

Abstract
The Ena-VASP family of proteins act as molecular adaptors linking the cytoskeletal system to signal transduction pathways. Their N-terminal EVH1 domains use groups of exposed aromatic residues to specifically recognize ‘FPPPP’ motifs found in the mammalian zyxin and vinculin proteins, and ActA protein of the intracellular bacterium Listeria monocytogenes. Here, evidence is provided that the affinities of these EVH1–peptide interactions are strongly dependent on the recognition of residues flanking the core FPPPP motifs. Determination of the VASP EVH1 domain solution structure, together with peptide library screening, measurement of individual Kds by fluorescence titration, and NMR chemical shift mapping, revealed a second affinity-determining epitope present in all four ActA EVH1-binding motifs. The epitope was shown to interact with a complementary hydrophobic site on the EVH1 surface and to increase strongly the affinity of ActA for EVH1 domains. We propose that this epitope, which is absent in the sequences of the native EVH1-interaction partners zyxin and vinculin, may provide the pathogen with an advantage when competing for the recruitment of the host VASP and Mena proteins in the infected cell.
Keywords: ActA/EVH1 domain/FPPPP motif/protein complex/zyxin
Introduction
The cytoskeletal and focal adhesion associated protein VASP (vasodilator-stimulated phosphoprotein) was initially characterized as a substrate of both cGMP- and cAMP-dependent protein kinases (cGPK, cAPK), and as a component of inhibitory signal transduction pathways in human platelets and other cells (Halbrügge and Walter, 1989; Halbrügge et al., 1990; Reinhard et al., 1999). It has since been shown to be necessary for the enhancement of spatially confined actin polymerization, thus modulating filament formation, and hence cytoskeletal rearrangements, in response to extracellular signals (Chakraborty et al., 1995; Pistor et al., 1995; Beckerle, 1998; Dramsi and Cossart, 1998; Laurent et al., 1999; Vasioukhin et al., 2000). A tripartite structural organization was originally predicted for VASP from the delineated amino acid sequences of human, canine and murine VASP cDNAs, and confirmed by the subsequent identification of the Drosophila Enabled (Ena) protein, its mammalian homologue Mena and the Ena-VASP-like protein, Evl (Haffner et al., 1995; Gertler et al., 1995, 1996; Zimmer et al., 1996). The proteins share highly homologous N- and C-terminal domains, known as the Ena-VASP homology domains 1 and 2 (EVH1 and EVH2), which are separated by a more divergent proline-rich region (Gertler et al., 1996; Reinhard et al., 1999). A number of cytoskeletal proteins have been identified as direct binding partners for each of the three domains. The N-terminal EVH1 domain is responsible for interactions with proteins associated with focal adhesion plaques (Reinhard et al., 1995a, 1996; Brindle et al., 1996; Gertler et al., 1996; Niebuhr et al., 1997), the central proline-rich region interacts with profilins and various SH3 domains (Reinhard et al., 1995b; Ahern-Djamali et al., 1998), and the C-terminal EVH2 domain interacts with polymerized actin fibres, as well as being involved in the tetramerization of the protein (Bachmann et al., 1999; Laurent et al., 1999).
The EVH1 domains of VASP and Mena are known to bind the mammalian focal adhesion proteins zyxin and vinculin, and the ActA surface protein of the intracellular pathogen Listeria monocytogenes (Reinhard et al., 1995a, 1996; Niebuhr et al., 1997). ActA is essential for actin-based motility in Listeria (Domann et al., 1992; Kocks et al., 1992; Sanger et al., 1992; Pistor et al., 1994, 1995), localizing VASP and Mena to the bacterial surface where they promote the rapid polymerization of an actin comet tail that propels the pathogen through the cytoplasm. Microinjection of FPPPPT-containing peptides displaces VASP and Mena from their normal locations in focal adhesions showing that both proteins recognize FPPPP motifs in their binding partners (Pistor et al., 1995; Gertler et al., 1996; Niebuhr et al., 1997). Deletion of the FPPPP repeat region in ActA leads to delocalization of VASP and Mena from the listerial surface, resulting in a visible reduction in intracellular motility and strongly attenuating Listeria virulence (Niebuhr et al., 1997). Peptide competition experiments have also revealed qualitative data showing that ActA binds VASP more strongly than zyxin (Niebuhr et al., 1997). Interestingly, the zyxin and ActA sequences contain four FPPPP motifs in close tandem repeat, whereas in vinculin oligomers, each monomer contains only one. Multiplicity of binding sites thus provides a possible mechanism by which the oligomeric state of VASP and Mena proteins could be used to regulate their affinity for zyxin, ActA or vinculin by co-operative binding (Bachmann et al., 1999; Laurent et al., 1999).
Multiple sequence alignments (Callebaut et al., 1998) show that the closest relatives to EVH1 domains are the WASP homology 1 (WH1) domain and the Ran-binding domain 1 (RanBD1). The NMR and X-ray structures of the EVH1 domain (this work; Fedorov et al., 1999; Prehoda et al., 1999) show a close structural relationship to the pleckstrin homology (PH) and phosphotyrosine-binding (PTB) domains, despite very low sequence similarities. The crystal structures of the Mena and Evl EVH1 domains in complex with the ActA-derived FPPPPT and FEFPPPPTDEE peptides yielded information on the interactions formed between the core FPPPPT motif and specific residues of the EVH1 domain, but no electron density was observed for the peptide residues flanking the FPPPPT core. Extended ActA-derived peptides are known to bind EVH1 domains more strongly than small, truncated subsequences (Niebuhr et al., 1997), suggesting that important specificity and affinity determinants exist within the residues flanking the FPPPPT core. These interactions in the complex may also determine the polarity of binding. Understanding the roles of the various core-flanking residues for EVH1 recognition are therefore of particular interest for elucidating the mechanisms by which the FPPPP motifs of host cell proteins compete for EVH1 binding within the cell.
To understand better the mode of peptide binding, we therefore solved the structure of the human VASP EVH1 domain by NMR spectroscopy. A complete dataset of NMR experiments were also acquired on a 1:1 complex of 13C,15N-labelled VASP EVH1 with the unlabelled ActA peptide, 332SFEFPPPPTEDEL344. However, protein– peptide NOEs in the standard set of 2D and 3D NOESY spectra were not detected. We therefore acquired a series of 2D 13C-half filtered NOESY experiments (Gemmecker et al., 1992; Ikura and Bax, 1992) at a range of different mixing times (50–200 ms) in an attempt to record specifically NOEs between the 13C-attached protons (of the protein) and the 12C-attached protons (of the peptide). In addition, a complete series of transfer NOE experiments were acquired at a number of different peptide:protein ratios (from 1:1 to 50:1) and over a range of mixing times (30–500 ms), but direct NOEs between the protein and peptide in the complex were never observed. The reason for the absence of direct intermolecular NOEs no doubt lies in the very weak binding affinity and fast exchange properties of the EVH1–peptide interaction. This behaviour is not unusual in interactions between proteins and proline-rich ligands. The presence of multiple species in the equilibrium of a fast, multi-step binding process would not allow sufficient time for protein–peptide NOEs to build up to detectable levels.
Whilst the observation of NOEs between peptide and protein is desirable for determining efficiently and precisely the interactions between ligand and domain, similar information can be obtained by combining a number of alternative methods. The interaction surface can also be defined by mapping the chemical shift perturbations of individual nuclei from each of the interacting partners. In order to pinpoint which residues are responsible for which interactions, it is necessary to map a domain surface with several variants of the same peptide and to observe the differences in chemical shift perturbations arising with each change. In this work, 15N-HSQC spectra were used to map chemical shift perturbations of the backbone and sidechain protein 15N–1HN resonances in the presence of three different peptides, derived from the third FPPPP repeat of the ActA protein. The interaction sites of the peptide were defined by monitoring the chemical shift perturbations of the peptide 1H signals in a separate set of 13C-(ω1)–13C-(ω2) double half-filtered 1H TOCSY and NOESY experiments. The chemical shift mapping, taken together with the results of peptide library scans, which showed the effect of each peptide residue on the binding affinity, and the determination of Kds for individual peptides of significant interest by fluorescence spectroscopy, enabled us to model the ActA peptide onto our structure of the EVH1 domain and thus draw conclusions about the interactions and functions of the FPPPP core-flanking residues.
Results
Solution structure of the EVH1 domain from human VASP
The structure of the VASP EVH1 domain (Figure 1) was determined using triple-resonance, multidimensional heteronuclear NMR spectroscopy on 15N- and 13C,15N-labelled protein samples. Complete assignment of carbon, proton and nitrogen resonances was achieved as described previously (Ball et al., 1997), and the final ensemble of structures was calculated by simulated annealing on the basis of 3117 NOE distance, dihedral angle and hydrogen-bond restraints (see Table I). The molecule possesses a β-barrel fold similar to that of PH and PTB domains (Saraste et al., 1995), with seven β-strands packed together to form an antiparallel β-sandwich closed along one edge by a long, C-terminal α-helix. The hydrophobic core is formed by the conserved residues Ile6, Ala11, Val13, Val36, Ile38, Phe47, Val49, Val61, Ile62, Ile66, Val70, Tyr72, Trp82, Trp89, Leu91, Phe93, Phe102, Met106, Ala109 and Leu110 (VASP numbering). The very highly conserved, non-core aromatic residues Tyr16, Trp23 and Phe79 form a triad, with their exposed rings arranged in a ladder on the surface of the protein, presenting a peptide recognition groove between the loops joining β1–β2 and β5–β6. In contrast, PH and PTB domains each possess an additional element of secondary structure that occupies this groove via an intramolecular interaction. The groove remains unoccupied in EVH1 domains and is therefore available for peptide binding.

Fig. 1. Superposition of the backbone (N, Cα and C′) atoms for the 15 lowest energy structures of the VASP EVH1 domain. Core residues are shown in orange and the residues of the aromatic triad, Tyr16, Trp23, Phe79 are shown in red. Figures were created using the program Molscript (Kraulis, 1991). The structural statistics are given in Table I.
Table I. Restraints and structural statistics for the final VASP EVH1 ensemble.
| Restraints | ||
| total experimental restraints | 3117 | |
| total NOE restraints | 3043 | |
| intraresidue (i = j) | 1161 | |
| sequential (|i – j| = 1) | 637 | |
| medium range (1 < |i – j| ≤ 4) | 323 | |
| long range (|i – j| > 4) | 922 | |
| H-bond restraints | 25 | |
| dihedral angle restraints | 49 | |
| average NOEs per residue | 26.5 | |
| NOE violations >0.3 Å | 0 | |
| dihedral angles >5° | 0 | |
| accepted structures out of 100 | 75 | |
| Energies | ||
| final energies (kcal/mol) | (SA)ensemblea | <SA>s.c.m.b |
| Etotal | 128.53 ± 1.30 | 126.21 |
| Ebonds | 5.44 ± 0.25 | 5.05 |
| Eangles | 52.26 ± 1.20 | 52.08 |
| Eimpropers | 6.57 ± 0.46 | 5.87 |
| EvdW | 49.44 ± 1.38 | 48.56 |
| ENOE | 14.70 ± 0.60 | 14.41 |
| R.m.s.ds | ||
| Deviation from ideal values | ||
| bonds | 0.0017 ± 0.00004 | 0.0017 |
| angles | 0.3272 ± 0.0040 | 0.327 |
| impropers | 0.2115 ± 0.0074 | 0.200 |
| backbone Cα (2–114) (Å) | 0.369 ± 0.0684 | 0.267 |
| non-hydrogen atoms (Å) | 0.798 ± 0.0423 | 0.715 |
a(SA)ensemble represents the average r.m.s. deviations for the ensemble of 15 structures.
b<SA>s.c.m. are the statistics for the structure closest to the mean.
The VASP and Mena EVH1 domains show some small, but interesting differences. The insertion of a proline at position 56 (Figure 1) in the sequence of the VASP EVH1 domain results in a slightly larger loop differing in structure to the corresponding loop in both Mena and Ev1. Interestingly, this loop occurs in the region that interacts with the C-terminal leucine residue of the ActA peptide investigated in these studies (see below). The Mena EVH1 domain also shows a deletion at position Thr28 compared with the other EVH1 sequences, thereby shortening this loop slightly. Since VASP and Mena EVH1 domains are known to bind overlapping peptides, but with different affinities, it is possible that these small differences, which occur in loop regions close to the peptide-binding site, could provide some degree of ligand specificity. The structure also explains a point mutation in the EVH1 domain of Ena, A97V, which resulted in a lethal phenotype in Drosophila and in disturbed interactions with zyxin in transfected mammalian cells (Ahern-Djamali et al., 1998). Mutation of Ala97 to a larger amino acid would prevent correct folding of the EVH1 domain by precluding the close packing of the α-helix against the β-barrel.
The EVH1-binding affinities of ActA and zyxin peptides are determined by residues neighbouring the FPPPP sequence
To analyse specificity-determining residues flanking the core FPPPP motif, libraries of peptides derived from the overlapping sequences 332SFEFPPPPTEDEL344 and 334EFPPPPTEDELEII347 of the third ActA repeat were created using the previously described spot synthesis technique (Kramer and Schneider-Mergener, 1998). Residues in each peptide were substituted in turn for all 20 amino acids and screened for VASP EVH1 binding. The results for the more strongly binding 332SFEFPPPPTEDEL344 sequence are shown in Figure 2. The domain showed very strong preferences for Phe and Pro residues at the positions Phe335, Pro336 and Pro339. Exceptions were F335W, where strong binding was retained, and F335L and F335Y, for which binding was observed but very much reduced. P339F, P339W and P339Y substitutions also bound, but with significantly reduced affinities. Pro336, the first proline in the poly-l-proline series, was absolutely essential for binding to EVH1. In contrast, substitution of Pro337 for almost all other residues was well tolerated, whereas Pro338 could be replaced by any of the hydrophobic residues, Ala, Ile or Leu, Phe or Val. EVH1 favoured negatively charged, polar or hydrophobic residues at position Glu343 and, interestingly, displayed a considerable preference for Leu at the C-terminal position (Leu344). The reduction in binding affinity on substitution of Leu344 suggests that this residue may be a second, hydrophobic, specificity determinant. The hydrophobic substitutions L344I, L344F and L344V maintain significant but noticeably weaker binding affinities, whereas all other substitutions result in greatly weakened interactions with EVH1. Substitutions within the 334EFPPPPTEDELEII347 peptide (data not shown), showed the same general consensus, but demonstrated in addition that the residues C-terminal to Leu344 were completely unimportant for EVH1 binding.

Fig. 2. Mutational analysis of the native ActA-derived 332SFEFPPPPTEDEL344 peptide by screening of the VASP EVH1 domain against cellulose membrane-bound ActA peptide derivatives. Residues in the peptides were substituted each in turn by all 20 l-amino acids (rows) and assayed for binding to the EVH1 domain of human VASP. The left column is a control, showing the binding of the wild-type sequence. Each of the other positions in the grid represent peptides synthesized with single amino acid substitutions, with the spot intensities reflecting the binding affinities of each substituted peptide. Quantitative measurements of binding constants were later determined using fluorescence spectroscopy based on the results of these analyses.
EVH1–peptide-binding affinities were determined quantitatively by fluorescence spectroscopy. Binding constants were measured for a number of N- and C-terminally truncated/substituted peptides derived from the third tandem repeat of the listerial ActA protein, and additionally, for peptides derived from each of the four FPPPP-containing repeats of human zyxin; 69EDFPLP PPPLAG80, 91GAFPPPPPPIEE102, 102ESFPPAPLEEEI113 and 112EIFPSPPPPPEE123. The results are given in Table II. Examination of the results shows that the 12 residue ActA peptide 333FEFPPPPTEDEL344 binds with the highest affinity (Kd = 19 µM), whereas deletion of the N-terminal Phe333 reduced the affinity by a factor of 1.5. Removal of the next residue, Glu334, reduced the binding by an additional factor of 2, indicating that a charged residue at this position enhances binding. This is in agreement with previous observations (Carl et al., 1999). Truncation from the C-terminus showed that the 341EDEL344 residues were very important for maintaining a high binding affinity, with a >10-fold increase in the measured Kd from 19 to 214 µM upon their removal. The importance of the C-terminal 343EL344 was investigated further by measuring the affinity of the peptide, 334EFPPPPTED342. The binding of this peptide to VASP and Mena EVH1 domains was found to be more than five times weaker than that of 334EFPPPPTEDEL344. Significantly, replacement of Leu344 for Ala led to a 3-fold reduction in binding affinity (from 32 to 102 µM). Interestingly, substitution of the very highly conserved Phe335 for Leu did not completely abolish binding, but instead led to a 4-fold (from 32 to 118 µM) reduction in affinity. This suggests that the primary role of Phe335 is one of recognition rather than directly increasing binding affinity, with the aromatic sidechain providing a specific anchor to guide the positioning of the polyproline sequence in the peptide groove.
Table II. Binding constants of VASP and Mena EVH1 domains with ActA and zyxin peptides (µM).
| Peptide (Ac-peptide-CONH2) | VASP(1–115) | Mena(1–115) |
|---|---|---|
| ActA-derived peptides from repeat 3: | ||
| a333FEFPPPPTEDEL344 | 19 ± 3 | 17 ± 1 |
| 334EFPPPPTEDEL344 | 32 ± 2 | 27 ± 3 |
| 335FPPPPTEDEL344 | 58 ± 6 | 40 ± 6 |
| 334EFPPPPTEDEA344 | 102 ± 13 | 64 ± 6 |
| 334ELPPPPTEDEL344 | 118 ± 12 | 93 ± 10 |
| 334EFPPPPTED342 | 177 ± 36 | 151 ± 12 |
| 332SFEFPPPPT340 | 214 ± 32 | 201 ± 71 |
| 335FPPPPT340 | n.b. | 417 ± 82 |
| zyxin FPPPP repeats 1–4: | ||
| 69EDFPLPPPPLAG80 | 74 ± 16 | 49 ± 14 |
| 91GAFPPPPPPIEE102 | 61 ± 15 | 38 ± 7 |
| 102ESFPPAPLEEEI113 | 197 ± 31 | 193 ± 8 |
| 112EIFPSPPPPPEE123 | n.b. | n.b. |
n.b., no binding observed by fluorescence spectroscopy; apeptide not acetylated.
The EVH1 domains of VASP and Mena bind each of the four FPPPP-containing repeats of human zyxin between 3 and 10 times more weakly than the FEFPPPPTEDEL peptide derived from the third ActA repeat (Table II). It is interesting that the fourth zyxin repeat shows no EVH1 affinity by fluorescence titration when measured as an isolated peptide. Several of the FPPPP flanking residues N-terminal to the fourth zyxin repeat overlap with FPPPP flanking residues C-terminal to the third repeat, suggesting that repeats 3 and 4 share flanking interactions with the EVH1 domain. The ActA repeats, however, are more widely spaced, and bind VASP and Mena EVH1 domains independently of each another.
In all cases, the binding constants for the EVH1 domain of Mena followed the same trend, with Mena EVH1 exhibiting a slightly higher binding affinity in general for all the peptides. The core FPPPPT motif showed no affinity for VASP EVH1 in our assays, whereas binding to Mena EVH1 was very weak but nonetheless detectable (417 µM). According to these measurements, it could be concluded that N-terminal deletions up to and including Glu334 have little effect on binding affinity, whereas truncation of the C-terminal residues, particularly 343EL344, very strongly reduces it.
Definition of the ActA peptide-binding site on the EVH1 domain
To understand the structural basis of the EVH1–peptide interaction, a complex of the domain with the ActA-derived peptide 332SFEFPPPPTEDEL344 was formed and investigated by NMR chemical shift mapping. A series of 2D 15N-HSQC spectra were used to monitor chemical shift changes of protein 1H–15N resonances in separate titrations with the three ActA peptides, 332SFEFPPPPTEDEL344, 334EFPPPPTEDEL344 and 334EFPPPPTED342. Since the peptide–protein interaction was in the fast exchange regime, it was possible to follow the migration of 1H–15N crosspeaks as a function of peptide concentration during all titrations, without the need for resonance reassignment. For 1H–15N crosspeaks that migrated into overlapped regions, the assignments in the bound state were confirmed from 3D CBCA(CO)NH and 3D 15N-separated NOESY-HSQC spectra acquired on the final 1:1 complex. The sidechain resonances of two residues, Trp23 and Gln81, showed severe broadening at intermediate peptide:protein molar ratios, with crosspeaks completely vanishing around the midpoint (between 0.2:1 and 0.4:1) and returning to their original intensities at equimolar peptide:protein ratios. This suggests that these nuclei interact with the peptide with different exchange characteristics to the rest of the protein, indicating an involvement in direct protein–peptide interactions.
The combined chemical shift perturbations (ΔδTOTAL) of all 1H and 15N resonances were weighted according to ΔδTOTAL = Δδ[1H] + 0.2*Δδ[15N] (Hadjuk et al., 1997) and plotted for each titration, as a function of sequence (Figure 3). For simplicity only backbone resonances are shown in the figure. Figure 4A shows the most perturbed 1H and 15N resonances in relation to their location in the three dimensional structure. The backbone amides of Lys71, Tyr72 and Ala75 and the indole NH of Trp23 showed very large perturbations (ΔδTOTAL between 0.4 and 0.6 p.p.m.). The strongest perturbation in all peptide titrations (ΔδTOTAL of 1.2 p.p.m.) was observed for the sidechain NH resonance of Gln81 (data not shown). The large chemical shift changes and accompanying line-broadening of the Gln81 and Trp23 sidechains upon peptide binding suggest putative roles for these protons in hydrogen bond formation. Migrations of the backbone NHs of Lys71, Tyr72 and Ala75 may be explained by contacts to the peptide from these residues in the peptide-binding groove. Other large shift changes (0.1–0.25 p.p.m.) were observed for the backbone amides of residues Gln31 and Ala32, which form a loop at the top end of the groove, and Val67, Gly69 and Val70, which form a small hydrophobic patch at the bottom.

Fig. 3. Bar plot showing changes in chemical shifts of the backbone 1H and 15N resonances between the unbound VASP EVH1 domain and the domain following titration to an equimolar ratio with each of the three ActA-derived peptides discussed in the text. Shifts are displayed as a function of sequence position. Yellow bars represent the changes seen with addition of 332SFEFPPPPTEDEL344, pink bars with the shorter 334EFPPPPTEDEL344, and black bars with the C-terminally truncated peptide 334EFPPPPTED342. For clarity, the regions of interest (Gln31, Ala32 and Ala85, Arg86) are enlarged and displayed below the full sequence plot. The perturbations, measured from 15N-HSQC spectra, are shown as a combined, weighted function of 15N and 1H shifts: ΔδTOTAL = Δδ[1H] + 0.2*Δδ[15N] (Hadjuk et al., 1997). Changes in chemical shifts of the sidechain protons of the peptide, measured from 13C-(ω1)–13C-(ω2) double half-filtered 1H TOCSY spectra (Gemmecker et al., 1992; Ikura and Bax, 1992) on a sample containing fully 13C,15N-labelled protein bound to unlabelled peptide in a D2O solvent, are shown in Table III.



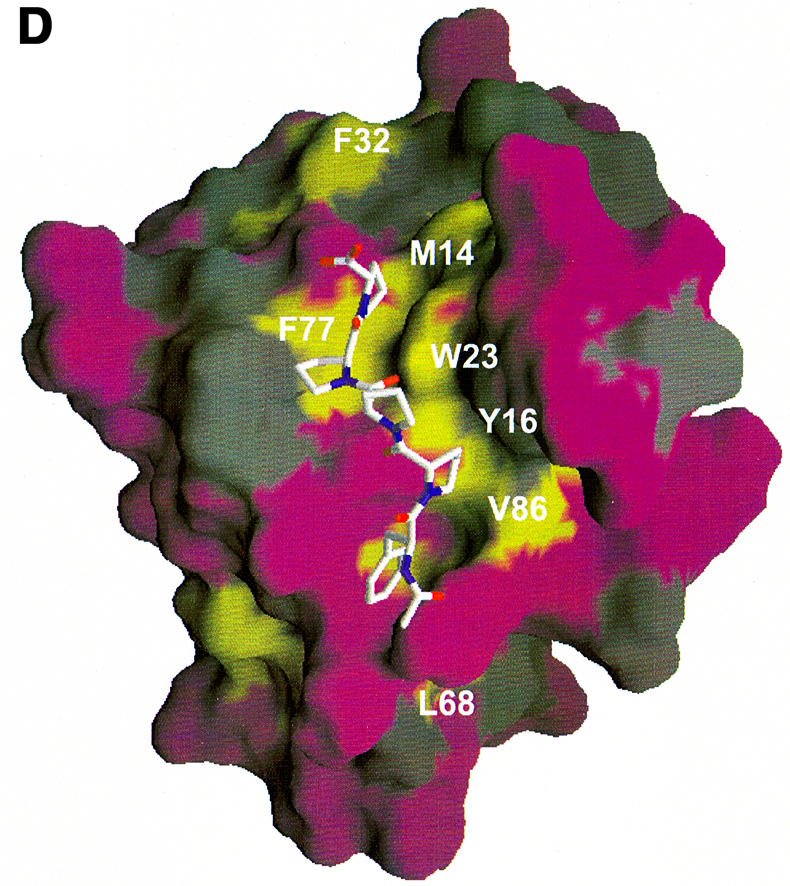
Fig. 4. (A) Combined chemical shift perturbations of residues in the EVH1 domain on binding to ActA SFEFPPPPTEDEL. Red: perturbations >0.4 p.p.m.; purple: perturbations between 0.25 and 0.4 p.p.m.; lilac: perturbations between 0.1 and 0.25 p.p.m. Spheres denote 15N/1H pairs which experience the greatest differences in chemical shift perturbation when either the peptide 343EL344 or 332SF333 residues are removed (specifically, these are the backbone N/H atoms of the EVH1 Gln31, Ala32 and Ala85, Arg86, respectively). The reduction in shift perturbations of the Trp23 indole N/H atoms result from the much weaker overall binding to a peptide lacking the ‘EL’ epitope. (B) Chemical shift perturbations of the ActA SFEFPPPPTEDEL peptide upon EVH1 binding (see Table III). The surface of the VASP EVH1 domain is coloured according to (A), with the difference that only surface exposed amides are shown. Residues in the peptide are coloured cyan (perturbations >0.2 p.p.m.) and green (perturbations between 0.15 and 0.2 p.p.m.). (C) and (D) show the hydrophobic surfaces of VASP and Mena EVH1 domains, respectively. It should be noted that the Mena EVH1 domain was co-crystallized using a much shorter peptide comprising just the core FPPPPT residues (Prehoda et al., 1999). The view of each molecule is rotated 45° forwards about the x-axis, with respect to the view in (A) in order to show the additional binding contacts of the longer peptide. Hydrophobic surfaces were calculated with the program GRASP (Nicholls et al., 1991) using hydrophobicity scales from Covell et al. (1994). Hydrophobic areas are shown in yellow and hydrophilic areas in purple. The yellow hydrophobic groove, running from top to bottom of the molecule, comprises the binding site for the ActA peptide. Contacts between the FPPPP motif and the residues in and around the triad region, Tyr16, Trp23, Phe79 and Met14 are clearly visible, as are secondary contacts between the peptide C-terminal leucine and Met54, Ala32 and Phe33 of the protein. The residues comprising the groove in each of the proteins are labelled, showing the distribution of surface hydrophobicities to be highly similar between the VASP and Mena domains. The SFEFPPPPTEDEL peptide was docked onto the domain as described in the Materials and methods.
Due to the high degree of symmetry of the poly- l-proline helix of the FPPPP motif in 332SFEFPPP PTEDEL344, peptide binding can in principle occur in two opposite orientations. This bi-directional binding ability has already been reported for polyproline ligands binding to profilins and SH3 domains (Mahoney et al., 1999). Hence, to determine which of the core-flanking residues were important for directing the binding orientation, titrations were carried out with N- and C-terminally truncated ActA peptides, EFPPPPTEDEL and EFPPPP TED. Titration with the peptide lacking the N-terminal 332SF333 residues resulted in only minor changes in the chemical shift perturbations of most residues, with the notable exceptions of two backbone amide resonances, Ala85 and Arg86, which were considerably less perturbed in the absence of the peptide Ser332 and Phe333 residues (Figure 3). This observation was complemented by the chemical shift perturbations observed on titration with EFPPPPTED, which lacks both N- and C-terminal residues. Apart from an expected overall reduction in the magnitudes of shift perturbations due to the weaker association of the protein with the shorter peptide, the general pattern of chemical shift perturbations for the majority of EVH1 residues was very similar to that observed with the full-length SFEFPPPPTEDEL. How ever, residues Gln31, and particularly Ala32, showed dramatic decreases in the magnitudes of their chemical shift perturbations (the Ala32 amide now almost totally unperturbed) upon loss of the 343EL344 residues (Figure 3). This indicates strongly that Gln31 and Ala32 interact with the C-terminal residues of the extended peptide. Together with the smaller, but significant, decreases in perturbations of Ala85 and Arg86 upon truncation of the N-terminal 332SF333 peptide residues, we were therefore able to define unambiguously the orientation of the bound peptide (Figure 4A–C).
Identification of ActA peptide residues involved in EVH1 binding
Chemical shift perturbations in the SFEFPPPPTEDEL peptide ligand were monitored to identify the peptide amino acids involved in binding. Assignment of the unbound peptide resonances was carried out using standard homonuclear TOCSY, NOESY and DQF-COSY spectra on a sample of pure peptide in H2O solution. The assignment of the peptide resonances in the bound form, however, required the detection of a small number of 12C-attached protons from the unlabelled ligand in the presence of a large number of 13C-attached protons from the protein. These assignments were therefore achieved using 13C-(ω1)–13C-(ω2) double half-filtered 1H TOCSY and NOESY experiments (Gemmecker et al., 1992; Ikura and Bax, 1992) acquired with different mixing times, on a complex of 13C,15N-labelled VASP EVH1 domain with equimolar unlabelled peptide, in 99% D2O solution.
The results are depicted in Figure 4B and summarized in Table III, with the numbers in bold type representing those nuclei that were most perturbed. The first two residues (Ser and Phe) could not be assigned unambiguously and the resonance frequencies of Glu334 showed no discernible differences to those of the unbound peptide. In contrast, the conserved residue Phe335 showed very large Hα and Hβ2 chemical shift perturbations, indicating a major role for this residue in forming peptide–protein contacts. This supports our results from the peptide amino acid substitution analysis (Figure 2), which show clearly that strong EVH1 binding only occurs when this position contains either Phe or Trp. A relatively large shift perturbation was also observed for the Hα of Pro336, which was also shown by substitution analysis to be essential for binding. It was not possible to obtain shift differences for the remaining proline residues due to excessive overlap in this region of the 13C–13C-filtered 1H TOCSY spectrum. The backbone Hα resonances of Thr340, Glu341, Asp342, Glu343 and Leu344 and sidechain Hβs and Hγs of Thr340, Glu341, Glu343 and Leu344 were all significantly shifted upon complexation, showing perturbations of between 0.1 and 0.2 p.p.m. This complements our peptide binding data, supporting the hypothesis that the EDEL epitope is important in increasing binding affinity by making many contacts to the EVH1 domain. The most marked sidechain perturbations, of >0.2 p.p.m., were seen for the Hδ1 and Hδ2 protons of the C-terminal Leu344. This further supports our data from substitution analysis and fluorescence spectroscopy, which strongly suggest that Leu344 is a major contributor to the binding affinity. The less conserved central pair of prolines, Pro337 and Pro338, could not be assigned sequence specifically from the 2D 1H spectra, but it could be seen that their resonances were largely unperturbed. The Ser332 and Phe333 residues showed more than one set of crosspeaks each, suggesting that these residues sample multiple conformations on a slow timescale.
Table III. ActA peptide chemical shift perturbations (p.p.m.) upon EVH1 binding.
| Residue | HA | HB1 | HB2 | HG1 | HG2 | HG2# | HD1 | HD2 | HD# | HE# |
|---|---|---|---|---|---|---|---|---|---|---|
| S332 | n.r. | n.r. | n.r. | – | – | – | – | – | – | – |
| F333 | n.r. | n.r. | n.r. | – | – | – | – | – | n.r. | n.r. |
| E334 | 0.01 | 0.00 | 0.04 | 0.01 | 0.00 | – | – | – | – | – |
| F335 | 0.34 | 0.04 | 0.18 | – | – | – | – | – | n.r. | n.r. |
| P336 | 0.12 | n.r. | n.r. | n.r. | 0.04 | – | 0.18 | n.r. | – | – |
| P337 | n.r. | n.r. | n.r. | n.r. | n.r. | – | n.r. | n.r. | – | – |
| P338 | n.r. | n.r. | n.r. | n.r. | n.r. | – | n.r. | n.r. | – | – |
| P339 | 0.05 | n.r. | n.r. | n.r. | n.r. | – | 0.11 | 0.05 | – | – |
| T340 | 0.19 | 0.03 | – | – | – | 0.09 | – | – | – | – |
| E341 | 0.14 | 0.11 | 0.06 | 0.09 | 0.13 | – | – | – | – | – |
| D342 | 0.11 | 0.04 | 0.03 | – | – | – | – | – | – | – |
| E343 | 0.22 | 0.02 | 0.00 | 0.05 | 0.11 | – | – | – | – | – |
| L344 | 0.18 | 0.03 | 0.09 | n.r. | n.r. | – | 0.23 | 0.21 | – | – |
n.r., resonance not resolved; –, resonance does not apply. Bold, peptide shifts >0.15 p.p.m.
Mutations of the EVH1 domain affecting folding, solubility and peptide binding
The roles of several residues in regions that showed large chemical shift perturbations upon peptide binding, were further investigated by mutational analysis. The mutants F33A and V67A showed 1.5- to 2-fold weaker binding to a peptide, FEFPPPPTEDEL (data not shown). Further mutations, W23L, Y72D, F79A and H80N, resulted in insoluble proteins, probably due to the roles of these sidechains in stabilizing the protein fold. Examination of the 3D structure, together with the large numbers of NOEs observed from these sidechains to other core residues located in strands β1, β6 and β7, provides strong evidence for this. Trp23, which is 100% conserved in all EVH1 domains, performs a dual function; first, using its indole ring to recognize the ligand FPPPP motif; and secondly, using its six-membered ring to participate in hydrophobic contacts which stabilize the EVH1 core.
Modelling the VASP EVH1 domain in complex with the SFEFPPPPTEDEL peptide of ActA
A complex of the VASP EVH1 domain and the peptide 332SFEFPPPPTEDEL344 based on the observed chemical shift perturbations and on the results of the peptide binding assays was modelled as described in the methods (see Figure 4). The core FPPPT motif was docked onto the domain maintaining the same poly-l-proline contacts as observed in the crystal structure of Mena (Prehoda et al., 1999). The remaining residues were restrained loosely, using two additional restraints derived from the chemical shift mapping studies as described in the methods.
From inspection of the protein chemical shift changes, it could be seen that contacts to the peptide were formed by: (i) the triad residues, Tyr16, Trp23 and Phe79; (ii) residues in the hydrophobic groove running along the top-facing β-sheet comprising strands β5–β6–β7 (see Figure 4A and C); (iii) the sidechain of Gln81, which resides exactly in the centre of the middle strand, β6; (iv) the hydrophobic patch above the groove comprising residues Gln31 to Phe33 (Figure 4C); and (v) a smaller hydrophobic patch below the groove comprising residues Val67–Val70 and Ala85, Arg86. Conversely, inspection of the peptide chemical shift perturbations showed that contacts to the domain were formed by: (i) the phenylalanine directly preceding the poly-l-proline helix, Phe335; (ii) Pro336 and Pro339; (iii) Thr340, particularly the α proton; and (iv) the Glu343 and Leu344 EL motif, most notably the delta protons of the leucine (Figure 4B).
The binding of the extended SFEFPPPPTEDEL peptide into the hydrophobic groove explains the large chemical shift perturbations of the backbone amides of Lys71, Tyr72 and Ala75 (Figures 3 and 4A), since these amino acids are in close proximity to the highly perturbed Gln81, the sidechain of which is probably involved in hydrogen bond formation. It appears from sequence alignments that hydrogen bond formation between a proton donating sidechain at this position and a peptide proline carbonyl oxygen could be a general feature of EVH1 polyproline recognition. Gln81 is completely conserved amongst the Ena, VASP, Mena and Evl EVH1 domains, indicating that this residue is indeed important. A second hydrogen bond interaction between the Trp23 indole proton and the carbonyl oxygen of Pro337 is also likely, based on the very large shift changes of 0.5 p.p.m. for this indole, together with the accompanying line-broadening (observed for Trp23 and Gln81 sidechains only) during the titrations with the peptide. Perturbations of 0.3–0.4 p.p.m. observed for the Asn73 sidechain and Gln74 backbone amide (β5), the His80 and Trp82 backbone amides (β6), and the Leu91 backbone and Asn92 sidechain (β7) indicate that re-organization in this region occurs to accommodate peptide binding. These residues directly surround the peptide gorge formed by β5, β6 and β7, with their most highly perturbed nuclei oriented towards Gln81 (β6).
Inspection of the surface of the VASP EVH1 domain shows an intriguing distribution of hydrophobic patches along the prospective peptide-binding groove, running from the aromatic triad to the bottom of the domain, roughly parallel to the helix (Figure 4C). The view presented shows both the polypeptide-binding groove and the hydrophobic patch above it. The latter consists of residues Met54, Ala32 and Phe33, which are involved in contacts to the C-terminal Leu344. The binding interaction surface was calculated to be 901.5 Å2 using the program SYBYL (Tripos, Inc., 1999). Taking the chemical shift changes of protein and peptide signals together, these data suggest that the peptide C-terminal leucine is responsible for the chemical shift changes of the backbone Gln31 to Phe33 amides and that the peptide N-terminus then contacts the region comprising Ala85, Arg86 and the hydrophobic loop Val67–Val70. Only very minor differences were noted between complexation with SFEFPP PPTEDEL and EFPPPPTEDEL, showing that the residues Ser332 and Phe333 are relatively unimportant for peptide binding specificity. Modest decreases in the magnitudes of the shift perturbations of Ala85, Arg86 upon truncation of 332SF333 suggested a weak interaction between these two groups of residues. This agrees both with previously reported deletion studies (Niebuhr et al., 1997) and with our own binding constant measurements (see Table II). However, removal of the C-terminal Glu343 and Leu344 residues resulted in a dramatic reduction in the perturbations of Gln31 and Ala32 amides. The single substitution of Leu344 for an alanine at the C-terminus attenuated EVH1-binding affinity by a factor of three, further implying a role for this leucine as a specificity determinant.
Discussion
A conserved, second EVH1 specificity-determining binding site is exploited by ActA
EVH1 domains bind the FPPPPT motif with very low affinity (Kd >400 µM); therefore, additional affinity, hence specificity, determinants must exist to raise the binding strength to biologically significant levels. As shown in this study, the EVH1 domains of VASP and Mena possess a second hydrophobic region comprising residues Ala32, Phe33 and Met54 (VASP numbering), in addition to a hydrophobic, poly-l-proline-binding groove, which is ideally situated to offer an additional interaction site for residues C-terminal to the FPPPPT core motif. We have shown that interaction of the C-terminal residues of the peptide with this site is important for modulating peptide-binding affinity, and could also be a factor in determining ligand orientation. Figure 4C and D shows this hydrophobic region for VASP and Mena, respectively, whereas Figure 4B illustrates the interacting residues in the peptide. This second hydrophobic site is conserved in VASP, Mena and Evl EVH1 domains, and therefore similar interactions of all three EVH1 domains would be expected with the ActA peptides. This would explain why VASP and Mena EVH1 domains bind each of the ActA-derived peptides with highly similar (though not identical) affinities (Table II). The hydrophobic surface comprising Ala32, Phe33 and Met54 may thus play an important role in the discrimination of VASP and homologous proteins for their respective binding partners, contributing to the targeting of these proteins to their specific locations in the presence of competing intracellular sites.
In all four EVH1-binding motifs of the listerial ActA protein, we identified a strictly conserved peptide epitope (EL), C-terminal to the FPPPP core motif, the leucine of which efficiently utilizes the second, hydrophobic region present on the EVH1 domain surface. The mammalian VASP EVH1 target zyxin (Golsteyn et al., 1997) contains four less highly conserved FPPPP-type repeats (see Table II), with the second and third proline positions being more variable than their ActA counterparts and the equivalent C-terminal residues being AG, EE, EI and EE, respectively. In comparison with the 333FEFPPPPTEDEL344 peptide derived from the third ActA repeat, the first three zyxin peptide repeats showed a range of reduced binding affinities (from 3- to 10-fold) for the VASP and Mena EVH1 domains (Table II). No EVH1-binding was detectable by fluorescence spectroscopy for the fourth zyxin repeat. Of all four zyxin repeats, only the third (containing EI) could make a hydrophobic interaction with the second hydrophobic binding site on the EVH1 domain. However, this repeat shows the weakest binding affinity of the three peptides that bound; an effect likely to be due to the replacement of the third proline in the core motif of this repeat by an alanine, FPPAP. The Kd for interaction with the VASP EVH1 domain is 197 µM, compared with 74 and 61 µM for the first and second zyxin repeats. It is possible that the presence of the Ile at the C-terminal position in this peptide serves to rescue the attenuated binding caused by loss of the third proline.
Previous workers have also demonstrated that EVH1-binding peptides from a number of cytoskeletal proteins bind with higher efficiencies when their core flanking residues are present and that isolated core regions generally bind very weakly. Peptides derived from each of the four proline-rich repeats of ActA, which included the EL residues plus two additional residues C-terminal to these, were observed to bind VASP and Mena EVH1 domains more strongly than the truncated core regions (Niebuhr et al., 1997). However, the increase in binding affinity of these longer peptides relative to the core FPPPPT motifs was not quantified by these workers and was never attributed specifically to the EL epitope identified in our study. We have shown by amino acid substitution analysis of the ActA peptides, 332SFEFPPPPTEDEL344 (Figure 2) and 334EFPPPPTEDELEII347 (data not shown), that the residues C-terminal to Leu344 and N-terminal to Phe335 could be replaced by any other residues with no noticeable loss in binding affinity. Substitution of the EL residues, however, leads to substantial decreases in peptide-binding affinity (Figure 2). This observation is supported quantitatively by our measurements of individual EVH1:peptide Kds from fluorescence titration curves (Table II). In particular, the importance of the C-terminal leucine (Leu344) is clearly highlighted by the 3-fold reduction in peptide-binding affinity when this residue is replaced by an alanine. The analysis in Figure 2 combined with the Kd measurements from fluorescence thus enabled us to identify the EL motif. Chemical shift mapping of peptides with and without this motif then clearly identified the EVH1 residues with which it specifically interacted.
The analysis shown in Figure 2 also provides new information on the proline specificity of the EVH1 interaction with the FPPPP core. Previous semi-quantitative results from a peptide, SFEFPPPPTD, had suggested that the central two prolines were the most important for EVH1 binding (Niebuhr et al., 1997). Figure 2, however, clearly shows that the first and fourth prolines are essential for EVH1 binding and that the central two prolines may be substituted by a considerable number of other amino acid types without loss of binding. This is in good agreement with the consensus Ena-VASP EVH1-binding sequence, FPXØP, (where X is any residue and Ø is hydrophobic) and also agrees very well with the precise protein–peptide interactions observed in the available structures, in which the first and fourth proline sidechains make close contacts with the peptide-binding groove of the EVH1 domain and the central two proline sidechains are exposed. Notably, the SFEFPPPPTD peptide studied by Niebuhr and co-workers lacked the EL epitope identified in our study. From our fluorescence measurements it can be seen that such a peptide would bind the VASP and Mena EVH1 domains with Kd in the range of 150–214 µM (Table II). In contrast, our studies were based on the 332SFEFPPPPT EDEL344 peptide, which binds with a Kd of 19 µM, thus increasing dramatically the intrinsic signal:noise ratio and subsequent accuracy of our measurements.
The cumulative evidence from all of our observations offers some explanation as to why the EVH1 domains of VASP and Mena bind cytoplasmic zyxin less strongly than the foreign ActA protein of invading Listeria. The individual ActA repeats may exploit the small, second hydrophobic site in EVH1 domains far more efficiently than the corresponding repeats in zyxin to increase their EVH1-binding affinity. Interestingly, the third and fourth zyxin repeats overlap in sequence, and thus share a number of flanking residues necessary for interactions with the EVH1 domain. This reduces the number of potential interactions of the third and fourth repeats with their target sites, possibly even excluding their simultaneous accessibility. If zyxin could bind only three repeats simultaneously, this would offer further explanation for its weaker EVH1-binding ability compared with ActA. The presence of four closely spaced proline-rich repeats in both zyxin and ActA hints at co-operative EVH1 binding, with each repeat binding to one of the four EVH1 domains available in tetrameric VASP or Mena (Bachmann et al., 1999; Laurent et al., 1999). Alternatively, each proline-rich repeat could dock a separate VASP or Mena tetramer in order to maximally increase the local concentration of VASP or Mena proteins wherever zyxin (or ActA) is present. To understand these complexes in greater detail, further work will be required to determine their precise stoichiometries.
Like zyxin, the FPPPP motif present in the human vinculin sequence (PDFPPPPPDLEQ) also lacks the C-terminal hydrophobic residue present in each of the ActA repeats, suggesting a similarly weak interaction between the vinculin motif and EVH1. It is believed for vinculin (Brindle et al., 1996; Reinhard et al., 1996), which possesses only a single EVH1-binding motif per polypeptide chain, that binding strength is increased by a mechanism of oligomerization that compensates for the lack of multiple EVH1-binding sites in each monomer. Furthermore, the oligomerization state of vinculin, and hence complex formation with EVH1 domains, is controlled by the conformational state of the vinculin protein (Hüttelmaier et al., 1998), which is capable of adopting open and closed forms. Binding of phosphatidylinositol-4,5-bisphosphate, promotes the open form, thus exposing the otherwise protected proline-rich binding site. It therefore appears that EVH1-binding affinity is sensitive not only to the number of binding motifs in the ligand and their precise sequences, but also to the oligomerization states of both interaction partners.
The structural basis of the difference in binding strengths of various EVH1-binding motifs leads us to the biological interpretation that the ActA protein of Listeria may have evolved a further highly efficient, EVH1-binding epitope, containing a leucine residue that exploits the second hydrophobic surface present in the EVH1 domains, in order to successfully displace the natural EVH1 ligands of the host. The host zyxin and vinculin proteins rely on weak EVH1 interactions in order to maintain a sensitive and dynamic response to continuously changing extracellular signals. During the course of infection, the pathogen must recruit EVH1 domains more efficiently than the host EVH1-binding proteins in order to successfully pirate the host cell actin polymerization machinery. Increasing the binding affinity for EVH1 domains by the use of additional affinity increasing epitopes, together with strict conservation of the FPPPP motif, achieves this in a simple and elegant manner. This functional adaptation of major bacterial virulence factors, like ActA, may have contributed, during the course of evolution, to providing intracellular pathogens such as Listeria with a selective advantage, allowing them to exploit host cell systems that must themselves strictly maintain specific and low affinity binding interactions in order to fulfil their cellular functions.
Materials and methods
Preparation of recombinant VASP and Mena EVH1 domains
DNA sequences of VASP(1–115) and Mena(1–115) and mutants of these were cloned into the plasmids pGEX-4T-1 (Pharmacia). The domains were then expressed as glutathione S-transferase (GST) fusion proteins, induced at A600 of 0.5–1.0 using 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) and incubated for a further 5 h before harvesting. Cells were lysed by sonication and the desired protein purified on a glutathione–Sepharose 4B column. The GST moiety was cleaved using thrombin protease (Pharmacia), and separated from the protein by chromatography on a 120 ml Superdex 75 column (Pharmacia). Samples were then concentrated using Centriplus-10 concentrators (Amicon). Protein molecular masses, determined by mass spectrometry, agreed with those predicted.
Fluorescence titrations
Dissociation constants were determined from changes in fluorescence emission spectra of VASP and Mena EVH1 domains upon addition of peptide at defined concentrations. Fluorescence was measured at 20°C on an Aminco Bowman Series 2 luminescence spectrophotometer. Fluorescence intensities were obtained by determining the mean value of fluorescence emission at 335 nm over a time period of 10 s after addition of peptide, with an excitation of 295 nm and a bandwidth of 4 nm (Häfner et al., 2000). Binding curves were fitted based on the formation of a 1:1 complex.
NMR spectroscopy
NMR spectra were acquired at 300 K, using Bruker DRX600 and DMX750 spectrometers in standard configuration, with triple resonance probes equipped with self-shielded triple axis gradient coils. All spectra were recorded essentially as described previously (Ball et al., 1997; Smalla et al., 1999). Uniformly 15N- and 13C,15N-labelled samples of the VASP EVH1 domain were prepared by growing Escherichia coli BL21 cells transformed with the plasmid, in a MOPS minimal medium (Neidhardt et al., 1974) containing 15NH4Cl, with or without 0.2% 13C6-glucose and 0.2% 13C,15N labelled peptone from Chenopodium rubrum (EMBL, Heidelberg). The samples were purified as described above. All samples for NMR spectroscopy were prepared at pH 6.0, in a buffer containing 20 mM KH2PO4, 50 mM KCl and 0.2 mM NaN3. A 2 mM 15N-labelled sample in 90% H2O/10% D2O was prepared for 15N-edited TOCSY, NOESY, HNHB and HNHA experiments and later exchanged into D2O solution to determine the hydrogen-bond protected amides. A 1.3 mM 13C,15N-labelled sample in 90% H2O/10% D2O was used for the NH-detected triple resonance experiments, CBCA(CO)NNH and CBCANNH, and a second 1.3 mM 13C,15N-labelled sample in 99.8% D2O was prepared for HCCH-TOCSY and 13C-separated NOESY spectra. Protein–peptide complexes with EFPPPPTEDEL and EFPPPPTED peptides were formed by addition of equimolar amounts of peptide to 15N-labelled protein in 90% H2O. The complex with the longer SFEFPPPPTEDEL peptide was made using 13C,15N-labelled protein and unlabelled peptide. The complex was dissolved in 90% H2O for 15N-filtered spectra and then lyophilized and redissolved into 99.8% D2O for the 13C-filtered spectra (Gemmecker et al., 1992; Ikura and Bax, 1992), required for chemical shift measurements in the bound peptide 12C–H sidechains.
Data were processed using the XWIN-NMR program (version 1.3) of Bruker Analytik GmbH (Rheinstetten, Germany) and the AZARA pro gram (version 2.1) of W.Boucher (unpublished). Assignment was carried out on Silicon Graphics Indy workstations, using the interactive program, ANSIG 3.3 (Kraulis, 1989). AZARA and ANSIG are both available by anonymous ftp from ftp.bio.cam.ac.uk in the directory ftp/pub.
Structure calculations
Structures were calculated from the restraints listed in Table I with the program CNS version 0.9a (Brünger et al., 1998), together with the PARALLHDG v4.02 forcefield. Calculations were performed using a standard simulated annealing protocol starting from random co-ordinates. The force constants for the NOE and phi angle restraints were 150 kcal/mol/Å2 and 200 kcal/mol/rad2, respectively. Distance restraints were categorized as strong (≷2.7 Å), medium (≷3.5 Å), weak (≷5 Å) and very weak (≷6 Å). The lowest energy structures with no distance violations >0.3 Å and no angle violations >5° were accepted.
Modelling of a complex of VASP EVH1 with the ActA peptide, SFEFPPPPTEDEL
The EVH1 complex with SFEFPPPPTEDEL was modelled in a 250 ps molecular dynamics simulation at 300 K using Amber 5.0 (Weiner et al., 1986; Case et al., 1997). The simulation was carried out with a step width of 2 fs and a distance dependent dielectric function, to mimic the presence of a high dielectric solvent. A non-bonded cutoff of 12 Å was used and the non-bonded pair list was updated every 10 fs. During the calculation only those residues were allowed to move that showed chemical shift perturbations above 0.1 p.p.m. The starting structure of the complex was obtained by docking of the FPPPP motif of the peptide onto the VASP EVH1 surface as observed for the reported Mena EVH1 crystal structure (Prehoda et al., 1999). Mena co-ordinates were taken directly from the Protein Data Bank, accession code 1evh. Based on our experimental data, we defined two additional restraints, the first from Glu343 and Leu344 of the peptide to Gln31 and Ala32 of the protein, and the second from Phe333 of the peptide to Ala85, Arg86 of the protein. The complex structure after 250 ps was then minimized until an r.m.s.d. gradient of <0.05 was reached. The AMBER program package is available from http://www.amber.ucsf.edu/amber/amber.html.
VASP-EVH1 binding to cellulose membrane-bound peptides
Cellulose-bound peptides were prepared by automated spot synthesis as previously described (Kramer and Schneider-Mergener, 1998). The membrane-bound peptides were washed with ethanol and then with Tris-buffered saline, TBS [50 mM Tris–(hydroxymethyl)-aminomethane, 137 mM NaCl, 2.7 mM KCl, pH 8] before blocking overnight at 4°C with blocking buffer (blocking reagent: CRB, Northwich, UK, in TBS 1:10 containing 1% sucrose). After washing with TBS, 10 µg/ml of GST-fused VASP-EVH1 domain in blocking buffer was added and incubated at 4°C for 16 h. The membrane was then washed further with TBS before incubation with a GST antibody (polyclonal rabbit IgG, 1 µg/ml in blocking buffer, Calbiochem, Bad Soden, Germany) at 4°C for 16 h. Before detection, a peroxidase-labelled anti-rabbit antibody (Sigma, Munich, Germany, 1 µg/ml in blocking buffer) was applied at room temperature for 4 h and the membrane was washed again with TBS. Binding was detected by addition of the chemiluminescent substrate, Super Signal™ (Pierce, Rockford, IL) using a Lumi-Imager (Boehringer Mannheim, Mannheim, Germany).
Co-ordinates
Co-ordinates were deposited in the Protein Data Bank, accession code 1egx.
Acknowledgments
Acknowledgements
We thank M.Beyermann and co-workers for synthesis of peptides; E.Krause for mass spectrometry; F.Gertler for the gift of Mena cDNA; B.Schinke and S.Roth for their excellent technical assistance; G.Kristjansson for sequencing of constructs; and M.Kelly and G.Krause for insightful discussion. L.J.B., T.J. and M.H. were supported by EMBO, the Deutsche Forschungsgemeinschaft and the Czech Academy of Sciences, respectively.
References
- Ahern-Djamali S.M., Comer,A.R., Bachmann,C., Kastenmeier,A.S., Reddy,S.K., Beckerle,M.C., Walter,U. and Hoffmann,F.M. (1998) Mutations in Drosophila Enabled and rescue by human vasodilator-stimulated phosphoprotein (VASP) indicate important functional roles for Ena/VASP homology domain 1 (EVH1) and EVH2 domains. Mol. Biol. Cell, 9, 2157–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann C., Fischer,L., Walter,U. and Reinhard,M. (1999) The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding and actin bundle formation. J. Biol. Chem., 274, 23549–23557. [DOI] [PubMed] [Google Scholar]
- Ball L.J. et al. (1997) Structure of the chromatin-binding (chromo) domain from mouse modifier protein 1. EMBO J., 16, 2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerle M.C. (1998) Spatial control of actin filament assembly: Lessons from Listeria.Cell, 95, 741–748. [DOI] [PubMed] [Google Scholar]
- Brindle N.P.J., Holt,M.R., Davies,J.E., Price,C.J. and Critchley,D.R. (1996) The focal adhesion vasodilator-stimulated phosphoprotein (VASP) binds to the proline-rich domain in vinculin. Biochem. J., 318, 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system (CNS): A new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Callebaut I. Cossart,P. and Dehoux,P. (1998) EVH1/WH1 domains of VASP and WASP proteins belong to a large family including Ran-binding domains of the RanBP1 family. FEBS Lett., 441, 181–185. [DOI] [PubMed] [Google Scholar]
- Carl U.D., Pollmann,M., Orr,E., Gertler,F.B., Chakraborty,T. and Wehland,J. (1999) Aromatic and basic residues within the EVH1 domain of VASP specify its interaction with proline-rich ligands. Curr. Biol., 9, 715–718. [DOI] [PubMed] [Google Scholar]
- Case D.A. et al. (1997) AMBER 5. University of California, San Francisco, CA, http://www.amber.ucsf.edu/amber.
- Chakraborty T. et al. (1995) A focal adhesion factor directly linking intracellularly motile Listeria monocytogenes and Listeria ivanovii to the actin-based cytoskeleton of mammalian cells. EMBO J., 14, 1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covell D.G., Smythers,G.W., Gronenborn,A.M. and Clore,G.M. (1994) Analysis of hydrophobicity in the α-chemokine and β-chemokine families and its relevance to dimerization. Protein Sci., 3, 2064–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domann E., Wehland,J., Rohde,M., Pistor,S., Hartl,M., Goebel,W., Leimeister-Wächter,M., Wuenscher,M. and Chakraborty,T. (1992) A novel bacterial virulence gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO J., 11, 1981–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S. and Cossart,P. (1998) Intracellular pathogens and the actin cytoskeleton. Annu. Rev. Cell. Dev. Biol., 14, 137–166. [DOI] [PubMed] [Google Scholar]
- Fedorov A.A., Fedorov,E., Gertler,F.B. and Almo,S.C. (1999) Structure of EVH1, a novel proline-rich ligand-binding module involved in cytoskeletal dynamics and neural function. Nature Struct. Biol., 6, 661–665. [DOI] [PubMed] [Google Scholar]
- Gemmecker G., Olejniczak,E.T. and Fesik,S.W. (1992) An improved method for selectively observing protons attached to 12C in the presence of 1H–13C spin pairs. J. Magn. Res., 96, 199–204. [Google Scholar]
- Gertler F.B., Comer,A.R., Juang,J.-L., Ahern,S.M., Clark,M.J., Liebl,E.C. and Hoffmann,F.M. (1995) Enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain binding properties. Genes Dev., 9, 521–533. [DOI] [PubMed] [Google Scholar]
- Gertler F.B., Niebuhr,K., Reinhard,M., Wehland,J. and Soriano,P. (1996) Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell, 87, 227–239. [DOI] [PubMed] [Google Scholar]
- Golsteyn R.M., Beckerle,M.C., Koay,T., and Friederich,E. (1997) Structural and functional similarities between the human cytoskeletal protein zyxin and the ActA protein of Listeria monocytogenes. J. Cell Sci., 110, 1893–1906. [DOI] [PubMed] [Google Scholar]
- Hadjuk P.J. et al. (1997) NMR-based discovery of lead inhibitors that block DNA binding of the human papillomavirus E2 protein. J. Med. Chem., 40, 3144–3150. [DOI] [PubMed] [Google Scholar]
- Häfner A., Merola,F., Duportail,G., Hutterer,R., Schneider,F.W. and Hof,M. (2000) Calcium-induced conformational change in fragment 1–86 of factor Xa. Biopolymers, 57, 226–234. [DOI] [PubMed] [Google Scholar]
- Haffner C., Jarchau,T., Reinhard,M., Hoppe,J., Lohmann,S.M. and Walter,U. (1995) Molecular cloning, structural analysis and functional expression of the proline-rich focal adhesion and micro filament-associated protein VASP. EMBO J., 14, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrügge M. and Walter,U. (1989) Purification of a vasodilator-regulated phosphoprotein from human platelets. Eur. J. Biochem., 185, 41–50. [DOI] [PubMed] [Google Scholar]
- Halbrügge M., Friedrich,C., Eigenthaler,M., Schanzenbächer,P. and Walter,U. (1990) Stoichiometric and reversible phosphorylation of a 46 kDa protein in human platelets in response to cGMP- and cAMP-elevating vasodilators. J. Biol. Chem., 265, 3088–3093. [PubMed] [Google Scholar]
- Hüttelmaier S., Mayboroda,O., Harbeck,B., Jarchau,T., Jockusch,B.M. and Rüdiger,M. (1998) The interaction of the cell contact proteins VASP and vinculin is regulated by phosphatidylinositol-4,5-bisphosphate. Curr. Biol., 8, 479–488. [DOI] [PubMed] [Google Scholar]
- Ikura M. and Bax,A. (1992) Isotope-filtered 2D NMR of a protein–peptide complex: study of a skeletal muscle myosin light chain kinase fragment bound to calmodulin. J. Am. Chem. Soc., 114, 2433–2440. [Google Scholar]
- Kocks C., Gouin,E., Tabouret,M., Berche,P., Ohayon,H. and Cossart,P. (1992) Listeria monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell, 68, 521–531. [DOI] [PubMed] [Google Scholar]
- Kramer A. and Schneider-Mergener,J. (1998) Synthesis and screening of peptide libraries on continuous cellulose membrane supports. Methods Mol. Biol., 87, 25–39. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1989) ANSIG: A program for the assignment of protein 1H 2D NMR spectra by interactive graphics. J. Magn. Res., 24, 627–633. [Google Scholar]
- Kraulis P.J. (1991) Molscript—A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laurent V., Loisel,T.P., Harbeck,B., Wehman,A., Gröbe,L., Jockusch,B.M., Wehland,J., Gertler,F.B. and Carlier,M.-F. (1999) Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J. Cell Biol., 144, 1245–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney N.M., Rozwarski,D.A., Fedorov,E., Fedorov,A.A. and Almo,S.C. (1999) Profilin binds proline-rich ligands in two distinct amide backbone orientations. Nature Struct. Biol., 6, 666–671. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association—insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Niebuhr K. et al. (1997) A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena-VASP family. EMBO J., 16, 5433–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt F.C., Bloch,P.L. and Smith,D.F. (1974) Culture medium for Enterobacteria. J. Bacteriol., 119, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistor S., Chakraborty,T., Niebuhr,K., Domann,E. and Wehland,J. (1994) The ActA protein of Listeria monocytogenes acts as a nucleator inducing reorganization of the actin cytoskeleton. EMBO J., 13, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistor S., Chakraborty,T., Walter,U. and Wehland,J. (1995) The bacterial actin nucleator protein ActA of Listeria monocytogenes contains multiple binding sites for host microfilament proteins. Curr. Biol., 5, 517–525. [DOI] [PubMed] [Google Scholar]
- Prehoda K.E., Lee,D.J. and Lim,W.A. (1999) Structure of the Enabled/VASP homology 1 domain–peptide complex: A key component in the spatial control of actin assembly. Cell, 97, 471–480. [DOI] [PubMed] [Google Scholar]
- Reinhard M., Jouvenal,K., Tripier,D. and Walter,U. (1995a) Identification, purification and characterization of a zyxin-related protein which binds the focal adhesion and microfilament protein VASP. Proc. Natl Acad. Sci. USA, 92, 7956–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M., Giehl, K, Abel,K., Haffner,C., Jarchau,T., Hoppe,V., Jockusch,B.M. and Walter,U. (1995b) The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilin. EMBO J., 14, 1583–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M., Rüdiger,M., Jockusch,B.M., and Walter,U. (1996) VASP interaction with vinculin: a recurring theme of interactions with proline-rich motifs. FEBS Lett., 399, 103–107. [DOI] [PubMed] [Google Scholar]
- Reinhard M., Jarchau,T., Reinhard,K. and Walter,U. (1999) VASP. In Kreis,T. and Vale,R. (eds), Guidebook to the Cytoskeletal and Motor Proteins. Oxford University Press, Oxford, UK, pp. 168–171. [Google Scholar]
- Sanger J.M., Sanger,J.W. and Southwick,F.S. (1992) Host cell actin assembly is necessary and likely to provide the propulsive force for intracellular movement of Listeria monocytogenes. Infect. Immun., 60, 3609–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M. and Hyvönen,M. (1995) Pleckstrin homology domains—A fact file. Curr. Opin. Struct. Biol., 5, 403–408. [DOI] [PubMed] [Google Scholar]
- Smalla M., Schmieder,P., Kelly,M., Ter Laak,A., Krause,G., Ball,L., Wahl,M., Bork,P. and Oschkinat,H. (1999) Solution structure of the receptor tyrosine kinase EphB2 SAM domain and identification of the distinct homotypic interaction sites. Protein Sci., 8, 1954–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V., Bauer,C., Yin,M. and Fuchs,E. (2000) Directed actin polymerization is the driving force for epithelial cell–cell adhesion. Cell, 100, 209–219. [DOI] [PubMed] [Google Scholar]
- Weiner S.J., Kollman,P.A., Nguyen,D.T. and Case,D.A. (1986) An all atom forcefield for simulation of proteins and nucleic acids. J. Comp. Chem., 7, 230–252. [DOI] [PubMed] [Google Scholar]
- Zimmer M., Fink,T., Fischer,L., Hauser,W., Scherer,K., Lichter,P. and Walter,U. (1996) Cloning of the VASP (vasodilator-stimulated phosphoprotein) genes in human and mouse: structure, sequence and chromosomal localization. Genomics, 36, 227–233. [DOI] [PubMed] [Google Scholar]