypical RelA–p50 heterodimer, are critical regulators of genes involved in immune and inflammatory responses, as well as protecting against apoptosis (Ghosh et al., 1998; Foo and Nolan, 1999). NF-κB is normally sequestered in the cytoplasm of resting cells, where it is bound to I-κB inhibitory proteins (I-κBα, I-κBβ and I-κBε). This interaction masks the nuclear localization signal of NF-κB, preventing its nuclear translocation (Ghosh et al., 1998). Various extracellular stimuli, including pro-inflammatory cytokines such as tumor necrosis factor α (TNFα) and interleukin-1 (IL-1), virus infection, lipopolysaccharide (LPS) and lymphocyte activation, result in the phosphorylation of I-κB proteins on two conserved serine residues (Ser32 and -36 on I-κBα, and Ser19 and -23 on I-κBβ) (Ghosh et al., 1998). This phosphorylation targets I-κB for ubiquitylation and subsequent proteosome- mediated degradation, resulting in the release of NF-κB. NF-κB then translocates to the nucleus where it binds to specific κB sites in promoter sequences and activates transcription (Ghosh et al., 1998).
Phosphorylation of I-κB in response to extracellular stimuli is carried out by the I-κB kinase (IKK) complex, which is comprised of (at least) two catalytic subunits, IKKα and IKKβ (DiDonato et al., 1997; Mercurio et al., 1997; Regnier et al., 1997; Woronicz et al., 1997; Zandi et al., 1997), as well as the modulator NEMO (NF-κB essential modulator) (Rothwarf et al., 1998; Yamaoka et al., 1998). Although IKKα and IKKβ appear to have analogous activities in vitro, IKKα- and IKKβ-deficient mice have dramatically different phenotypes. IKKα–/– mice exhibit severe limb and skin abnormalities. Surprisingly, the induction of NF-κB in ikkα–/– murine embryonic fibroblasts (EFs) by either TNFα or IL-1 is normal (Hu et al., 1999; Takeda et al., 1999). In contrast, the phenotype of IKKβ-deficient mice resembles that of relA–/– mice (Beg et al., 1995) but is more severe. IKKβ–/– animals display massive liver degeneration due to apoptosis and die between E12.5 and E14.5 (Q.Li et al., 1999b; Z.W.Li et al., 1999; Tanaka et al., 1999). IKKβ–/– EFs show enhanced sensitivity to TNFα-induced apoptosis, and NF-κB activation in response to TNFα and IL-1 is significantly reduced compared with the wild type (Q.Li et al., 1999b; Z.W.Li et al., 1999; Tanaka et al., 1999). These results indicate that it is IKKβ that mediates responses to TNFα and IL-1 (Q.Li et al., 1999b; Z.W.Li et al., 1999; Tanaka et al., 1999), while IKKα may respond to an as yet uncharacterized morphogenetic signal (Hu et al., 1999; Q.Li et al., 1999a; Takeda et al., 1999). The phenotype of NEMO-deficient mice is similar to that of ikkβ–/– animals, although even more severe. Mutant mice display massive liver degeneration due to apoptosis and die between E12.5 and E13 (Makris et al., 2000; Rudolph et al., 2000). EFs from nemo–/– embryos show enhanced sensitivity to TNFα-induced apoptosis, and NF-κB activation in response to TNFα and IL-1 is dramatically reduced compared with wild-type EFs, demonstrating that NEMO is an essential component of the IKK complex (Makris et al., 2000; Rudolph et al., 2000).
Members of the TNF receptor-associated factor (TRAF) family of adaptor proteins are involved in coupling stimulation of receptors for pro-inflammatory cytokines, such as the IL-1 receptor (IL-1R) and the TNF receptors (TNFRs) to NF-κB activation, and other downstream events (Arch et al., 1998). TRAF2 plays a critical role in signal transduction mediated by both TNFR1 and TNFR2, and has been implicated in TNFα-induced activation of NF-κB and the stress-activated protein kinase (SAPK/JNK) (Rothe et al., 1995; Liu et al., 1996; Lee et al., 1997; Natoli et al., 1997; Yeh et al., 1997). Similarly, TRAF6 is important for the transduction of IL-1-induced signals, including those resulting in NF-κB activation (Cao et al., 1996b; Lomaga et al., 1999). However, the signaling pathway linking the TRAF proteins to NF-κB activation remains to be defined. A MAP-3K homolog termed NF-κB-inducing kinase (NIK) has been shown to bind both TRAF2 and TRAF6, linking these adaptors to downstream signaling (Song et al., 1997). However, recent studies highlighting the role of NIK in lymph node organogenesis and B-lymphocyte activation indicate that the function of NIK may be restricted to certain cell types, implying that additional molecules must also be involved in regulating NF-κB activation downstream of TRAF proteins (Shinkura et al., 1999; Garceau et al., 2000).
In this report, we describe the characterization of a novel TRAF2-associated kinase, T2K, and study its physiological function by gene targeting. T2K-deficient mice die of liver apoptosis, but t2k–/– EFs show normal sensitivity to TNFα-induced apoptosis, and normal I-κB degradation and NF-κB DNA binding in response to TNFα or IL-1 stimulation. Nevertheless, NF-κB-directed transcription in response to these pro-inflammatory cytokines is reduced in t2k–/– EF cells. These results indicate that T2K is critical in protecting embryonic liver from massive apoptosis, and that T2K may regulate NF-κB activation in a way that is distinct from the process involving I-κB degradation.
Results
Isolation of T2K
Since TRAF2 plays a role in the activation of NF-κB, it seemed reasonable that TRAF2 might interact, directly or indirectly, with a mediator involved in the signaling cascade leading to activation of the transcription factor. We investigated this possibility using the 293 cell line stably expressing a FLAG-tagged TRAF2 molecule. TRAF2 and associated proteins were immunoprecipitated and assayed for kinase activity by determining their ability to phosphorylate I-κBα in vitro. A kinase activity was indeed identified in TRAF2 complexes that could phosphorylate I-κBα in vitro. After further purification by gel filtration and monoQ column chromatography, the amino acid sequence of the putative kinase was determined and used to search EST sequences held in the National Center of Biological Information. A matching EST sequence (R19830) was used to clone a full-length cDNA encoding a putative protein kinase, which was named T2K. Both recombinant T2K and immunoprecipitated TRAF2 complexes could phosphorylate a wild-type I-κBα peptide (25–41) and a mutant peptide in which Ser32 was mutated to Ala, but not an altered pepide in which Ser36 was mutated to Ala (Figure 1A). These results indicate that the kinase activity is specific for Ser36 of I-κBα. Furthermore, since both recombinant T2K and the immunoprecipitated TRAF2 complexes had the same substrate specificity, T2K is likely to account for the kinase activity detected in the TRAF2 complexes. The identity of T2K was further confirmed by western blot analysis in which polyclonal rabbit antiserum raised against recombinant T2K recognized a band of ∼80 kDa only in anti-TRAF2 immunoprecipitates of untransfected 293 cells, not in anti-IKKβ or anti-TRAF6 immunoprecipitates (Figure 1B). While this manuscript was in preparation, two groups reported the isolation of molecules identical to T2K. Tojima et al. (2000) reported cloning of a cDNA encoding a kinase, called NAK, capable of phosphorylating IKKβ and I-κBα Ser36 in vitro. In addition, Pomerantz and Baltimore (1999) reported the cloning of a cDNA encoding TANK-binding kinase 1 (TBK1). TANK (I-TRAF) is a TRAF-binding protein with both stimulatory and inhibitory properties that has been implicated in NF-κB activation (Cheng and Baltimore, 1996; Rothe et al., 1996). These researchers also observed that TBK1/T2K is a kinase capable of forming a complex with TANK and TRAF2 (Pomerantz and Baltimore, 1999), consistent with our findings.
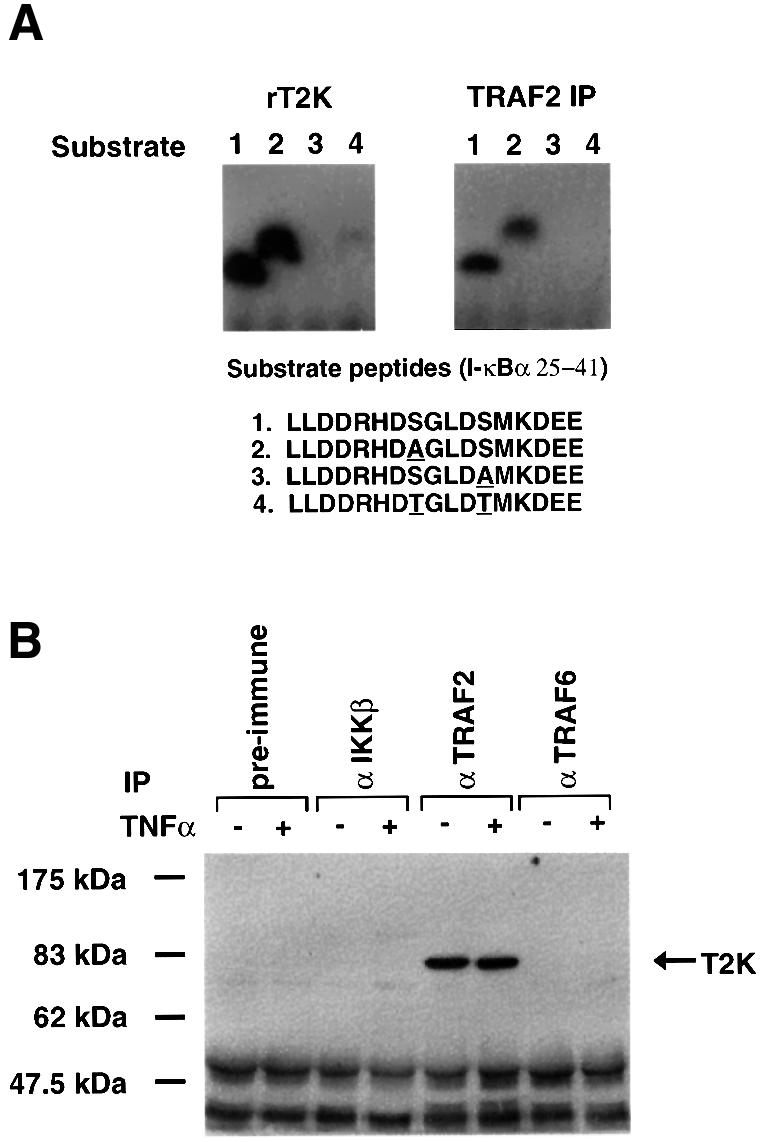
Fig. 1. Identification of a TRAF2-associated kinase (T2K). (A) TRAF2 complexes or recombinant T2K were used in in vitro kinase assays to assess the phosphorylation of peptide substrates encompassing amino acids 25–41 of either wild-type or mutated I-κBα (mutated amino acids are underlined). (B) Co-immunoprecipitation of endogenous T2K. Extracts of 293 cells either left untreated or stimulated with 100 ng/ml TNFα for 10 min were incubated with pre-immune rabbit serum or rabbit anti-serum specific for either IKKβ, TRAF2 or TRAF6 monoclonal antibody followed by western blotting using antiserum specific for recombinant T2K.
Generation of T2K-deficient mice
To characterize the function of T2K in vivo, we disrupted the murine t2k gene in embryonic stem (ES) cells, using a targeting vector designed to replace exons 1 and 2 of the endogenous locus with a PGK-neo cassette (Figure 2A). Correctly targeted ES cell clones were identified by Southern blotting and injected into C57BL/6 blastocysts to generate chimeric mice. Male chimeras with germline transmission were utilized to generate t2k+/– animals, which were interbred to generate t2k–/– mice. Genotypes were confirmed by Southern blotting (Figure 2B) and PCR analysis (Figure 2C). Although t2k+/– mice appeared normal and were fertile, no viable homozygous mutant mice were observed in the first 111 pups derived from t2k+/– intercrosses, suggesting that T2K deficiency results in embryonic lethality. EFs were subsequently derived from early mutant embryos and total RNA was prepared for northern blot analysis. The results confirmed the absence of detectable T2K mRNA in t2k–/– cells (Figure 2D).

Fig. 2. Targeted disruption of the murine t2k gene. (A) Top, a portion of the endogenous t2k locus containing three exons (shown as solid boxes). Middle, the targeting construct. Bottom, the mutant locus resulting from homologous recombination. The SacI (S) restriction site and the flanking probe used to detect homologous recombination are indicated. (B) Genomic Southern blot of SacI-digested DNA from E13.5 embryos of the genotypes indicated, showing the 6.6 kb wild-type band and the 14.6 kb mutant band detected by the flanking probe in (A). (C) Genotyping by PCR of DNA from E13.5 embryos using the primers described in Materials and methods. (D) Northern blot analysis of T2K RNA expression in E13.5 EFs of the genotypes indicated. The cells were either left untreated or stimulated with 10 ng/ml TNFα for the time points indicated.
Liver degeneration in T2K-deficient embryos
Embryos from timed matings of t2k+/– animals were analyzed to determine the time of lethality (Table I). Offspring of all three expected genotypes were present at the normal Mendelian ratio until E14.5 when t2k–/– embryos appeared pale and exhibited dramatic liver degeneration (Figure 3A–D). Development of all other organs appeared normal. Histological analysis of embryo sections revealed liver hemorrhage and severe tissue loss (Figure 3C–F). To determine whether the liver degeneration was attributable to apoptosis, TUNEL assays were performed on sections of E12.5, E13.5 and E14.5 wild-type and mutant embryos. Sections of wild-type livers at any of these stages showed very few TUNEL-positive cells (Figure 3G and data not shown). In contrast, while sections of E12.5 t2k–/– embryos appeared normal, livers of E13.5 t2k–/– embryos displayed mild, focal liver degeneration and apoptosis (data not shown). By E14.5, extensive apoptosis was detected throughout liver sections of t2k–/– embryos (Figure 3H), indicating that apoptosis is a major cause of liver degeneration in these mutants, and consequently, of embryonic death. The liver apoptosis occurring in the absence of T2K was strikingly similar to that observed in nemo–/–, ikkβ–/– and relA–/– animals (Beg et al., 1995; Q.Li et al., 1999b; Z.W.Li et al., 1999; Tanaka et al., 1999; Rudolph et al., 2000), prompting us to speculate that T2K might be involved in the activation of NF-κB.
Table I. Genotypes of embryos derived from t2k+/– intercrosses.
| Stage | +/+ | +/– | –/– |
|
Total |
|---|---|---|---|---|---|
| Normal | Abnormal | ||||
| E12.5 | 8 | 26 | 6 | 0 | 40 |
| E13.5 | 36 | 82 | 19 | 21 | 158 |
| E14.5 | 17 | 28 | 4 | 12 | 61 |
| E15.5 | 10 | 20 | 0 | 4a | 34 |
aIn resorption.

Fig. 3. Massive liver apoptosis in the absence of T2K. (A and B) Low power view of E14.5 wild-type (A) and t2k–/– (B) embryos. (C–F) Histological analysis of E14.5 liver sections. Views of H&E staining of wild-type (C and E) and mutant (D and F) livers at either low (C and D) or high (E and F) magnification are shown. (G and H) Enhanced apoptosis in livers of E14.5 T2K-deficient embryos. TUNEL assays were performed on sections of wild-type (G) and mutant (H) E14.5 embryos. A high power view of the liver is shown in both cases.
Normal sensitivity of thymocytes and EFs to TNFα-induced apoptosis
Stimulation of TNFR1 results in signal transduction that contributes both to apoptosis and cell survival (Hsu et al., 1996b; Yeh et al., 1999). TRAF2 and NF-κB play important roles in the survival pathway, as shown by the finding that relA–/–, ikkβ–/–, nemo–/– and traf2–/– cells display enhanced sensitivity to TNFα-induced apoptosis. Wild-type EFs are resistant to treatment with TNFα alone, but relA–/–, nemo–/– and ikkβ–/– EFs are exquisitely sensitive to TNFα-induced cell death (Beg et al., 1995; Q.Li et al., 1999b; Z.W.Li et al., 1999; Tanaka et al., 1999; Rudolph et al., 2000). TRAF2-deficient EFs are mildly sensitive to apoptosis induced by TNFα alone, but exhibit a dramatic increase in sensitivity in the presence of TNFα combined with low concentrations of the protein synthesis inhibitor cycloheximide (Yeh et al., 1997). At higher concentrations, cycloheximide can also render wild-type EFs sensitive to TNFα treatment. In this context, we assessed the response of t2k–/– EFs to TNFα alone or in combination with various concentrations of cycloheximide. Sensitivity to staurosporine was used as a positive control. T2K-deficient EFs behaved like wild-type EFs rather than relA–/–, ikkβ–/–, nemo–/– or traf2–/– EFs, being resistant to apoptosis induced by TNFα at all but the highest concentrations of cycloheximide (Figure 4A).

Fig. 4. Normal sensitivity of thymocytes and EFs to TNFα-induced cell death in the absence of T2K. (A) TNFα-induced apoptosis of EFs. Wild-type and T2K-deficient EFs were either left untreated or stimulated with staurosporine (2 µM) or TNFα (10 ng/ml) in combination with the indicated concentrations of cycloheximide for 18 h. Cells were harvested and stained for viability as described in Materials and methods. Values shown are the percentage of viable cells after treatment relative to untreated controls. Individual treatments were measured in triplicate. (B) TNFα-induced apoptosis of CD4+CD8+ (DP) thymocytes. Thymocytes from the reconstituted mice described in the text were cultured at 37°C for 20 h in the presence of either 1 µM dexamethasone or the indicated concentrations of TNFα. Cell viability was determined by 7-amino actinomycin D (7-AAD) staining. Results are expressed as the percentage of total viable DP cells remaining after treatment relative to the untreated control of the same genotype. Individual treatments were measured in quadruplicate.
The importance of the role of TRAF2 in protection against TNFα-induced apoptosis was highlighted further by the demonstration that traf2–/– thymocytes undergo enhanced apoptosis in response to TNFα (Yeh et al., 1997). In order to obtain t2k–/– thymocytes to assess their sensitivity to apoptotic stimuli, we used fetal liver cells from either wild-type or t2k–/– E12.5 to reconstitute the lymphoid compartment of irradiated rag1–/– mice. Bone marrow, thymus, lymph nodes, spleens and peripheral blood of all reconstituted animals had normal proportions of immature, and mature, T- and B-cell populations (data not shown). T2K-deficient double positive (DP) thymocytes were as sensitive as wild-type DP thymocytes to dexamethasone-induced apoptosis. In contrast to traf2–/– DP thymocytes, t2k–/– DP thymocytes were as resistant to TNFα treatment as were wild-type DP thymocytes (Figure 4B).
Importantly, the embryonic lethality and liver apoptosis of animals lacking either RelA or IKKβ can be attributed to TNFR1 signaling. Thus, animals lacking both RelA and TNFR1 (Beg and Baltimore, 1996), like those lacking both IKKβ and TNFR1 (Q.Li et al., 1999b), develop to term. Results from Table II show that six of the first 94 pups born from intercrossing t2k+/– tnfr1+/– mice were t2k–/– tnfr1–/– mice (expected Mendelian ratio 6.4%, 1:16). These double mutant animals developed to term rather than dying at ∼E14.5. The apparent rescue of embryonic lethality by inactivation of the TNFR1 gene supports the hypothesis that embryonic lethality of t2k–/– animals is due to TNFα-induced liver apoptosis, and that T2K is an important mediator in TNFR1 signaling. Together, these results indicate that although T2K plays an important role in protection against TNFα-induced apoptosis, it is dispensable for resistance of certain cell types, such as thymocytes and EFs, to TNFα-induced apoptosis.
Table II. Genotypes of embryos derived from t2k+/– tnfr1+/– intercrosses.
| t2k+/+ tnfr1+/+ | t2k+/+ tnfr1+/– | t2k+/+ tnfr1–/– | t2k+/– tnfr1+/+ | t2k+/– tnfr1+/– | t2k+/– tnfr1–/– | t2k–/– tnfr1+/+ | t2k–/– tnfr1+/– | t2k–/– tnfr1–/– | Total |
|---|---|---|---|---|---|---|---|---|---|
| 6 | 18 | 12 | 16 | 18 | 18 | 0 | 0 | 6 | 94 |
| 6.4% | 19.1% | 12.8% | 17% | 19.1% | 19.1% | 6.4% | 100% |
Normal I-κBα phosphorylation/degradation and κB-binding activity in t2k–/– cells
Given the ability of T2K to phosphorylate I-κBα in vitro, and the similarities in liver phenotype observed in t2k–/–, relA–/–, nemo–/– and ikkβ–/– mice, we investigated various steps leading to activation of NF-κB-dependent transcription in t2k–/– EFs. Cytoplasmic and nuclear protein extracts were prepared from wild-type and T2K-deficient EFs stimulated with either TNFα or IL-1 for 1, 3, 9, 27 or 90 min. Phosphorylation of Ser32 of I-κBα and degradation of I-κBα, I-κBβ and I-κBε were assessed by probing western blots of cytoplasmic extracts with either a phospho-I-κBα-specific antibody, or I-κBα-, I-κBβ- and I-κBε-specific antibodies, respectively. Rapid phosphorylation of I-κBα Ser32, comparable to that in the wild type, was observed in t2k–/– EFs in response to either TNFα or IL-1 (Figure 5A). Degradation of I-κBα (Figure 5A), I-κBβ and I-κBε (data not shown) in response to either stimulus was also normal in the mutant cells.

Fig. 5. Normal I-κBα phosphorylation, degradation and NF-κB DNA binding activity in t2k–/– EFs. (A) I-κBα phosphorylation/degradation. Wild-type and T2K-deficient EFs were treated with either TNFα (10 ng/ml) or IL-1β (10 ng/ml) for the times indicated. Cytoplasmic extracts were analyzed by western blotting using monoclonal antibodies specific for either phospho-I-κBα (top), I-κBα (middle) or the loading control actin (bottom). (B) κB binding activity. Wild-type and t2k–/– EFs were incubated with either mouse recombinant TNFα (10 ng/ml) (top) or IL-1β (10 ng/ml) (bottom) for the times indicated. NF-κB activation in 10 µg of nuclear extract was determined by EMSA as described in Materials and methods. For both (A) and (B), one result representative of three independent experiments is shown. (C) RelA binding activity. Wild-type and t2k–/– EFs were incubated with 10 ng/ml of either mouse recombinant TNFα or IL-1β for 27 min. RelA binding activity was assessed by incubation of 10 µg of nuclear extract with RelA-specific antibody followed by electrophoretic mobility supershift assay, as described in Materials and methods.
NF-κB DNA binding activity in nuclear extracts of wild-type and t2k–/– EFs was then assessed by electrophoretic mobility shift assay (EMSA) using a labeled oligonucleotide containing κB binding sites. Stimulation with either TNFα or IL-1, as above, resulted in a rapid induction of κB binding activity in nuclear extracts from cells of both genotypes (Figure 5B). Together these data show that, at least in EFs, T2K is not essential for either phosphorylation-induced degradation of I-κB or NF-κB DNA binding in response to TNFα or IL-1.
To assess the contribution of RelA to the κB DNA binding activity observed in Figure 5B, nuclear extracts from either wild-type or t2k–/– EFs, stimulated with either TNFα or IL-1, were pre-incubated with RelA-specific Ab prior to EMSA analysis. The κB DNA binding activity induced by either cytokine was reactive to the RelA-specific antibody, causing a supershift (Figure 5C). Furthermore, RelA-reactive κB DNA binding activity could account for the majority of the activity detected in extracts from both wild-type and t2k–/– EFs (Figure 5C), indicating that T2K is not essential for RelA DNA binding in response to TNFα or IL-1.
Impaired NF-κB-dependent gene expression in t2k–/– cells
NF-κB-dependent gene expression was assessed both by a reporter assay and by northern blot analysis of the expression of endogenous NF-κB target genes. To assess reporter activity, protein extracts were prepared from wild-type and T2K-deficient EFs that had been transfected with either the NF-κB-dependent E-selectin promoter-luciferase (pELAM-luc) or the 6XκB promoter-luciferase (6XκB-luc) reporter construct, and stimulated with TNFα, IL-1 or PDGF for 2 or 6 h. An expression vector encoding the β-galactosidase gene was co-transfected as a control for transfection efficiency. A robust induction of pELAM-luc reporter activity was observed in wild-type EFs stimulated for 2 h with either TNFα or IL-1. However, pELAM-luc activity was only marginally induced in t2k–/– EFs (Figure 6A). After 6 h stimulation with TNFα or IL-1, reporter activity in wild-type EFs was increased a total of 18- and 11-fold, respectively, compared with untreated controls. In contrast, only a 3-fold induction of reporter activity was observed in T2K-deficient cells after 6 h with either TNFα or IL-1 (Figure 6A).

Fig. 6. Impaired NF-κB-dependent gene expression in t2k–/– EFs. (A) Impaired transactivation of NF-κB-dependent reporter activity in t2k–/– EFs. Wild-type and t2k–/– EFs were transfected with the pELAM-luc reporter and CMV-lacZ vectors. After stimulation with 10 ng/ml TNFα or IL-1, or 100 ng/ml PDGF for the times indicated, lysates were assayed for luciferase activity as described in Materials and methods. Individual treatments were measured in triplicate, and results representative of four independent experiments are shown. (B) Impaired induction of the endogenous NF-κB target genes ICAM-1 and TLR-2. Wild-type and t2k–/– EFs were stimulated with 10 ng/ml TNFα or IL-1 for the times indicated followed by northern blot analysis of 20 µg of total RNA using a 32P-labeled random priming probe corresponding to the full-length ICAM-1 or TLR-2 cDNA. GAPDH expression was analyzed to control for RNA loading. In the bottom panel, wild-type and relA–/– EFs were stimulated with 10 ng/ml for the times indicated followed by northern blot analysis of 2 µg of mRNA, as above.
Results from the T2K/NAK study (Tojima et al., 2000) highlighted a role for T2K/NAK in PDGF-mediated NF-κB activation, prompting us to compare PDGF-induced NF-κB-dependent reporter activity in wild-type and t2k–/– EFs. The maximum induction of pELAM-luc reporter activity observed in wild-type EFs in response to PDGF stimulation (after 2 h) was much weaker (3.3-fold) than that observed in response to either TNFα or IL-1. Furthermore, the reporter activity induced in t2k–/– EFs in response to PDGF stimulation was only slightly lower (1.4-fold) than wild-type controls. After 6 h of PDGF stimulation, both wild-type and t2k–/– EFs exhibit weak and comparable reporter activity (Figure 6A). Similar results were obtained with all the above stimuli using the 6XκB-luc reporter construct (data not shown).
We next assessed induction of endogenous NF-κB target gene expression in the absence of T2K. To this end, RNA samples prepared from wild-type and T2K-deficient EFs that had been stimulated with IL-1 or TNFα for 0.5, 2, 6, 12 or 24 h were subjected to northern blot analysis to assess the levels of Toll-like receptor-2 (TLR-2) and intercellular adhesion molecule-1 (ICAM-1). To confirm that TLR-2 is indeed an NF-κB target, we compared mRNA expression of TLR-2 in wild-type and relA–/– EFs. The levels of mRNA for TLR-2 are strongly induced after 2 h of TNFα stimulation in wild-type cells, but there is no detectable TLR-2 mRNA in the absence of RelA (Figure 6B). Furthermore, the levels of TNFα-induced mRNA for TLR-2 were significantly reduced in t2k–/– EFs compared with wild-type controls. Moreover, the levels of mRNA for the NF-κB target gene ICAM-1 (Chen et al., 1995) induced after 2 h of IL-1 (Figure 6B, right) or TNFα (data not shown) stimulation were also substantially reduced in t2k–/– EFs compared with wild-type controls. Together, these results indicate that T2K-deficient EFs are impaired in expression of certain NF-κB-dependent genes induced by pro-inflammatory cytokines.
Discussion
In this study, we have demonstrated that, like IKKβ, NEMO and the RelA subunit of NF-κB, T2K is crucial in protecting embryonic liver from devastating apoptosis. However, analysis of NF-κB activation in EFs indicates that, in contrast to these mediators, T2K plays a novel role in the activation of NF-κB-dependent transcription, independently of I-κBα phosphorylation and κB DNA binding. Much attention has been focused on the process by which extracellular signals induce phosphorylation of I-κB by the IKK complex, leading to degradation of I-κB and release of NF-κB from cytoplasmic sequestration. However, I-κB phosphorylation is not the sole means of regulating NF-κB transcriptional activity. Several recent studies have demonstrated that signal-induced phosphorylation of the RelA subunit of NF-κB is critical for the induction of NF-κB-dependent transcription (Bird et al., 1997; Zhong et al., 1997, 1998; Wang and Baldwin, 1998; Anrather et al., 1999; Egan et al., 1999; Sizemore et al., 1999). In fact, signals such as TNFα, LPS and IL-1, which induce phosphorylation of I-κB proteins, can also cause phosphorylation of NF-κB proteins (Bird et al., 1997; Zhong et al., 1997; Wang and Baldwin, 1998; Egan et al., 1999; Sizemore et al., 1999). Importantly, while the inducible phosphorylation of RelA increases the transcriptional activity of NF-κB, it does not appear to affect its nuclear translocation or DNA binding activity (Wang and Baldwin, 1998). Therefore, it is possible that T2K may regulate the trans-activation potential of NF-κB through phosphorylation of NF-κB proteins, either directly or indirectly.
It is also possible that T2K plays a role in inducing coactivators of NF-κB-dependent transcription. Previous studies have shown that LPS-induced phosphorylation of RelA Ser276 stimulates trancriptional activity by promoting the interaction of NF-κB with the coactivator CBP/p300 (Zhong et al., 1998). Different stimuli, including LPS and TNFα, induce phosphorylation of RelA on different residues (Zhong et al., 1997, 1998; Wang and Baldwin, 1998), implying that different coactivators, with distinct effects on different promoters, may be recruited to the transcriptional complex. Of note in this context is that while the results presented in this study demonstrate that T2K is critical in NF-κB-dependent transcription, expression of certain NF-κB target genes such as I-κBα and IL-6 does not seem to be affected by the lack of T2K. In protein extracts from cells stimulated with either TNFα or IL-1, I-κBα is first degraded, then re-expressed at comparable levels and with similar kinetics in wild-type and T2K-deficient EF cells (Figure 5A). Furthermore, IL-1-induced expression of IL-6 by t2k–/– EFs is comparable to that of wild-type EFs (data not shown). These seemingly paradoxical results are consistent with a role for T2K as a gene-specific regulator of NF-κB-dependent transcription.
The role of T2K in activation of NF-κB may be, in part, independent of its kinase activity, as is the case for RIP and IRAK, components of the TNFR1 and IL-1 receptor signaling complexes, respectively (Cao et al., 1996a; Hsu et al., 1996a; Knop and Martin, 1999; Vig et al., 1999). Results from the study by Pomerantz and Baltimore (1999) indicated that a mutated form of TBK1/T2K in which the kinase activity was abrogated was able to inhibit TANK-mediated NF-κB reporter activity, but not that induced by TNFα, IL-1 or CD40. The study by Tojima et al. (2000) also shows that a kinase-inactive form of NAK/T2K is unable to inhibit TNFα- and IL-1-induced NF-κB reporter activity. While the precise role of TANK in NF-κB activation remains to be elucidated, these data suggest that the function of TBK1/T2K in TNFα- or IL-1-induced NF-κB-dependent transcription may be partially independent of its kinase activity.
The phenotype of t2k–/– animals has striking similarities to that of nemo–/–, ikkβ–/– or relA–/– animals, since all these animals die of profound liver degeneration and apoptosis during embryonic development (Beg et al., 1995; Q.Li et al., 1999b; Z.W.Li et al., 1999; Tanaka et al., 1999; Rudolph et al., 2000). Furthermore, like ikkβ–/– or relA–/– animals, embryonic lethality and liver apoptosis of t2k–/– can be attributed to TNFR1 signaling (Beg and Baltimore, 1996; Q.Li et al., 1999b). However, there are some important differences. RelA-deficient, nemo–/– and ikkβ–/– EF cells exhibit enhanced sensitivity to TNFα-induced apoptosis, implying that the liver cell death observed in these mutant animals can be attributed to a lack of NF-κB-mediated protection from TNFα-mediated cytotoxicity (Beg and Baltimore, 1996; Q.Li et al., 1999b; Z.W.Li et al., 1999; Tanaka et al., 1999; Rudolph et al., 2000). In contrast, t2k–/– EFs are not sensitive to TNFα-induced apoptosis, even in the presence of moderate concentrations of cycloheximide. These results suggest that there may be cell type-specific molecules involved in both the activation of NF-κB-dependent transcription and protection against TNFα-mediated cytotoxicity. Thus, the uncoupling of cytoprotection in fetal liver and EFs may be caused by differential roles of T2K in different cell types. Alternatively, mediators such as T2K may have partially redundant functions that can be compensated for by other molecules in some cell types. Further investigation is required to address these possibilities.
Two independent studies published during the preparation of this manuscript reported the identification and characterization of a novel kinase termed IKKi or IKKε (Shimada et al., 1999; Peters et al., 2000). IKKε/i is 48% identical and 64% similar to T2K, and is activated by several inducers of NF-κB, including LPS and phorbol 12-myristate 13-acetate (PMA) (Shimada et al., 1999; Peters et al., 2000). While IKKε/i can phosphorylate I-κBα Ser36 in vitro, it apparently functions in vivo as part of a larger complex (which does not include IKKα/β) that can phosphorylate both Ser32 and Ser36 of I-κBα in response to PMA (Peters et al., 2000). Furthermore, a kinase-inactive form of IKKε/i inhibited NF-κB reporter activity induced by PMA or a T-cell activation stimulus, but not that induced by TNFα or IL-1 stimulation (Peters et al., 2000). T2K and IKKε/i may thus have partially overlapping functions, but additional studies are needed to determine whether there is a functional relationship between these two kinases in vivo.
In conclusion, in this study we have isolated and characterized T2K, a novel kinase associated with TRAF2 that is essential for the protection of embryonic liver from apoptosis. Analysis of T2K-deficient EF cells has shown that T2K plays a unique role in NF-κB transcriptional activation in response to pro-inflammatory cytokines that is at least partly independent of I-κB degradation and κB binding. Further studies of the role of T2K in NF-κB transcriptional activation will help define the molecular network governing this important process.
Materials and methods
Identification and purification of T2K
Cells of the 293 cell line stably expressing FLAG-tagged TRAF2 were lysed in lysis buffer [50 mM HEPES pH 7.6, 250 mM NaCl, 1 mM EGTA, 1 mM dithiothreitol (DTT), protease inhibitor cocktail (Boehringer Mannheim), 20 mM β-glycerophosphate, 1 mM sodium orthovanadate, 5 mM p-nitrophenyl phosphate, 10% glycerol, 0.1% NP-40] for 20 min on ice. After centrifugation of lysates at 100 000 g for 30 min, supernatants were incubated with anti-Flag M2 beads (Sigma; 10 µl/ml of cell lysate) overnight at 4°C with rocking. M2 beads were washed extensively in lysis buffer and used as ‘TRAF2 complexes’ in kinase assays as described below. The kinase activity found to phosphorylate recombinant I-κBα was purified first by gel filtration, and then by monoQ column chromatography. The peptide sequence of a protein band of ∼80 kDa that co-fractionated with I-κBα kinase activity was used to identify several EST sequences in the National Center of Biological Information (Bethesda, MD). One EST sequence (R19830) was used to clone a full-length cDNA encoding an apparent protein kinase that we named T2K. The same kinase has been identified as TANK-interacting protein (I-TRAF) and named as TANK-binding kinase (TBK1) (Pomerantz and Baltimore, 1999) (DDBJ/EMBL/GenBank accession No. AF191839).
Co-immunoprecipitation
Recombinant T2K was generated in Escherichia coli (BL21), and was used to induce the production of a rabbit polyclonal anti-T2K antiserum using standard protocols. For co-immunoprecipitations, 1 × 107 293 cells were either left untreated or treated for 5 min with 10 ng/ml TNFα (recombinant mouse TNFα; R&D Systems Inc.). Cells were harvested, lysed as above and centrifuged at 100 000 g for 30 min to yield extracts used in experiments. Extract samples (1.0 ml; 107 cell equivalents) were incubated with 1 µl of either control pre-immune rabbit serum or rabbit antisera specific for either IKKβ, TRAF6 (generated as described above) or TRAF2 (generated against a peptide derived from TRAF2 73–106 amino acids) and 10 µl of protein A–Sepharose beads (Pharmacia) overnight at 4°C with rocking. After washing, the beads were boiled in 20 µl of SDS sample buffer and 10 µl of the eluate were fractionated by 10% SDS–PAGE followed by western blotting with the rabbit polyclonal antiserum to T2K. The reactive bands were detected with horseradish peroxidase-conjugated protein A (Bio-Rad) and an enhanced chemiluminescence sytem (Amersham) used according to the manufacturer’s instructions.
Kinase assays
TRAF2 complexes (from 107 cell equivalents) bound to anti-FLAG M2 beads were washed extensively, then eluted using 20 µl of FLAG peptide in lysis buffer at 200 µg/ml. The eluate (10 µl) was incubated at 37°C for 30 min with 1 µg of bacterially expressed I-κBα (1–250 amino acids) as substrate in a 20 µl kinase reaction mixture containing 20 mM HEPES pH 7.6, 125 mM NaCl, 1 mM EGTA, 1 mM DTT, 10 mM MgCl2, protease inhibitor cocktail (Boehringer Mannheim), 20 mM β-glycerophosphate, 1 mM sodium orthovanadate, 5 mM p-nitrophenyl phosphate and [γ-32P]ATP. The reactions were stopped by adding 10 µl of SDS sample buffer. Phosphorylated proteins were resolved by SDS–PAGE and visualized by autoradiography. To determine kinase activity toward wild-type and mutant I-κBα-derived peptides, FLAG-tagged T2K was transiently expressed in 293 cells and immunopurified with M2 beads. Approximately 0.2 µg of recombinant T2K were incubated with 50 µM I-κBα-derived peptides in a 20 µl kinase assay as described above. The phosphorylated peptides were resolved on a pre-cast tricine gel (Bio-Rad) and visualized by autoradiography.
Generation of t2k–/– mice by gene targeting
A genomic DNA clone for t2k was isolated by screening a 129/J mouse genomic DNA library using a probe derived from the 5′ end of the human t2k cDNA. The targeting construct was designed to replace most of exon 1, all of exon 2 and the intervening intron with the PGK-neo gene cassette in reverse orientation to the endogenous t2k gene. The linearized targeting construct was transfected into ES cells (E14 clone, derived from 129/Ola mouse embryos) by electroporation as described previously (Yeh et al., 1997). Neomycin-resistant ES clones were selected in 300 µg/ml G418 (Gibco-BRL) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 15% fetal bovine serum and leukemia inhibitory factor. Homologous recombinants were identified by PCR and further analyzed by Southern blot hybridization using the flanking probe depicted in Figure 2A or a neo-specific probe. Chimeric mice were produced by microinjection of heterozygous ES cells into E3.5 C57BL/6J blastocysts that were subsequently transferred to CD1 pseudopregnant foster mothers. Male chimerae were backcrossed with C56BL/6J females and germline transmission in F1 t2k+/– mice was verified by Southern blot analysis. Heterozygous mice were interbred to obtain t2k–/– mice. Genotyping of the F2 mice was performed by PCR on tail genomic DNA and verified by Southern blot analysis. The primer 5′-GGCTCT CTGTTCAGTTAGGATGTCAA-3′ was used in conjunction with either the primer 5′-TTCTCATTGTCTTACGAGATGTGG-3′ for the wild- type t2k allele, or the primer 5′-AGAGGCCACTTGTGTAGCGCC AAGTGCC-3′ for the mutant allele. Mice were maintained in the animal facility of the Ontario Cancer Institute in accordance with its ethical guidelines.
Histology
Embryos were fixed with 4% buffered formalin at 4°C for 12 h and embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E) according to standard protocols. For apoptosis determination, TUNEL (terminal deoxytransferase-mediated deoxyuridine triphosphate nick-end labeling) assays were performed using an in situ cell death detection kit (Boehringer Mannheim) according to the manufacturer’s instructions.
Adoptive transfer and flow cytometry
Six-week-old rag1–/– mice were purchased from Taconic. Prior to cell transfer, recipient mice were irradiated once with 900 rads of γ-rays. Approximately 2 × 106 freshly isolated fetal liver cells from E12.5 t2k+/+ or t2k–/– embryos were injected intravenously into recipients. After transfer, mice were treated with Clavulin-125F (1.25 mg/ml; SmithKline Beecham) for 2 weeks and monitored daily for survival.
Apoptosis assays
Single-cell suspensions of thymocytes were prepared as previously described (Amakawa et al., 1996) from rag1–/– mice 6 weeks after reconstitution with E12.5 fetal liver, and cells were plated at 5 × 106 cells/ml in 96-well plates in RPMI supplemented with 5% fetal bovine serum and containing either no apoptotic stimulus, or dexamethasone (1 µM; Sigma) or TNFα at 1.0, 10 or 100 µg/ml. After 20 h incubation at 37°C in 5% CO2, the cells were harvested and stained with PE-labeled anti-CD8α, FITC-labeled anti-CD4 (both from Pharmingen) and 1 µg/ml of the vital dye 7-AAD (Sigma), and analyzed by flow cytometry as previously described (Nishina et al., 1997). The proportions of viable DP (CD4+CD8+) thymocytes were displayed on a dot plot, gating on the 7-AAD negative population. Individual treatments were measured in quadruplicate.
EFs (1.2 × 105) were plated onto 6-well plates in DMEM supplemented with 10% fetal bovine serum and β-mercaptoethanol. After 24 h incubation, TNFα (10 ng/ml) alone or TNFα in combination with the concentrations of cycloheximide (Sigma) indicated in Figure 4C was added to induce cell death. After 18 h culture, cells were harvested, stained with propidium iodide (PharMingen) and analyzed by flow cytometry to determine cell viability.
Gel mobility shift assay and western blot analysis
Nuclear and cytoplasmic extracts were harvested from EF cells according to previously described protocols (Pfeffer et al., 1993). Recombinant mouse TNFα and IL-1β were purchased from R&D Systems Inc. The Bio-Rad protein assay was used to adjust for equal amounts of nuclear or cytoplasmic proteins in each sample. For supershift EMSA, nuclear extracts were pre-incubated with 1 µg of RelA-specific antibody for 15 min at room temperature prior to EMSA analysis. Nuclear extracts were utilized in gel mobility shift assay, as previously described (Yeh et al., 1997). Cytoplasmic extracts were utilized in western blotting and were incubated with anti-phospho-I-κBα antibody, anti-I-κBα antibody (both from New England Biolabs) or anti-actin antibody (Fisher Scientific).
Reporter assays
Cells were plated at 1.3 × 105 in 6-well plates 24 h prior to transfection using the polycationic transfection reagent Superfect (Qiagen), with a total of 2 µg of DNA. In order to normalize results to transfection efficiency, each transfection included 0.2 µg of CMV-lacZ, which constitutively expresses β-galactosidase (Shu et al., 1997), in addition to either the pELAM-luc reporter, which has been previously described (Hsu et al., 1996b), or empty parental expression vector as the control. The medium was changed after 3 h, and 42 h after transfection cells were stimulated with 10 ng/ml TNF-α or IL-1β, or 100 ng/ml PDGF. After 2 or 6 h, cells were washed twice with phosphate-buffered saline and lysed in 200 µl of reporter lysis buffer (Promega) at room temperature for 30 min. Luciferase activity in 5 µl of extract was measured immediately using the Luciferase Assay System (Promega) and a luminometer (BioOrbiter, Mandel Scientific) according to the manufacturers’ instructions. β-galactosidase activity was determined in 5 µl of extract with 4 mg/ml ONPG (o-nitrophenyl-β-galactopyranoside; Sigma) in 0.067 M sodium phosphate buffer pH 7.5 by measuring the optical density at 420 nm. Fold stimulation of reporter activity was calculated for each sample by dividing the luciferase activity in the experimental sample (normalized to β-galactosidase activity) by the luciferase activity in the unstimulated control.
Acknowledgments
Acknowledgements
The authors thank Hien Chau, Seiko Sugimoto and Jennifer Tsang for excellent technical assistance, and Dr David Goeddel and members of W.-C.Y.’s laboratory for helpful discussions. We also thank Mary Saunders for scientific editing and Irene Ng for administrative assistance. This work was supported in part by the National Cancer Insitute of Canada (NCIC) with funds from the Terry Fox Run (#010028, W.-C.Y.).
References
- Amakawa R. et al. (1996) Impaired negative selection of T cells in Hodgkin’s disease antigen CD30-deficient mice. Cell, 84, 551–562. [DOI] [PubMed] [Google Scholar]
- Anrather J., Csizmadia,V., Soares,M.P. and Winkler,H. (1999) Regulation of NF-κB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Cζ in primary endothelial cells. J. Biol. Chem., 274, 13594–13603. [DOI] [PubMed] [Google Scholar]
- Arch R.H., Gedrich,R.W. and Thompson,C.B. (1998) Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev., 12, 2821–2830. [DOI] [PubMed] [Google Scholar]
- Beg A.A. and Baltimore,D. (1996) An essential role for NF-κB in preventing TNF-α-induced cell death. Science, 274, 782–784. [DOI] [PubMed] [Google Scholar]
- Beg A.A., Sha,W.C., Bronson,R.T., Ghosh,S. and Baltimore,D. (1995) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kB. Nature, 376, 167–170. [DOI] [PubMed] [Google Scholar]
- Bird T.A., Schooley,K., Dower,S.K., Hagen,H. and Virca,G.D. (1997) Activation of nuclear transcription factor NF-κB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J. Biol. Chem., 272, 32606–32612. [DOI] [PubMed] [Google Scholar]
- Cao Z., Henzel,W.J. and Gao,X. (1996a) IRAK: a kinase associated with the interleukin-1 receptor. Science, 271, 1128–1131. [DOI] [PubMed] [Google Scholar]
- Cao Z., Xiong,J., Takeuchi,M., Kurama,T. and Goeddel,D.V. (1996b) TRAF6 is a signal transducer for interleukin-1. Nature, 383, 443–446. [DOI] [PubMed] [Google Scholar]
- Chen C.C., Rosenbloom,C.L., Anderson,D.C. and Manning,A.M. (1995) Selective inhibition of E-selectin, vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression by inhibitors of IκB-α phosphorylation. J. Immunol., 155, 3538–3545. [PubMed] [Google Scholar]
- Cheng G. and Baltimore,D. (1996) TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-κB activation. Genes Dev., 10, 963–973. [DOI] [PubMed] [Google Scholar]
- DiDonato J.A., Hayakawa,M., Rothwarf,D.M., Zandi,E. and Karin,M. (1997) A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature, 388, 548–554. [DOI] [PubMed] [Google Scholar]
- Egan L.J., Mays,D.C., Huntoon,C.J., Bell,M.P., Pike,M.G., Sandborn,W.J., Lipsky,J.J. and McKean,D.J. (1999) Inhibition of interleukin-1-stimulated NF-κB RelA/p65 phosphorylation by mesalamine is accompanied by decreased transcriptional activity. J. Biol. Chem., 274, 26448–26453. [DOI] [PubMed] [Google Scholar]
- Foo S.Y. and Nolan,G.P. (1999) NF-κB to the rescue: RELs, apoptosis and cellular transformation. Trends Genet., 15, 229–235. [DOI] [PubMed] [Google Scholar]
- Garceau N., Kosaka,Y., Masters,S., Hambor,J., Shinkura,R., Honjo,T. and Noelle,R.J. (2000) Lineage-restricted function of NF-κB-inducing kinase (NIK) in transducing signals via CD40. J. Exp. Med., 191, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., May,M.J. and Kopp,E.B. (1998) NF-κB and Rel proteins. Annu. Rev. Immunol., 16, 225–260. [DOI] [PubMed] [Google Scholar]
- Hsu H., Huang,J., Shu,H.B., Baichwal,V. and Goeddel,D.V. (1996a) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity, 4, 387–396. [DOI] [PubMed] [Google Scholar]
- Hsu H., Shu,H.B., Pan,M.G. and Goeddel,D.V. (1996b) TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell, 84, 299–308. [DOI] [PubMed] [Google Scholar]
- Hu Y., Baud,V., Delhase,M., Zhang,P., Deerinck,T., Ellisman,M., Johnson,R. and Karin,M. (1999) Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science, 284, 316–320. [DOI] [PubMed] [Google Scholar]
- Knop J. and Martin,M.U. (1999) Effects of IL-1 receptor-associated kinase (IRAK) expression on IL-1 signaling are independent of its kinase activity. FEBS Lett., 448, 81–85. [DOI] [PubMed] [Google Scholar]
- Lee S.Y., Reichlin,A., Santana,A., Sokol,K.A., Nussenzweig,M.C. and Choi,Y. (1997) TRAF2 is essential for JNK but not NF-κB activation and regulates lymphocyte proliferation and survival. Immunity, 7, 703–713. [DOI] [PubMed] [Google Scholar]
- Li Q., Lu,Q., Hwang,J.Y., Buscher,D., Lee,K.F., Izpisua-Belmonte,J.C. and Verma,I.M. (1999a) IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev., 13, 1322–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Van Antwerp,D., Mercurio,F., Lee,K.F. and Verma,I.M. (1999b) Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science, 284, 321–325. [DOI] [PubMed] [Google Scholar]
- Li Z.W., Chu,W., Hu,Y., Delhase,M., Deerinck,T., Ellisman,M., Johnson,R. and Karin,M. (1999) The IKKβ subunit of IκB kinase (IKK) is essential for NF-κB activation and prevention of apoptosis. J. Exp. Med., 189, 1839–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.G., Hsu,H., Goeddel,D.V. and Karin,M. (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell, 87, 565–576. [DOI] [PubMed] [Google Scholar]
- Lomaga M.A. et al. (1999) TRAF6-deficiency results in osteopetrosis and defective interleukin-1, CD40 and LPS signaling. Genes Dev., 13, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris C., Godfrey,V.L., Krahn-Senftleben,G., Takahashi,T., Roberts,J.L., Schwartz,T., Feng,L., Johnson,R.S. and Karin,M. (2000) Female mice heterozygous for IKKγ/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell, 5, 969–979. [DOI] [PubMed] [Google Scholar]
- Mercurio F. et al. (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science, 278, 860–866. [DOI] [PubMed] [Google Scholar]
- Natoli G., Costanzo,A., Ianni,A., Templeton,D.J., Woodgett,J.R., Balsano,C. and Levrero,M. (1997) Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science, 275, 200–203. [DOI] [PubMed] [Google Scholar]
- Nishina H. et al. (1997) Stress-signaling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature, 385, 350–353. [DOI] [PubMed] [Google Scholar]
- Peters R.T., Liao,S.-M. and Maniatis,T. (2000) IKKε is part of a novel PMA-inducible IκB kinase complex. Mol. Cell, 5, 513–522. [DOI] [PubMed] [Google Scholar]
- Pfeffer K. et al. (1993) Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L.monocytogenes infection. Cell, 73, 457–467. [DOI] [PubMed] [Google Scholar]
- Pomerantz J.L. and Baltimore,D. (1999) NF-κB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J., 18, 6694–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier C.H., Song,H.Y., Gao,X., Goeddel,D.V., Cao,Z. and Rothe,M. (1997) Identification and characterization of an IκB kinase. Cell, 90, 373–383. [DOI] [PubMed] [Google Scholar]
- Rothe M., Sarma,V., Dixit,V.M. and Goeddel,D.V. (1995) TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science, 269, 1424–1427. [DOI] [PubMed] [Google Scholar]
- Rothe M., Xiong,J., Shu,H.B., Williamson,K., Goddard,A. and Goeddel,D.V. (1996) I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc. Natl Acad. Sci. USA, 93, 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwarf D.M., Zandi,E., Natoli,G. and Karin,M. (1998) IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature, 395, 297–300. [DOI] [PubMed] [Google Scholar]
- Rudolph D., Yeh,W.C., Wakeham,A., Rudolph,B., Nallainathan,D., Potter,J., Elia,A.J. and Mak,T.W. (2000) Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev., 14, 854–862. [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Kawai,T., Takeda,K., Matsumoto,M., Inoue,J., Tatsumi,Y., Kanamaru,A. and Akira,S. (1999) IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IκB kinases. Int. Immunol., 11, 1357–1362. [DOI] [PubMed] [Google Scholar]
- Shinkura R., Kitada,K., Matsuda,F., Tashiro,K., Ikuta,K., Suzuki,M., Kogishi,K., Serikawa,T. and Honjo,T. (1999) Alymphoplasia is caused by a point mutation in the mouse gene encoding NF-κB-inducing kinase. Nature Genet., 22, 74–77. [DOI] [PubMed] [Google Scholar]
- Shu H.B., Halpin,D.R. and Goeddel,D.V. (1997) Casper is a FADD- and caspase-related inducer of apoptosis. Immunity, 6, 751–763. [DOI] [PubMed] [Google Scholar]
- Sizemore N., Leung,S. and Stark,G.R. (1999) Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-κB p65/RelA subunit. Mol. Cell. Biol., 19, 4798–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.Y., Regnier,C.H., Kirschning,C., Goeddel,D. and Rothe,M. (1997) Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of NF-κB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc. Natl Acad. Sci. USA, 94, 9792–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K. et al. (1999) Limb and skin abnormalities in mice lacking IKKα. Science, 284, 313–316. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Fuentes,M.E., Yamaguchi,K., Durnin,M.H., Dalrymple,S.A., Hardy,K.L. and Goeddel,D.V. (1999) Embryonic lethality, liver degeneration and impaired NF-κB activation in IKK-β-deficient mice. Immunity, 10, 421–429. [DOI] [PubMed] [Google Scholar]
- Tojima Y. et al. (2000) NAK is an IκB kinase-activating kinase. Nature, 404, 778–782. [DOI] [PubMed] [Google Scholar]
- Vig E., Green,M., Liu,Y., Donner,D.B., Mukaida,N., Goebl,M.G. and Harrington,M.A. (1999) Modulation of tumor necrosis factor and interleukin-1-dependent NF-κB activity by mPLK/IRAK. J. Biol. Chem., 274, 13077–13084. [DOI] [PubMed] [Google Scholar]
- Wang D. and Baldwin,A.S.,Jr (1998) Activation of NF-κB-dependent transcription by TNFα is mediated through phosphorylation of RelA/p65 on serine 529. J. Biol. Chem., 273, 29411–29416. [DOI] [PubMed] [Google Scholar]
- Woronicz J.D., Gao,X., Cao,Z., Rothe,M. and Goeddel,D.V. (1997) IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science, 278, 866–869. [DOI] [PubMed] [Google Scholar]
- Yamaoka S., Courtois,G., Bessia,C., Whiteside,S.T., Weil,R., Agou,F., Kirk,H.E., Kay,R.J. and Israel,A. (1998) Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell, 93, 1231–1240. [DOI] [PubMed] [Google Scholar]
- Yeh W.C. et al. (1997) Early lethality, functional NF-κB activation and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity, 7, 715–725. [DOI] [PubMed] [Google Scholar]
- Yeh W.C., Hakem,R., Woo,M. and Mak,T.W. (1999) Gene targeting in the analysis of mammalian apoptosis and TNF receptor superfamily signaling. Immunol. Rev., 169, 283–302. [DOI] [PubMed] [Google Scholar]
- Zandi E., Rothwarf,D.M., Delhase,M., Hayakawa,M. and Karin,M. (1997) The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell, 91, 243–252. [DOI] [PubMed] [Google Scholar]
- Zhong H., SuYang,H., Erdjument-Bromage,H., Tempst,P. and Ghosh,S. (1997) The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell, 89, 413–424. [DOI] [PubMed] [Google Scholar]
- Zhong H., Voll,R.E. and Ghosh,S. (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell, 1, 661–671. [DOI] [PubMed] [Google Scholar]