

Abstract
Ectopic expression of a defined set of transcription factors chosen from Oct3/4, Sox2, c-Myc, Klf4, Nanog, and Lin28 can directly reprogram somatic cells to pluripotency. These reprogrammed cells are referred to as induced pluripotent stem cells (iPSCs). To date, iPSCs have been successfully generated using lentiviruses, retroviruses, adenoviruses, plasmids, transposons, and recombinant proteins. Nucleic acid-based approaches raise concerns about genomic instability. In contrast, a protein-based approach for iPSC generation can avoid DNA integration concerns as well as provide greater control over the concentration, timing, and sequence of transcription factor stimulation. Researchers recently demonstrated that polyarginine peptide conjugation can deliver recombinant protein reprogramming factor (RF) cargoes into cells and reprogram somatic cells into iPSCs. However, the protein-based approach requires a significant amount of protein for the reprogramming process. Producing fusion reprogramming factors in the large amounts required for this approach using traditional heterologous in vivo production methods is difficult and cumbersome since toxicity, product aggregation, and proteolysis by endogenous proteases limit yields. In this work, we show that cell-free protein synthesis (CFPS) is a viable option for producing soluble and functional transducible transcription factors for nuclear reprogramming. We used an E. coli-based cell-free protein synthesis system to express the above set of six human RFs as fusion proteins, each with a nona-arginine (R9) protein transduction domain. Using the flexibility offered by the CFPS platform, we successfully addressed proteolysis and protein solubility problems to produce full-length and soluble R9-RF fusions. We subsequently showed that R9-Oct3/4, R9-Sox2, and R9-Nanog exhibit cognate DNA binding activities, R9-Nanog translocates across the plasma and nuclear membranes, and R9-Sox2 exerts transcriptional activity on a known downstream gene target.
Keywords: cell-free protein synthesis, induced pluripotent stem cells (iPSC), transduction, nona-arginine, Nanog, Oct3/4, Sox2
Introduction
The Yamanaka and Thomson groups were the first to show that viral expression of a few human transactivating factors is capable of reprogramming human somatic cells into a pluripotent state (Takahashi et al. 2007; Yu et al. 2007). These reprogrammed cells are referred to as induced pluripotent stem cells (iPSCs). The iPSCs self-renew and differentiate into a wide variety of cell types, making them an appealing option for disease- and patient- specific regenerative medicine therapies. Furthermore, iPSCs generated from diseased cells can serve as useful tools for studying disease mechanisms and potential therapies.
iPSCs can be generated by ectopic expression of a set of reprogramming factors (RFs) chosen from Oct3/4, Sox2, c-Myc, Klf4, Lin28, and Nanog (Takahashi et al. 2007; Yu et al. 2007). Oct3/4 and Sox2 are transcription factors that maintain pluripotency in embryonic stem (ES) cells while Klf4 and c-Myc are transcription factors thought to boost iPSC generation efficiency (Jaenisch and Young 2008). The transcription factor c-Myc is believed to modify chromatin structure to allow Oct3/4 and Sox2 to more efficiently access genes necessary for reprogramming while Klf4 enhances the activation of certain genes by Oct3/4 and Sox2 (Nakatake et al. 2006; Jaenisch and Young 2008). Similarly, Nanog and Lin28 may be utilized in iPSC generation. Nanog, like Oct3/4 and Sox2, is a transcription factor that maintains pluripotency in ES cells while Lin28 is an mRNA-binding protein thought to influence the translation or stability of specific mRNAs during differentiation (Balzer and Moss 2007; Jaenisch and Young 2008).
The first iPSCs were made using retroviral and lentiviral constructs (Takahashi et al. 2007; Yu et al. 2007). These viral strategies for overexpressing reprogramming factors can allow integration of exogenous DNA into the genome and may silence indispensable genes and/or induce tumorogenicity (Maherali and Hochedlinger 2008). Though the initial Yamanaka and Thomson iPSCs were unfit for clinical use, they sparked a race in the scientific community to find safer, alternative methods for iPSC generation. There are now a myriad of ways to generate iPSCs. iPSCs have subsequently been produced using adenoviruses (Stadtfeld et al. 2008), plasmids (Okita et al. 2008), and transposons (Kaji et al. 2009; Woltjen et al. 2009). Though the use of adenoviruses, plasmids, and transposons reduce the risk of insertional mutagenesis, they can still potentially modify the genome. Recently, two groups showed that recombinant proteins can be used to generate iPSCs (Zhou et al. 2009; Kim et al. 2009). Zhou et al. expressed murine reprogramming factors fused to an eleven arginine (R11) protein transduction domain (PTD) to reprogram mouse embryonic fibroblasts into iPSCs. Kim et al. used human reprogramming factors fused to a nine arginine (R9) PTD to reprogram human newborn fibroblasts into iPSCs. These recent studies suggest that transient administration of defined amounts of fusion proteins in a controllable fashion provide the most promising route for safer and clinically-feasible iPSC generation from somatic cells.
Arginine-rich PTDs such as HIV Tat and polyarginines have been widely used in the literature to transduce a variety of conjugated fusion proteins across the cell membrane (Nagahara et al. 1998; Cao et al. 2002; Kwon et al. 2005; Landry et al. 2008). Prior to the protein iPSC studies, polyarginines had already been demonstrated to effectively deliver protein cargoes such as RNaseA and p53 into cells (Michiue et al. 2005; Fuchs and Raines 2005). Both R9-RNaseA and R9-p53 traversed the plasma membrane and triggered their designed function, cell death. It is believed that basic residues on the PTDs interact non-specifically with the negatively charged heparan sulfate on cell surfaces (Edenhofer 2008). While smaller PTD peptides can directly transduce the cell membrane, larger PTD conjugates, such as fusion proteins, enter the cell via endocytotic mechanisms (Tunnemann et al. 2006). Substitution experiments have shown that arginine contributes the most toward the internalization process (Wender et al. 2000; Rothbard et al. 2004) and cells deficient in heparan sulfate exhibit impaired protein transduction (Mai et al. 2002). An extensive discussion about the entry mechanisms of arginine-rich PTDs and their applications in protein transduction can be found in a recent review by Frank Edenhofer (Edenhofer 2008).
Many of the PTD fusions described in the literature have been heterologously expressed using Escherichia coli. Expression yields of fusion proteins in vivo are typically low due to product toxicity as well as the inherent challenges of synthesizing and folding a more complex polypeptide. Transcription/transactivating factors add another level of difficulty as heterologous expression often produces low soluble yields due to extensive product aggregation. Glutathione S-Transferase (GST) and Maltose Binding Protein (MBP) solubilization tags provide little benefit (Dyson et al. 2004). Recombinant Oct3/4 and Nanog as well as HIV Tat fusions of Oct3/4 and Sox2 were expressed in E. coli for functional studies, but protein yields were not reported (Loh et al. 2006; Bosnali and Edenhofer 2008). Zhou et al generated murine iPSCs using defined amounts of four R11-fused murine RFs that were refolded from E. coli inclusion bodies. Kim et al generated human iPSCs using crude cell extract from a transformed human cell line that had been transfected with retroviruses encoding the genes of the four R9-fused human RFs. Again, neither group reported protein yields. Thus, the literature suggests that transducible RFs are difficult to produce. Since the protein approach for generating iPSCs inherently requires a significant amount of protein due to extended dosing schedules, alternative production methods are desirable.
We have applied cell-free protein synthesis (CFPS) technology to facilitate the production of transducible, nona-arginine fusion RFs for protein-based iPSC generation (Swartz 2006). CFPS offers an E. coli-based production platform that bypasses the refolding step during the production of transducible RFs. Briefly, E. coli are lysed at high pressures to extract both protein production machinery as well as inner membrane vesicles for energy regeneration. This extract is incubated with template DNA encoding the protein of interest in a chemical environment that mimics the E. coli cytoplasm to yield the protein of interest (Jewett and Swartz 2004). Decoupling protein production from maintenance of host cell health permits production of toxic proteins and also concentrates energy and resources toward synthesis of the protein of interest (Wuu and Swartz 2008). In addition, the cell-free platform is an established system for efficiently producing effective therapeutic fusion protein cancer vaccines (Kanter et al. 2007). Thus, CFPS can potentially address toxicity and aggregation, which are two main roadblocks in fusion protein expression. The open nature of CFPS also enables easy and high-throughput manipulation of protein production parameters for optimizing protein expression conditions.
In this study, we take advantage of the flexibility offered by CFPS and present a platform for the production of fusion proteins for the six RFs described by Thompson and Yamanaka as useful in generating iPSCs, using nona-arginine as the protein transduction domain. The CFPS-produced R9-RF fusion proteins were full-length and soluble. We performed functional studies for a few R9-RFs and showed that they bound cognate DNA in vitro, translocated across cellular membranes, and exhibited transcriptional activity within cells.
Materials and Methods
Plasmid Construction
Genes for six human RFs as well as the DNA sequence encoding the N-terminal R9 fusion peptide (Figure 1) were codon-optimized for expression in E. coli using DNAworks (Hoover and Lubkowski 2002). Full codon-optimized gene sequences can be found in the supplementary materials. In addition, formation of stable mRNA secondary structures near the start codon was minimized using MFOLD (Zuker 2003). MFOLD was used to choose the first nine codons encoding the chloramphenicol acetyltransferase translation enhancer placed at the 5′end of the coding sequence. Overlapping oligonucleotides were designed for PCR-based gene synthesis using DNAworks. A two-step gene synthesis PCR method was utilized to generate RF coding sequences (Reisinger et al. 2006). The N-terminal R9 fusion peptide sequence (Figure 1) was synthesized as part of the Nanog gene, with a 5′ NdeI restriction site containing the start codon and 3′ NheI and SalI sites following the stop codon. The remaining genes were synthesized with 5′ BamHI sites near the start codon and 3′ NheI sites following the stop codon.
Figure 1. Modular Vector Design.

(A) Schematic depiction of the R9 fusion protein design. CAT9: 5′ translation enhancer, consisting of the first nine amino acids of chloramphenicol acetyltransferase (CAT) sequence to destabilize mRNA secondary structure for more efficient initiation of translation (Son et al. 2006), STREP: Strep Tag II purification tag, PCS: Protease cleavage site, Factor Xa was chosen for this construct to enable removal of the translation enhancer, if deemed necessary. R9: Nona-arginine translocation signal, HIS: Hexa-histidine purification tag, LNK: Linker sequence, GGGGS, to physically separate the N-terminal R9 fusion peptide from the RF cargo. Reprogramming Factor Coding Sequence: coding sequence for human transcription factors Oct3/4, Sox2, c-Myc, Klf4, Lin28, and Nanog. (B) DNA and amino acid sequences for the N-terminal R9 fusion peptide on the modular plasmid.
PCR products were first cloned into pCR2.1TOPO (Invitrogen) following the manufacturer’s instructions. The R9-Nanog fusion gene was then cloned into a pET24a (Novagen) expression vector between the T7 promoter and terminator using NdeI and SalI restriction sites. The modular design of the R9-RF fusions enabled facile assembly of the other expression plasmids by replacing the Nanog gene in pET-24a-R9-Nanog with the other RF coding sequences using BamHI and NheI restriction sites. The resulting pET24a-based expression vectors for R9-RF fusions were verified by DNA sequencing (Table 1). Milligram quantities of plasmid were isolated from E. coli cultures grown in Terrific Broth (Invitrogen) using Maxiprep and Gigaprep kits (QIAGEN) according to the manufacturer’s instructions.
Table 1.
List of plasmids used in this study.
| Plasmid name | Gene description | Native protein MW (kDa) | Fusion protein MW (kDa) |
|---|---|---|---|
| pET24a-R9-Nanog | CAT9-StrepTagII-Xa-R9-His6-G4S-Nanog | 34.6 | 39.7 |
| pET24a-R9-Oct3/4 | CAT9-StrepTagII-Xa-R9-His6-G4S-Oct3/4 | 38.4 | 43.5 |
| pET24a-R9-Sox2 | CAT9-StrepTagII-Xa-R9-His6-G4S-Sox2 | 34.3 | 39.4 |
| pET24a-R9-Lin28 | CAT9-StrepTagII-Xa-R9-His6-G4S-Lin28 | 22.7 | 27.8 |
| pET24a-R9-c-Myc | CAT9-StrepTagII-Xa-R9-His6-G4S-c-Myc | 48.8 | 53.9 |
| pET24a-R9-Klf4 | CAT9-StrepTagII-Xa-R9-His6-G4S-Klf4 | 50.1 | 55.2 |
Native MW refers to the molecular weight of the wild-type protein. Fusion MW refers to the molecular weight of the human R9 fusion reprogramming factors with the 5.1 kDa N-terminal R9 fusion peptide and the desired additional conjugates. (See Fig. 1 legend for definitions of abbreviations used in the gene descriptions.)
E. coli Cell Extract Preparation
E. coli KC6 (Calhoun and Swartz 2006), and BL21(DE3)Star (Invitrogen) cell extracts were prepared from cells grown in one of two formats: (1) small-scale shake flasks and (2) 10-liter high-density fermentations. In the shake flask format, cells were grown in 0.5L of defined media (Zawada and Swartz 2005) or 2YT media in 2L shake flasks and harvested during mid-late logarithmic growth at 3 to 6 OD600. In the high-density fermentation format, cells were grown in a B. Braun C-10-2 No. 153 15-L fermentor on defined media with glucose and amino acid feeds using a procedure that promotes logarithmic growth to moderate cell density while avoiding acetate accumulation (Zawada and Swartz 2005). The fermentation was harvested at approximately 30 OD600. Cell growth conditions (shake flask v. high-density, temperatures ranging from 20 °C to 37 °C, defined media v. 2YT) were observed to produce comparable extracts in terms of protein productivity and protease activities at a cell-free expression temperature of 25 °C. In BL21(DE3)Star cultures, 1mM IPTG was added at 0.6 OD600 to induce T7 RNA polymerase (RNAP) expression for CFPS.
Immediately after the growth/expression period, cultures were immediately centrifuged at 5000–8000g for 30 min at 4 °C and washed at 4 °C by resuspending in cold S30 buffer (10 mM Tris-acetate pH 8.2, 14 mM magnesium acetate, and 60 mM potassium acetate) and recentrifuged. The centrifugation and wash procedure was repeated one to two additional times, and the resulting cell paste was stored at −80 °C until it was processed into S30 cell extract. Frozen cell paste was thawed in 0.8 mL of S30 buffer per 1 gram of cell paste and suspended to homogeneity with a Model 700 rotary homogenizer (Fisher Scientific). The cells in suspension were lysed by a single pass through an Emulsiflex C-50 (Avestin) high-pressure homogenizer at 17,500 to 25,000 psi. The homogenate was clarified by centrifugation at 30,000 × g at 4 °C, twice for 30 min each, and the resulting pellets were discarded. The supernatant was then incubated for 80 minutes at 37 °C in the dark on a rotary shaker at 120 rpm. After this incubation, the cell extract was flash-frozen and stored at −80 °C.
Cell-free Production of Fusion Reprogramming Factors
CFPS was conducted using the PANOx-SP (PEP, Amino Acids, nicotinamide adenine dinucleotide (NAD), Oxalic Acid, Spermidine, and Putrescine) system as described previously with minor changes in component concentrations (Jewett and Swartz 2004). The modifications were: 20 mM magnesium glutamate, 0.17 mg/mL folinic acid, 85.3 μg/mL E. coli tRNA mixture (Roche Molecular Biochemicals), 2.7 mM potassium oxalate. Reagents were obtained from Sigma-Aldrich unless otherwise noted. For protease inhibitor studies, one Protease Inhibitor Cocktail tablet (Roche Molecular Biochemicals) was dissolved in 500-μL of sterile water. Protease inhibitor solution was added in place of water typically used to bring CFPS reactions up to volume. CFPS reactions were conducted at 20-μL volumes in 1.5-mL Eppendorf tubes for small-scale diagnostic purposes and at 1-mL volumes in 6-well tissue culture plates (BD Falcon) for preparative purposes. Reactions were carried out at 37 °C, 30 °C or 25 °C for 3 hours.
Quantification of protein yields by liquid scintillation counting
Following the cell-free reaction period, samples were placed on ice to stop the reaction. 3–5 μL of cold reaction mixture was spotted on filter paper and allowed to dry. The remainder of the CFPS reaction was centrifuged at 20,800 × g for 15 minutes at 4 °C to isolate the soluble fraction of the protein product. An equal volume of the supernatant was spotted on filter paper and allowed to dry. Total and soluble protein concentrations were then estimated using the trichloroacetic acid procedure described previously to precipitate the synthesized protein (Calhoun and Swartz 2005). The L-[U-14C]-Leucine radioactivity was quantified by a LS3801 liquid scintillation counter (Beckman Coulter). Total and soluble protein yields were calculated based on incorporated radioactivity and the leucine content of the protein of interest.
Assessment of Proteolysis and Solubility by SDS-PAGE and Autoradiography
3–5 μL of total and soluble protein fractions were loaded onto NuPAGE 10% Bis-Tris gels (Invitrogen) for protein quantification and characterization. Samples were run in MOPS running buffer (Invitrogen) under non-reducing conditions as RFs are non-disulfide bonded. Gels were stained, dried, and exposed to a storage phosphor screen (Molecular Dynamics) which was subsequently scanned using a Typhoon Scanner (GE Healthcare).
Ni-NTA Affinity Chromatography of Fusion Reprogramming Factors for Characterization
Following preparative-scale CFPS reactions, the soluble protein fraction was obtained after centrifugation at 20,800 × g for 15 minutes at 4 °C. The supernatant was dialyzed against 100 volumes of loading buffer (LB, 300 mM NaCl, 10 mM imidazole, 50 mM phosphate buffer, pH 8.0) with 2 buffer changes for 3 hours each at 4 °C and loaded on a 1-mL Ni-NTA column equilibrated with wash buffer (WB, 30 mM imidazole in LB). The column was washed with 6 column volumes (CV) of WB and eluted with increasing imidazole concentrations (1 CV each of 100, 175 and 250 mM imidazole in WB). After pooling appropriate elution fractions, eluate was simultaneously concentrated and buffer-exchanged into a 20% sucrose-PBS formulation using a 10-kDa molecular weight cutoff Amicon Ultra-4 centrifugal device. Protein concentrations were quantified using a DC Protein Assay (BioRad). Protein solutions were flash-frozen in liquid nitrogen and stored at −80 °C until characterization or use.
Assessment of the DNA Binding Activities of the Fusion Reprogramming Factors
The DNA-binding activities of the R9-Nanog, R9-Oct3/4, and R9-Sox2 fusion proteins were assayed by colorimetry utilizing the NoShift Transcription Factor Assay Kit (Novagen) according to the manufacturer’s instructions. To assess sequence-specific binding activity, 1–2μg of the R9-Nanog fusion protein, 2–10μg of the R9-Oct3/4 fusion protein or 1–4μg of the R9-Sox2 fusion protein were each incubated in 20μL with 0.5 μM of their respective biotinylated consensus dsDNA binding targets. The oligos used were: Tcf3 enhancer, Nanog cognate consensus sequence: ACC TGT TAA TGG GAG CGC (Jauch et al. 2008); POU binding site from the construct Conserved Region (CR) 4 in POU5f, Oct3/4 consensus sequence: GCA GAG AGA TGC ATG TGC CGT (Remenyi et al. 2003); HMG binding site from the construct CR4 in POU5f, Sox2 consensus sequence: GCA GAG GAC AAA GGT GCC GTG (Remenyi et al. 2003). Non-biotinylated competitor consensus dsDNA and scrambled dsDNA used to assess competitive binding were added at 2.5 μM. As a positive control, recombinant human (rh) Nanog (Abcam) and rhSox (Peprotech) were used. HRP-conjugated anti-mouse immunoglobulin G was used as a secondary antibody to recognize the anti–Nanog (Abcam), anti-Oct3/4 (R&D Systems), and anti-Sox2 (R&D Systems) mouse monoclonal antibodies. All assays were performed in duplicate. Binding activity was measured via colorimetric absorbance at 450 nm on a Tecan multiwell spectrophotometer using 3,3,5,5-tetramethylbenzidine as the substrate.
Assessment of the Cell Transducibility of the Fusion Reprogramming Factors
Mouse embryonic fibroblasts (MEFs) were seeded on gelatin-coated coverslips (VWR micro cover glass) and grown in fully supplemented Dulbecco’s Modified Eagle Medium (DMEM; GIBCO) overnight at 37 °C in a 5% CO2 incubator. MEFs have a large cytoplasm-to-nucleus ratio that allows for better visualization of protein localization. MEFs have been used to study the cellular uptake of arginine-rich peptides, but the level of heparan sulfate on their cell surfaces has not been quantified (Ziegler et al. 2005; Stewart et al. 2008). After 24h, media was aspirated and cells were washed with phosphate buffered saline (PBS; GIBCO) twice. The R9-Nanog fusion protein was diluted to a final concentration of 500nM with serum-free DMEM and added to the cells. Control groups were either treated with equimolar concentrations rhNanog (Abcam) diluted in serum-free DMEM or simply with serum-free DMEM of equal volume. The cells were incubated at 37 °C for 2 h to allow protein uptake (translocation), washed with PBS and 1 mg/mL heparin (Sigma H4784) to remove proteins attached to the cell surface and then immunostained by fixing them with 4% paraformaldehyde (Alfa Aesar) for 10 min at room temperature (RT). It has been reported in the literature that fixation using 4% formaldehyde or 100% methanol can lead to the redistribution of PTD-constructs within cells, generating fixation-induced artifacts that falsely suggest transduction (Richard et al. 2003). However, fixation using 4% paraformaldehyde does not result in PTD-related fixation-induced artifacts (Shen et al. 2007). Shen et al first incubated cells with fluorescently-labeled arginine-rich peptides. Then, they compared internalization patterns of live and fixed cells. The internalization pattern of cells fixed with 4% paraformaldehyde appeared identical to that of cells. However, fixation with 100% methanol did result in fixation-induced artifacts. Thus, we fixed our cells with 4% paraformaldehyde. Subsequently, the cells were washed with ice-cold PBS twice and permeabilized with 0.25% Triton-X (in PBS) for 10 min at RT. The permeabilized cells were then washed in PBS three times and blocked with a 2% non-fat milk and 10% normal goat serum (Chemicon) mixture for 1 hr at RT. Finally, cells were incubated overnight at 4 °C with mouse anti-human Nanog monoclonal antibody (Abcam; ab62734) diluted (1:250) in blocking solution. In parallel, negative assay controls with no primary antibodies were included. To remove unbound primary antibody, the cells were washed in PBS three times. The cells were then incubated with Alexa-Fluor 594 conjugated goat anti-mouse (1:200) secondary antibody (Molecular Probes; A11032) for 1hr at RT. Excess antibody was washed off and slides were mounted with mounting media containing DAPI (Santa Cruz Biotechnology; sc-24941) and examined by both fluorescence (Nikon Eclipse TE2000-U) and confocal (Zeiss LSM 510 Dual Photon) microscopy.
Assessment of the Transcriptional Activity of the Fusion Reprogramming Factors
The human neonatal foreskin BJ fibroblast cell line (passage ~6) was cultured in DMEM with 10% FBS and 1% penicillin/streptomycin (pen-strep) antibiotics (Invitrogen) in a humidified 5% CO2 incubator at 37°C. The respective cDNAs for Sox2 and GFP were cloned into the retroviral pMX vector and separately transfected into 293FT cells using lipofectamine 2000 (Invitrogen). Viral supernatants were harvested 3 days later, concentrated, and used to infect human BJ fibroblasts in DMEM with 10% FBS and 1% pen/strep. At 80% confluency, BJ fibroblast cells were serum-starved using 1% serum to induce G1 cell cycle arrest. The synchronized BJ fibroblasts were then treated with 200 nM R9-Sox2 fusion protein at 0 hours, 24 hours, 48, and 72 hours. Cellular RNA was extracted for real-time RT-PCR analysis at 0hr and 72hr.
Cultured BJ fibroblasts were collected using TrypLE EXPRESS (Invitrogen) and treated with TRIzol® (Invitrogen). Cellular RNA was purified using an RNeasy Mini Kit (QIAGEN) according to the manufacturer’s recommendations. Purified RNA was then treated by DNase I (QIAGEN) to remove genomic DNA contamination. First-strand cDNA synthesis was performed with 2μg total RNA for each sample in a total volume of 20μl. The reverse transcription reaction was performed with random primers and incubated at 25°C for 10min followed by 42°C for 50 min. Real-time RT-PCR analysis of Myb mRNA was performed using Gene Expression Assays using Taqman assay primers (Applied Biosystems) with 18S serving as an internal control. The TaqMan assay IDs are as follows: Myb assay ID: Hs00193527_m1, and 18S assay ID: Hs99999901_s1. All PCR reactions were performed in a total volume of 20 μl containing 2x TaqMan Universal PCR Master Mix (Applied Biosystems), 20x Gene Expression Assay Mix, and 40ng cDNA. All assays were performed in duplicate and run on an 7300 ABI Real time PCR System using the following conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 sec and 60°C for 1 min. Relative quantitation of the amplified products was based upon Ct values.
Results
Initial Expression of the Fusion Reprogramming Factors
Successful generation of iPSCs requires both intracellular as well as intranuclear delivery of RFs. The six RFs examined here are all transcription factors possessing native nuclear localization sequences (NLS) for nuclear entry. But, they do not contain a sequence for cell entry. Our modular pET24a-based R9-RF expression vector was designed to produce cell transducible proteins from each sub-cloned entity (Figure 1). The R9 sequence is believed to bind to heparan sulfate on the cell surface, thereby triggering cell uptake (Fuchs and Raines 2004). Fusion RFs were initially screened for expression at the 20-μL diagnostic reaction scale using the PanOx-SP CFPS system with E. coli cell extract derived from KC6, an amino acid-stabilized A19 strain, at the standard protein production temperature of 37 °C. Scintillation counting and autoradiograms of CFPS-produced proteins analyzed by SDS-PAGE indicated the accumulation of mostly truncated and insoluble products below the expected molecular weight. Since R9-Nanog exhibited both truncation and solubility problems, it was selected as the model protein for troubleshooting.
Identification and Abrogation of Proteolysis
Production of R9-Nanog using standard conditions yielded two polypeptide populations: one with the expected molecular weight and one with a prominent band approximately 2 kDa below the expected molecular weight (Figure 2A). Lowering the production temperature from 37 °C to 25 °C reduced the intensity of the truncated band, but it was still unclear whether truncation was due to (1) incomplete translation or (2) proteolysis. Suspecting that the R9 sequence offered a vulnerable protease target, we took advantage of the open cell-free system and supplemented the 25 °C reaction with Roche Protease Inhibitor Cocktail. As a result, a single band corresponding to the full-length R9-Nanog fusion protein appeared on the autoradiogram, thereby implicating proteolysis. Though protease inhibitor was an effective diagnostic tool, it was not a practical solution. Not only did it reduce total protein yield, but it also added a significant expense, especially for downstream processing since its removal by dialysis allowed proteases to regain activity. Thus, bacterial cell extract was prepared from E. coli BL21(DE3)Star, a commercially-available strain deficient in OmpT and Lon proteases. By substituting the standard extract with the protease-deficient extract, we significantly reduced proteolysis without the use of protease inhibitors (Figure 2).
Figure 2. Autoradiography analysis of fusion reprogramming factor proteolysis.
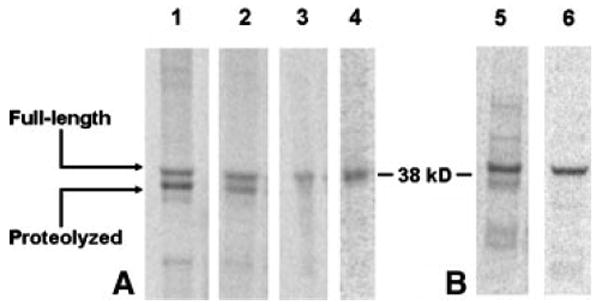
4-μL samples of soluble CFPS product were separated by SDS-PAGE. (A) Analysis of R9-Nanog proteolysis. 1: KC6 extract, expressed at 37 °C. 2: KC6 extract, 25 °C. 3: KC6 + Roche protease inhibitor, 25 °C. 4: BL21(DE3)Star protease-deleted extract, 25 °C. (B) Insight gained from solving R9-Nanog proteolysis is transferable to R9-Sox2. 5: KC6 extract, 37 °C. 6: BL21(DE3)Star protease-deleted extract, 25 °C. Representative images of individual lanes were selected from n=3 experiments.
We were able to apply conditions for producing full-length R9-Nanog to successfully produce other proteolysis-prone R9-RF fusion proteins. R9-Sox2 produced under standard conditions also exhibited lower molecular weight products (Figure 2B). Use of the BL21(DE3)Star extract at 25 °C also effectively curbed R9-Sox2 proteolysis.
Improving Protein Solubility
With proteolysis resolved, we moved on to solubility. Solubility is a problem that all six RFs share. Thus, we evaluated a variety of production schemes for enhancing solubility such as (1) supplementing CFPS reactions with molecular chaperones, (2) supplying the CFPS reaction with the cognate consensus dsDNA of respective RFs, and (3) lowering production temperature.
Often, aggregation of incorrectly-folded polypeptides is the root cause of poor solubility. Overexpression of molecular chaperones has been reported to improve soluble yields (de Marco 2007), but supplementing our RF CPFS reactions with chaperones such as BiP, DnaK, and GroES/GroEL did not improve solubility (data not shown). Further, many proteins undergo conformational changes upon binding their consensus dsDNA (Spolar and Record 1994). However, adding consensus dsDNA to respective R9-Nanog, R9-Oct3/4, and R9-Sox2 CFPS reactions also had no effect on solubility (data not shown). Production of soluble R9-Nanog was screened at 37 °C, 30 °C, and 25 °C. The 25 °C production temperature yielded the best results for R9-Nanog (Figure 3). Combinations of the above remedies did not improve production of soluble protein. In fact, the combinations were deleterious to protein production (data not shown). Of the various permutations tested, only lowering production temperature yielded modest improvements in solubility. Again, we sought to apply this solubility-improving condition to other proteins experiencing similar problems with solubility. R9-Oct3/4 was one of the most recalcitrant proteins in terms of solubility; a major portion of the synthesized product formed insoluble aggregates. As with R9-Nanog, lowering production temperature yielded a nearly two-fold improvement in the accumulation of soluble R9-Oct3/4 (Figure 3).
Figure 3. Autoradiography and scintillation counting analysis of temperature-dependent effects on soluble protein production.
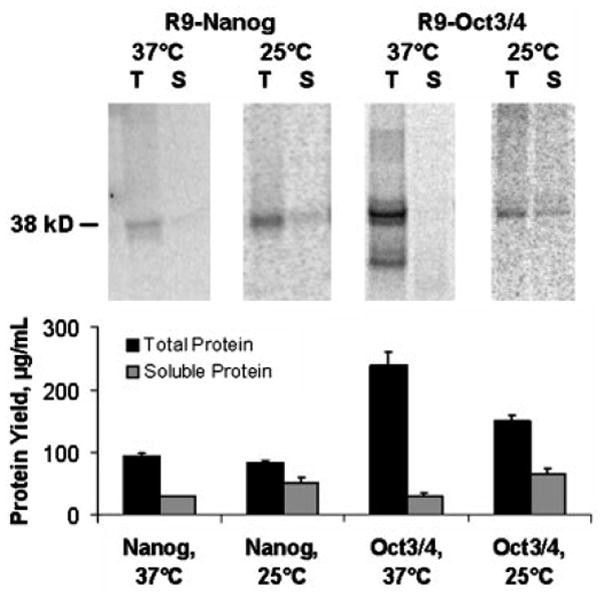
4-μL samples of total (T) and soluble (S) CFPS final reaction mixture product are separated by SDS-PAGE. Lowering R9-Nanog production temperature from 37° C to 25° C resulted in a slight improvement in soluble protein yield. This insight was also applied to R9-Oct3/4 and resulted in a significant improvement in soluble protein yield. Liquid scintillation data was averaged from n=3 experiments while the autoradiogram was selected from a representative experiment among the three.
Generation of Full-length and Soluble Fusion Reprogramming Factors
The proteolysis and solubility studies on R9-Nanog yielded an optimized set of production conditions for the synthesis of full-length and soluble fusion RFs. We applied the optimized conditions to our set of six R9-RFs and were able to curb proteolysis and improve solubility for each RF (Figure 4). While total protein yields range from 100–200 μg/mL, percent solubilities range from 20–40%. Thus, solubility problems still persist.
Figure 4. Autoradiography for Yamanaka and Thomson reprogramming factor R9 fusion proteins produced using CFPS.
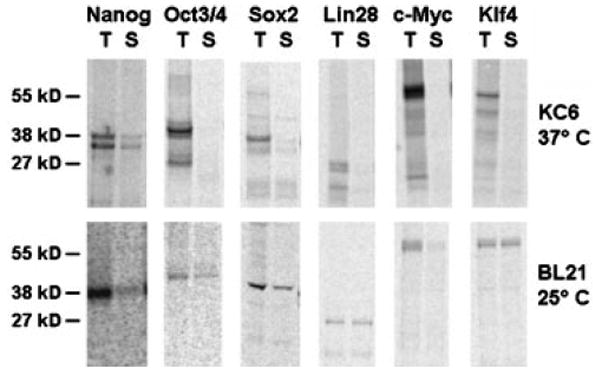
4-μL samples of total (T) and soluble (S) CFPS reaction products were separated by SDS-PAGE. All transcription factors are nona-arginine fusion proteins as described in Figure 1. The top row shows initial results obtained with KC6 extract and 37°C production temperature. The same panel of reprogramming factors was produced with BL21(DE3)Star cell extract at 25°C to generate improve yields of soluble and full-length proteins as shown in the bottom row. Representative images of individual lanes were selected from n=3 experiments.
While the R9- RFs are full-length and soluble, we must verify that (1) the N-terminal R9 fusion peptide does not interfere with the RFs’ DNA binding function and (2) that R9 confers cell transducibility. In order to produce sufficient amounts of protein for characterization studies, we scaled up the production of R9-Nanog, R9-Sox2, and R9-Oct3/4 using the optimized conditions described above. Scintillation counting of the product showed comparable soluble protein yields from 20-μL and 1-mL reactions (Figure 5). In order to produce sufficient amounts of protein, multiple 1-mL reactions could be easily co-processed. In this work, 80-mL batches were processed. Following protein synthesis, reaction mixtures were first centrifuged and product was isolated using a Ni-NTA column. Eluted protein samples were dialyzed into 20% sucrose-PBS, flash frozen, and stored at −80 °C before further characterization.
Figure 5. Scalable cell-free production of soluble R9-Nanog, R9-Sox2, and R9-Oct3/4 for characterization studies.
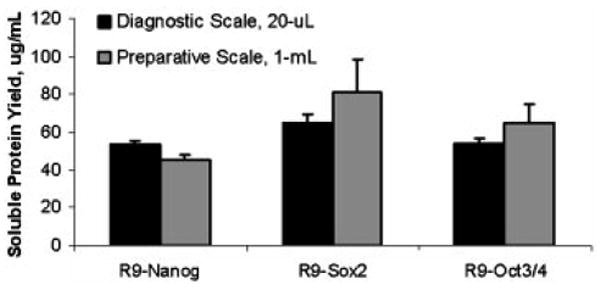
Multiple 1-mL reactions were co-processed at up to 80-mL total volumes to obtain greater amounts of fusion protein for characterization studies (n=3).
Fusion Reprogramming Factors Exhibit DNA Binding Activity
The NoShift Assay (Novagen) was used to verify the DNA binding activities of R9-Nanog, R9-Sox2, and R9-Oct3/4. The No-Shift Assay is a plate-based alternative to the electrophoretic mobility shift assay (EMSA), and is based on the same principles as EMSA. Briefly, fusion RFs and commercial recombinant protein positive controls were each incubated with their corresponding biotinylated cognate consensus dsDNA binding sequences. Protein-DNA complexes were then bound on a streptavidin-coated 96-well plate, and unbound complexes were washed away. Monoclonal primary antibodies specific for the RFs and fluorescently-labeled polyclonal secondary antibodies were used to probe for the bound complexes.
Fusion RFs behave similarly to their corresponding commercial recombinant proteins (Figure 6). The cell-free extract, which served as a negative control, did not yield a signal (data not shown). Co-incubation with non-biotinylated consensus DNA sequences lowered the binding signal obtained with the proteins and biotinylated consensus sequence, which suggests that the DNA binding was specific and competitive. The binding of R9-RFs to their consensus sequences was comparable to that of the commercially available transcription factors. Further, co-incubation of the biotinylated consensus DNA with non-biotinylated scrambled nonsense DNA did not diminish the level of the protein-DNA complex. These results show that the R9-RFs exhibit DNA binding specificity.
Figure 6. Competitive analysis (NoShift assay) of R9-Nanog (A), R9-Oct3/4 (B), R9-Sox2 (C) showing that nona-arginine fusion proteins exhibit cognate DNA binding activity.
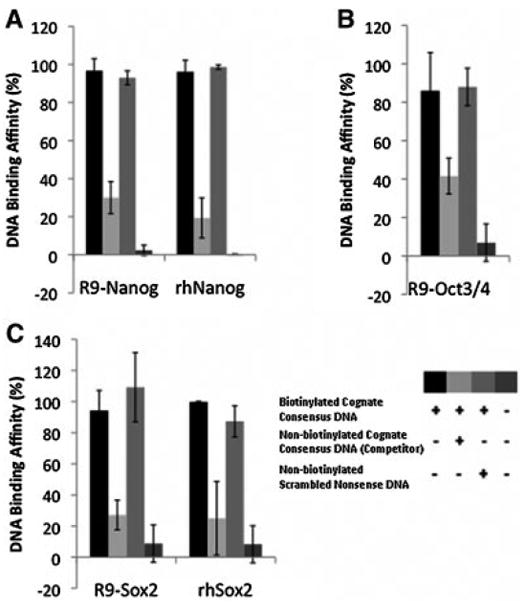
When incubated with a biotinylated cognate consensus sequence, R9-fusion protein-DNA binding was observed. Specific non-biotinylated competitor DNA with the cognate consensus sequence significantly reduced the binding activity in each R9-fusion protein, confirming sequence-specificity of the assay for R9-fusion protein binding. Non-biotinylated scrambled nonsense sequences had no effect on R9-fusion protein binding. As a positive control, human recombinant proteins (i.g. rhNanog and rhSox2) were used.
R9 Fusion Construct Successfully Transduces the Plasma Membrane
Cellular translocation studies were performed to verify that the N-terminal R9 confers cell transducibility. R9-Nanog was chosen to demonstrate the cell-transducing ability of our R9 fusion construct. Briefly, R9-Nanog was incubated with MEFs for 2h after which non-internalized R9-Nanog was washed away. Cells were fixed and stained with primary antibodies specific for Nanog and fluorescently-labeled secondary antibodies. Internalization was visualized using fluorescence and confocal microscopy.
Fluorescence and confocal microscopy analyses suggest that the R9-Nanog effectively enters the cells (Figure 7). We estimate that 85% of the cells contain fluorescent signal, which is in agreement with transducible protein literature (Tunnemann et al. 2006; Bosnali and Edenhofer 2008). Neither commercial recombinant Nanog nor the DMEM-treated control cells showed staining in any cellular compartments. In the time course used in these studies, the R9-Nanog signal was predominantly observed as granular dots in cytoplasmic and perinuclear locations.
Figure 7. R9-Nanog translocation in mouse embryonic fibroblasts.
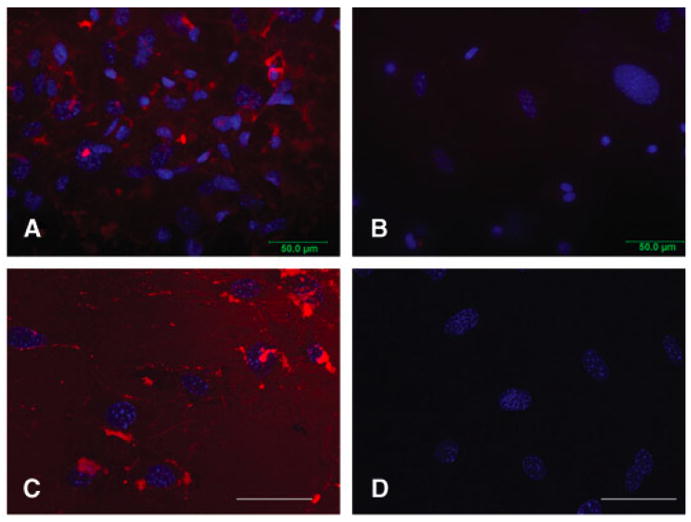
Cells were treated with 0.5μM R9-Nanog for 2 hrs at 37 °C. Red indicates Nanog by Alexa-Fluor 594 labeled antibody detection and blue indicates nuclei by DAPI stain. (A) Fluorescence microscopy of R9-Nanog-treated cells. (B) Fluorescence microscopy of cells treated with commercial rh-Nanog. Fluorescence Microscopy; 40X magnification. (C) Confocal microscopy of R9-Nanog-treated cells. (D) Confocal microscopy of cells treated with commercial rh-Nanog. Confocal Microscopy; 63X magnification; scale bar denotes 50 μm.
R9 Fusion Construct Exhibits Transcriptional Activity
To assess the biological activity of our R9-RF construct within cells, we assessed the effect of R9-Sox2 on the transcription of one of its known downstream gene targets, Myb. Sox2 has been shown to upregulate Myb, which plays an important role in maintaining pluripotentiality (Sharov et al. 2008). We exposed human fibroblasts to the R9-Sox2 fusion protein and detected the change in the mRNA level of Myb using quantitative real-time RT-PCR. We measured effect of R9-Sox2, vehicle, empty retroviral vector (pMX-GFP), and retroviral Sox2 (pMX-Sox2) on BJ fibroblast expression of Myb for 72 hrs. Transfection with a retroviral construct encoding Sox2 served as a positive control and transfection with a retroviral construct encoding green fluorescent protein (GFP) served as a negative control. R9-Sox2 treatment and positive control retroviral Sox2 transfection both upregulated Myb gene expression, while the negative control retroviral GFP transfection had no effect on Myb expression (Figure 8). Thus, these gene expression data confirm that transducible R9-Sox2 fusion proteins produced by CFPS are capable of nuclear-entry and are biologically active in BJ fibroblast cells.
Figure 8. R9-Sox2 transcriptional activity.
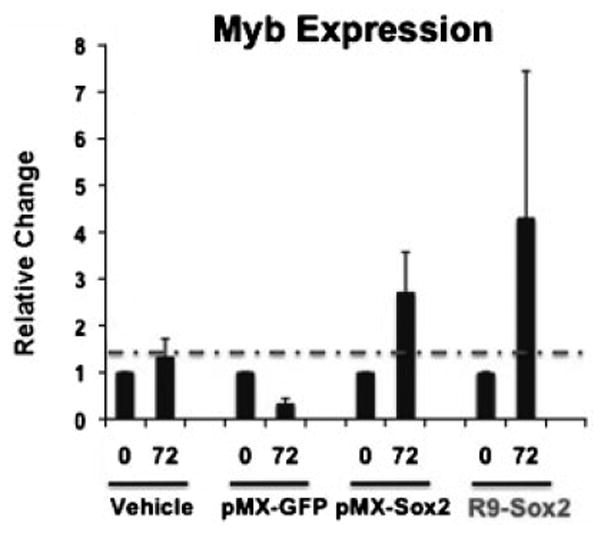
Fold differences in Myb gene expression were detected using real-time RT-PCR on RNA isolated from human BJ fibroblast cells that had been treated with vehicle, pMX-GFP (negative control), pMX-Sox2 (positive control), and 200 nM R9-Sox2 transducible transcription factor (n=2).
Discussion
Producing transducible transcription factors using traditional E. coli recombinant protein methods is difficult. Evidence from the literature supports this claim, and we have experienced it firsthand as well. We have also attempted to express our R9-RFs in vivo using E. coli strain BL21(DE3)Star (Invitrogen) LB cultures grown at temperatures ranging from 18 °C to 37 °C and induced with IPTG concentrations ranging from 0.1 to 1 mM. After cell harvest and lysis, we applied centrifuged lysate supernatants containing the soluble protein fraction to Ni-NTA resin. However, we were not able to recover any soluble R9-RFs.
In the past, CFPS has enabled the soluble production of proteins that were difficult to make in E. coli (Yang et al. 2005; Goerke et al. 2008). In this study, we were able to use CFPS to produce appreciable amounts of full-length, soluble, and transducible nona-arginine fusion RFs that exhibit DNA-binding activity. Proteolysis was curtailed while solubility was enhanced. Lowering temperature reduced proteolysis and also improved product solubility. Transcription and translation rates are slower at lower temperatures, providing the growing polypeptide more time to explore the protein folding landscape and find its correct conformation (Kiefhaber et al. 1991).
Though our results are encouraging, there is room for improvement. Enhancing solubility is a key priority as we are currently losing 60–80% of the total protein produced to insoluble aggregates. Thus, protein refolding studies are underway in order to recover functional R9-RF fusion proteins from insoluble aggregates. We hope to gain insights into measures that will improve proper folding, which can be transferred into our cell-free production environment.
Despite these solubility limitations, cell-free technology has enabled us to obtain appreciable amounts of protein for characterization studies. We used the NoShift Assay to show that R9 fusions of Nanog, Oct3/4, and Sox2 can bind cognate DNA. TAT fusions of Oct3/4 and Sox2 have been shown to bind cognate DNA using EMSA (Bosnali and Edenhofer 2008), but we chose to use the EMSA-based NoShift Assay because it is relatively rapid and avoids the use of strong ionizing radiation. The results with non-biotinylated consensus DNA and the scrambled DNA show the proteins’ specificity for their respective binding partners. The data also show that the N-terminal R9 fusion peptide does not affect DNA-binding.
Successful translocation of R9-Nanog into MEFs shows the R9 portion of the fusion protein is functional. Confocal microscopy shows that R9-Nanog crosses the plasma membrane. We estimate 85% of the cells contain R9-Nanog, but it is not known what fraction of the 500nM R9-Nanog that was administered transduced the cell membrane. To the best of our knowledge, there have been no reports in the literature that quantify the fraction internalized. However, determining this fraction is the subject of on-going studies in our laboratories. Intracellular delivery of R9-Nanog implies that the R9 internalization tag is accessible on the protein construct and has served its purpose. The cytoplasmic and perinuclear localization observed with R9-Nanog is in agreement with the PTD literature that describes the use of reprogramming factors. In the two recent successful reprogramming studies involving R11 and R9 fusions of Oct3/4, Sox2, c-Myc, and Klf4, the fusion transcription factors also localize near the nucleus (Kim et al. 2009; Zhou et al. 2009). TAT is also an arginine-rich PTD; thus, TAT fusions of Oct3/4 and Sox2 similarly localize in granular pockets of fluorescent signal in and around the nucleus (Bosnali and Edenhofer 2008). It is hypothesized that the positively-charged R9 and other PTDs bind non-specifically with heparan sulfate on cell surfaces (Fuchs and Raines 2004). This binding event triggers macropinocytosis of the PTD fusions, thereby placing the internalized PTD fusions into endosomal vesicles (Bosnali and Edenhofer 2008; Caron et al. 2004; Tunnemann et al. 2006). The granular appearance of our R9-Nanog signal suggests that the fusion protein follows the proposed endosomal sequestration model.
Nevertheless, preliminary functional studies in our laboratories indicate that a small fraction of the sequestered protein escapes the endosomes and alters gene transcription. Sox2 has been shown to upregulate the expression of Myb (Sharov et al. 2008). In this work, we showed that exogenously delivered R9-Sox2 can also upregulate the expression of Myb. Although we observed quantitative differences in the level of gene expression induced by the viral or protein delivery of exogenous Sox2, the directional changes in gene expression were the same and significantly different from the cells treated with media alone (vehicle control) or the empty vector (pMX-GFP). These data imply that the R9-RF construct shown in this work is capable of translocating across the cell membrane and is functional in a cellular context.
In order to enhance our non-viral fusion protein approach for generating iPSCs, methods to increase endosomal escape are under investigation. Endosomal escape is possible with the addition of endosomolytic chemicals (Shiraishi et al. 2005), but it is unclear what effects the chemicals will exert on the reprogramming process. The optimal scenario calls for an endosomolytic domain to be present in the N-terminal R9 fusion peptide. Endosomal escape via fusion partners appears possible. For example, fusogenic influenza HA2 facilitated endosomal escape for its fusion cargoes (Michiue et al. 2005; Wadia et al. 2004). However, the efficacy of such endosomolytic domains, and their effect on the other functions of the fusion proteins, is unpredictable. Accordingly, a strategy of directed empiricism is required to engineer nuclear reprogramming factors that can translocate across the cell membrane, escape the endosome, enter the nucleus, and interact properly with their respective consensus sequences.
Our findings achieve the first step for realizing a non-viral fusion protein approach for iPSC generation. We have developed CFPS reactions that enable the production of significant quantities of fusion R9-RFs. We encountered considerable proteolysis and solubility problems and addressed them to produce full-length and soluble fusion R9-RFs. We have shown that a subset of these fusion R9-RFs exhibit specific binding to their consensus dsDNA sequences, translocate across cellular membranes, and exert transcriptional influence within cells. Work is already underway in our laboratories to further enhance R9-RF solubility as well as enhance R9-RF endosomal escape so as to effectively and safely generate iPSCs using fusion protein RFs.
Acknowledgments
The authors would like to thank Carol Toman and Sharon Long for providing the GroESL plasmid, John Welsh for help with solubilization experiments, and Jon Kuchenreuther for providing the initial batch of BL21(DE3) extract. WCY is a recipient of a NDSEG fellowship and a NSF fellowship. This study was also supported by grants to JPC from the National Institutes of Health (R01 HL75774, R01 CA098303, R03 HL096238, R21 HL085743, 1K12 HL087746, 1P50HL083800), the California Tobacco Related Disease Research Program of the University of California (1514RT-0169 ), the California Institute for Regenerative Medicine (RS1-00183), and the Stanford Cardiovascular Institute.
References
- Balzer E, Moss EG. Localization of the developmental timing regulator Lin28 to mRNP complexes, P-bodies and stress granules. RNA Biol. 2007;4(1):16–25. doi: 10.4161/rna.4.1.4364. [DOI] [PubMed] [Google Scholar]
- Bosnali M, Edenhofer F. Generation of transducible versions of transcription factors Oct4 and Sox2. Biol Chem. 2008;389(7):851–61. doi: 10.1515/BC.2008.106. [DOI] [PubMed] [Google Scholar]
- Calhoun KA, Swartz JR. An economical method for cell-free protein synthesis using glucose and nucleoside monophosphates. Biotechnol Prog. 2005;21(4):1146–53. doi: 10.1021/bp050052y. [DOI] [PubMed] [Google Scholar]
- Calhoun KA, Swartz JR. Total amino acid stabilization during cell-free protein synthesis reactions. J Biotechnol. 2006;123(2):193–203. doi: 10.1016/j.jbiotec.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Cao G, Pei W, Ge H, Liang Q, Luo Y, Sharp FR, Lu A, Ran R, Graham SH, Chen J. In Vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J Neurosci. 2002;22(13):5423–31. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron NJ, Quenneville SP, Tremblay JP. Endosome disruption enhances the functional nuclear delivery of Tat-fusion proteins. Biochem Biophys Res Commun. 2004;319(1):12–20. doi: 10.1016/j.bbrc.2004.04.180. [DOI] [PubMed] [Google Scholar]
- de Marco A. Protocol for preparing proteins with improved solubility by co-expressing with molecular chaperones in Escherichia coli. Nat Protoc. 2007;2(10):2632–9. doi: 10.1038/nprot.2007.400. [DOI] [PubMed] [Google Scholar]
- Dyson MR, Shadbolt SP, Vincent KJ, Perera RL, McCafferty J. Production of soluble mammalian proteins in Escherichia coli: identification of protein features that correlate with successful expression. BMC Biotechnol. 2004;4:32. doi: 10.1186/1472-6750-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenhofer F. Protein transduction revisited: novel insights into the mechanism underlying intracellular delivery of proteins. Curr Pharm Des. 2008;14(34):3628–36. doi: 10.2174/138161208786898833. [DOI] [PubMed] [Google Scholar]
- Fuchs SM, Raines RT. Pathway for polyarginine entry into mammalian cells. Biochemistry. 2004;43(9):2438–44. doi: 10.1021/bi035933x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SM, Raines RT. Polyarginine as a multifunctional fusion tag. Protein Sci. 2005;14(6):1538–44. doi: 10.1110/ps.051393805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goerke AR, Loening AM, Gambhir SS, Swartz JR. Cell-free metabolic engineering promotes high-level production of bioactive Gaussia princeps luciferase. Metab Eng. 2008;10(3–4):187–200. doi: 10.1016/j.ymben.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30(10):e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132(4):567–82. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauch R, Ng CK, Saikatendu KS, Stevens RC, Kolatkar PR. Crystal structure and DNA binding of the homeodomain of the stem cell transcription factor Nanog. J Mol Biol. 2008;376(3):758–70. doi: 10.1016/j.jmb.2007.11.091. [DOI] [PubMed] [Google Scholar]
- Jewett MC, Swartz JR. Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol Bioeng. 2004;86(1):19–26. doi: 10.1002/bit.20026. [DOI] [PubMed] [Google Scholar]
- Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458(7239):771–5. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter G, Yang J, Voloshin A, Levy S, Swartz JR, Levy R. Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 2007;109(8):3393–9. doi: 10.1182/blood-2006-07-030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefhaber T, Rudolph R, Kohler HH, Buchner J. Protein aggregation in vitro and in vivo: a quantitative model of the kinetic competition between folding and aggregation. Biotechnology (N Y) 1991;9(9):825–9. doi: 10.1038/nbt0991-825. [DOI] [PubMed] [Google Scholar]
- Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4(6):472–6. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YD, Oh SK, Kim HS, Ku SY, Kim SH, Choi YM, Moon SY. Cellular manipulation of human embryonic stem cells by TAT-PDX1 protein transduction. Mol Ther. 2005;12(1):28–32. doi: 10.1016/j.ymthe.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Landry JR, Kinston S, Knezevic K, de Bruijn MF, Wilson N, Nottingham WT, Peitz M, Edenhofer F, Pimanda JE, Ottersbach K, et al. Runx genes are direct targets of Scl/Tal1 in the yolk sac and fetal liver. Blood. 2008;111(6):3005–14. doi: 10.1182/blood-2007-07-098830. [DOI] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38(4):431–40. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Maherali N, Hochedlinger K. Guidelines and techniques for the generation of induced pluripotent stem cells. Cell Stem Cell. 2008;3(6):595–605. doi: 10.1016/j.stem.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Mai JC, Shen H, Watkins SC, Cheng T, Robbins PD. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J Biol Chem. 2002;277(33):30208–18. doi: 10.1074/jbc.M204202200. [DOI] [PubMed] [Google Scholar]
- Michiue H, Tomizawa K, Wei FY, Matsushita M, Lu YF, Ichikawa T, Tamiya T, Date I, Matsui H. The NH2 terminus of influenza virus hemagglutinin-2 subunit peptides enhances the antitumor potency of polyarginine-mediated p53 protein transduction. J Biol Chem. 2005;280(9):8285–9. doi: 10.1074/jbc.M412430200. [DOI] [PubMed] [Google Scholar]
- Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4(12):1449–52. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- Nakatake Y, Fukui N, Iwamatsu Y, Masui S, Takahashi K, Yagi R, Yagi K, Miyazaki J, Matoba R, Ko MS, et al. Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol Cell Biol. 2006;26(20):7772–82. doi: 10.1128/MCB.00468-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322(5903):949–53. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- Reisinger SJ, Patel KG, Santi DV. Total synthesis of multi-kilobase DNA sequences from oligonucleotides. Nat Protoc. 2006;1(6):2596–603. doi: 10.1038/nprot.2006.426. [DOI] [PubMed] [Google Scholar]
- Remenyi A, Lins K, Nissen LJ, Reinbold R, Scholer HR, Wilmanns M. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev. 2003;17(16):2048–59. doi: 10.1101/gad.269303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126(31):9506–7. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- Sharov AA, Masui S, Sharova LV, Piao Y, Aiba K, Matoba R, Xin L, Niwa H, Ko MS. Identification of Pou5f1, Sox2, and Nanog downstream target genes with statistical confidence by applying a novel algorithm to time course microarray and genome-wide chromatin immunoprecipitation data. BMC Genomics. 2008;9:269. doi: 10.1186/1471-2164-9-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Liang K, Ye Y, Tetteh E, Achilefu S. Modulation of nuclear internalization of Tat peptides by fluorescent dyes and receptor-avid peptides. FEBS Lett. 2007;581(9):1793–9. doi: 10.1016/j.febslet.2007.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi T, Pankratova S, Nielsen PE. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chem Biol. 2005;12(8):923–9. doi: 10.1016/j.chembiol.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Son JM, Ahn JH, Hwang MY, Park CG, Choi CY, Kim DM. Enhancing the efficiency of cell-free protein synthesis through the polymerase-chain-reaction-based addition of a translation enhancer sequence and the in situ removal of the extra amino acid residues. Anal Biochem. 2006;351(2):187–92. doi: 10.1016/j.ab.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263(5148):777–84. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322(5903):945–9. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart KM, Horton KL, Kelley SO. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org Biomol Chem. 2008;6(13):2242–55. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- Swartz J. Developing cell-free biology for industrial applications. J Ind Microbiol Biotechnol. 2006;33(7):476–85. doi: 10.1007/s10295-006-0127-y. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tunnemann G, Martin RM, Haupt S, Patsch C, Edenhofer F, Cardoso MC. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006;20(11):1775–84. doi: 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10(3):310–5. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97(24):13003–8. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hamalainen R, Cowling R, Wang W, Liu P, Gertsenstein M, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–70. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuu JJ, Swartz JR. High yield cell-free production of integral membrane proteins without refolding or detergents. Biochim Biophys Acta. 2008;1778(5):1237–50. doi: 10.1016/j.bbamem.2008.01.023. [DOI] [PubMed] [Google Scholar]
- Yang J, Kanter G, Voloshin A, Michel-Reydellet N, Velkeen H, Levy R, Swartz JR. Rapid expression of vaccine proteins for B-cell lymphoma in a cell-free system. Biotechnol Bioeng. 2005;89(5):503–11. doi: 10.1002/bit.20283. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zawada J, Swartz J. Maintaining rapid growth in moderate-density Escherichia coli fermentations. Biotechnol Bioeng. 2005;89(4):407–15. doi: 10.1002/bit.20369. [DOI] [PubMed] [Google Scholar]
- Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4(5):381–4. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler A, Nervi P, Durrenberger M, Seelig J. The cationic cell-penetrating peptide CPP(TAT) derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochemistry. 2005;44(1):138–48. doi: 10.1021/bi0491604. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]