

Abstract
The Snf1 kinase and its mammalian homolog, the AMP-activated protein kinase, are heterotrimeric enzymes composed of a catalytic α-subunit, a regulatory γ-subunit and a β-subunit that mediates heterotrimer formation. Saccharomyces cerevisiae encodes three β-subunit genes, SIP1, SIP2 and GAL83. Earlier studies suggested that these subunits may not be required for Snf1 kinase function. We show here that complete and precise deletion of all three β-subunit genes inactivates the Snf1 kinase. The sip1Δ sip2Δ gal83Δ strain is unable to derepress invertase, grows poorly on alternative carbon sources and fails to direct the phosphorylation of the Mig1 and Sip4 proteins in vivo. The SIP1 sip2Δ gal83Δ strain manifests a subset of Snf phenotypes (Raf+, Gly–) observed in the snf1Δ 10 strain (Raf–, Gly–), suggesting that individual β-subunits direct the Snf1 kinase to a subset of its targets in vivo. Indeed, deletion of individual β-subunit genes causes distinct differences in the induction and phosphorylation of Sip4, strongly suggesting that the β-subunits play an important role in substrate definition.
Keywords: β-subunits/Gal83/Sip1/Sip2/Snf1 protein kinase
Introduction
The Snf1 protein is a member of a highly conserved subfamily of serine/threonine protein kinases found in fungi, plants, Drosophila, Caenorhabditis elegans and mammals (Hardie et al., 1998). Members of the Snf1 subfamily appear to be central components of kinase cascades that function as metabolic sensors in eukaryotic cells. A mammalian Snf1 homolog, the AMP-activated protein kinase (AMPK), is activated by increases in the AMP:ATP ratio, a property that has led this kinase to be called the ‘fuel gauge of the mammalian cell’ (Hardie and Carling, 1997). In yeast, SNF1 (CCR1, CAT1) was first identified as a gene required for growth on alternative carbon sources such as sucrose, glycerol and ethanol (Ciriacy, 1977; Zimmermann et al., 1977; Carlson et al., 1981). Additional studies have now shown that SNF1 is also required for sporulation, glycogen storage and the transcriptional induction of glucose-repressed genes (Hardie et al., 1998).
Members of the Snf1 kinase family are heterotrimeric enzymes with the three subunits designated α, β and γ. The α-subunit contains the 300 amino acid serine/threonine protein kinase domain at the N-terminus and a regulatory domain at the C-terminus. The presence and sequence of the regulatory domain is one feature that distinguishes the Snf1 family from other protein kinases. The kinase domain of Snf1 is most closely related to the CaMK subfamily of protein kinases, which includes kinases regulated by calcium/calmodulin and the phosphorylase kinases (Hanks and Hunter, 1995). The CaMK subfamily of kinases phosphorylates peptide substrates that contain basic residues, two to three residues N-terminal to the target serine or threonine. The substrate specificity of Snf1 and AMPK has been determined and a strong preference for a basic residue at position P-3 was noted (Dale et al., 1995; Smith et al., 1999). Mammals encode at least two α-subunits and these show differences in tissue and developmental expression patterns as well as distinct substrate specificities (Verhoeven et al., 1995; Michell et al., 1996; Woods et al., 1996; Salt et al., 1998). The yeast genome encodes the SNF1 gene as the only α-subunit gene. The regulation of Snf1 activity is complex but probably involves binding of the catalytic domain to an autoinhibitory sequence present in the C-terminal regulatory domain of the protein (Jiang and Carlson, 1996).
In yeast, the γ-subunit of the Snf1 kinase is encoded by the SNF4 gene. In mammals, there are at least three γ-subunit isoforms that have been identified to date (Hardie et al., 1998). It is likely that the γ-subunit stimulates the catalytic activity by direct binding to the autoinhibitory domain in the α-subunit, thereby displacing and freeing the catalytic domain (Jiang and Carlson, 1996). The means by which glucose levels in yeast affect the competition between the γ-subunit and the catalytic domain for binding to the autoinhibitory domain is not understood, but may involve phosphorylation of the α-subunit by an upstream kinase (Wilson et al., 1996).
The focus of this paper is on the role played by the β-subunits of the Snf1 kinase complex. At a minimum, the β-subunit plays a direct role in heterotrimer formation. The β-subunits of AMPK and Snf1 are able to interact directly and independently with the α- and γ-subunits (Wilson et al., 1996; Jiang and Carlson, 1997). Further more, heterotrimer formation is essential for the reconstitution of kinase activity in co-transfection experiments (Dyck et al., 1996). The β-subunits may also play a role in substrate definition either directly through recruitment of substrate molecules (Vincent and Carlson, 1999) or indirectly through effects on subcellular localization. The mammalian β1-subunit is N-terminally myristoylated (Mitchelhill et al., 1997) and likewise, the mammalian β2-subunit contains a consensus myristoylation sequence at its N-terminus. One of the three yeast β-subunits contains the glycine residue at position 2, which is required for myristoylation, and a peptide derived from this β-subunit is myristoylated in vitro (Ashrafi et al., 1998). N-terminal myristoylation targets proteins to membrane compartments within the cell. Since one of the β-subunits in both yeast and mammals is myristoylated, it is reasonable to think that the β-subunits may also play a role in controlling the subcellular localization of alternate forms of the kinase in vivo.
Yeast encode three β-subunit genes, SIP1, SIP2 and GAL83. Immunoprecipitation studies have shown that the yeast β-subunits are indeed associated with the Snf1 and Snf4 proteins in vivo (Yang et al., 1992, 1994). What is most puzzling, however, are the genetic studies that have shown that gene disruptions of one, two or all three β-subunits have no apparent effect on Snf1 kinase function in vivo (Erickson and Johnston, 1993; Yang et al., 1994). At the time these reports were made, the yeast genome had not been completely sequenced and it was possible that additional β-subunit genes were yet to be discovered, thus explaining the lack of a phenotype in the β-subunit mutants. However, it is now certain that yeast encode three and only three β-subunit genes. We noted that the β-subunit domains that interact with the α- and γ-subunits were located in the C-terminus of the β-subunits (Jiang and Carlson, 1997). Furthermore, all three disruptions of the SIP1, SIP2 and GAL83 genes left at least parts of these domains intact, leaving open the possibility that one or more of the β-subunit disruptions encoded partially functional proteins. With the advent of PCR methods for making complete and precise deletions of yeast genes (Wach, 1996), we decided to re-examine the requirement of the β-subunit genes for Snf1 kinase function in vivo. We report here that the β-subunits are indeed required for Snf1 kinase function and that they play a role in substrate definition.
Results
The β-subunits of Snf1 kinase are required for growth on alternative carbon sources
We re-examined the yeast requirement for the β-subunits by making precise and complete deletions of each of the β-subunit genes. A PCR-based method was used to replace the entire open reading frames of each β-subunit gene with the HIS3 gene (Wach, 1996). Gene replacements were confirmed by Southern blotting of genomic DNA and the double and triple mutants were obtained by genetic crosses.
A complete set of mutations affecting each subunit of the Snf1 kinase complex was examined for growth on media containing different carbon sources (Figure 1). The snf1Δ10 allele, a deletion of all but the last five codons of the SNF1 open reading frame (Celenza and Carlson, 1989), manifests a severe Snf– phenotype. The snf1Δ10 strain grows slowly on galactose media and is unable to grow on raffinose–antimycin media or on non-fermentable carbon sources such as glycerol–ethanol. The snf4Δ1 allele, a 0.15 kb deletion within the reading frame made by Bal31 digestion (Celenza et al., 1989), displays a Snf– phenotype that is slightly less severe than that observed in the snf1Δ10 strain. A complete deletion of SNF4 made by PCR does not produce a more severe Snf phenotype than that observed with the snf4Δ1 allele (data not shown). Deletion of any one of the β-subunit genes failed to produce a detectable growth phenotype. However, deletion of all three β-subunit genes produced a severe Snf– phenotype indistinguishable from that observed with the deletion of the catalytic subunit (snf1Δ10). The sip1Δ sip2Δ gal83Δ mutant grew slowly on galactose and was unable to grow on raffinose–antimycin or glycerol–ethanol media (Figure 1). Furthermore, both the snf1Δ10 and the sip1Δ sip2Δ gal83Δ mutants grow more slowly than wild-type cells on rich media with glucose as the carbon source and both strains displayed partial inositol auxotrophy (data not shown). We conclude that the β-subunits are in fact required in vivo for Snf1 kinase function. We also examined the growth properties of the β-subunit double mutants. The sip1Δ sip2Δ and the sip1Δ gal83Δ strains grew normally on all media tested. However, the sip2Δ gal83Δ strain displayed a novel synthetic phenotype not observed for any other mutation affecting the Snf1 kinase. The sip2Δ gal83Δ strain grew poorly on glycerol–ethanol media but normally on raffinose–antimycin media, demonstrating that these two growth phenotypes are distinct and can be separated genetically.

Fig. 1. Growth properties of strains lacking Snf1 kinase subunits. Serial dilutions of wild-type cells and cells with the mutations indicated were spotted onto agar plates containing YEP media with glucose (YEP Glu) or galactose (YEP Gal), or synthetic complete media with raffinose (SC-Raf) or glycerol–ethanol (SC-GE) as the carbon source. The strains used were MSY182 (wild type), FY1193 (snf1Δ10), FY454 (snf4Δ1), MSY528 (sip1Δ), MSY520 (sip2Δ), MSY522 (gal83Δ), MSY545 (sip1Δ sip2Δ), MSY552 (sip1Δ gal83Δ), MSY543 (sip2Δ gal83Δ) and MSY558 (sip1Δ sip2Δ gal83Δ).
The C-terminal domains of the β-subunits are functional in vivo
The C-terminal domains of the β-subunits contain two conserved sequences that mediate interactions with the other subunits of the Snf1 kinase enzyme. The KIS domain (amino acids 198–350 of Gal83) interacts with the C-terminus of the Snf1 protein and the ASC domain (amino acids 344–417 of Gal83) interacts with the Snf4 protein (Jiang and Carlson, 1997). Since the C-terminal regions of all three β-subunits were left intact in the gene disruptions studied previously (Erickson and Johnston, 1993; Yang et al., 1994), we hypothesized that these fragments of the β-subunits were somehow expressed and were functional. We tested whether the gal83::URA3 allele (Erickson and Johnston, 1993) was functional by introducing it into our deletion strains on a single copy plasmid (Figure 2A). The glycerol–ethanol growth defect observed in the sip2Δ gal83Δ strain was complemented by both a wild-type GAL83 gene and also by the gal83::URA3 disruption (Figure 2B). Similarly, the triple mutant sip1Δ sip2Δ gal83Δ was also complemented for growth on raffinose and glycerol–ethanol media by the gal83::URA3 disruption allele. Therefore, the gal83::URA3 allele expresses a Gal83 derivative that is at least partially functional. The fact that the complementation was not as complete as was observed with the wild-type GAL83 gene suggests that the other disruption alleles studied previously, sip1Δ1::URA3 and sip2Δ3::LEU2, may also express functional protein fragments that contributed to the lack of an observed phenotype (Erickson and Johnston, 1993; Yang et al., 1994). The sip2Δ3::LEU2 disruption and the sip1Δ::URA3 disruption retained the C-terminal 120 and 297 codons, respectively (Yang et al., 1994). The gal83::URA3 allele retained fragments of both the N- and C-terminal domains of the GAL83 open reading frame (98 and 139 codons, respectively). In order to determine which domain was functional, plasmids were engineered to express these domains of Gal83. In addition, we also tested the homologous regions of Sip2 protein. The C-terminal 139 amino acids of Gal83 and the C-terminal 138 amino acids of Sip2 were expressed under the control of the MET3 promoter and both were found to complement the glycerol–ethanol growth defect of the gal83Δ sip2Δ strain (Figure 2C). No complementation was detected for the N-terminal domains of either protein (not shown). Thus the C-terminal domains of the Gal83 and Sip2 proteins are by themselves sufficient to confer β-subunit function.

Fig. 2. The C-terminal domains of Gal83 and Sip2 are functional. (A) Structure of the genomic GAL83 locus, its disruption and deletion alleles are shown drawn to scale. The GAL83 open reading frame is represented as an arrow (middle) with the position of the ClaI and XhoI sites, and the KIS and ASC domains indicated. The gal83Δ::HIS3 allele made by PCR in this study (top) is a complete replacement of the entire GAL83 open reading. The gal83Δ::URA3 allele was made by replacing the ClaI–XhoI fragment of GAL83 with the ClaI–SalI fragment of YEp24 (Erickson and Johnston, 1993). This construction places the C-terminal 139 amino acids of the Gal83 protein, including part of the KIS domain and all of the ASC domain, downstream but not in-frame with the N-terminal 189 amino acids of the Tet resistance protein. (B) Complementation of growth phenotypes by the gal83Δ::URA3 allele. Serial dilutions of cells transformed with URA3 CEN plasmids were spotted onto agar plates containing synthetic complete media lacking uracil with either glucose, glycerol–ethanol or raffinose as the carbon source. The strains used were MSY520 (sip2Δ), MSY543 (sip2Δ gal83Δ), MSY545 (sip1Δ sip2Δ) and MSY558 (sip1Δ sip2Δ gal83Δ). The plasmids used were pRS316 (vector), pBM2431 (GAL83) or pBM2460 (gal83Δ::URA3) (Erickson and Johnston, 1993). (C) MSY543 (sip2Δ gal83Δ) was transformed with the MET3 expression plasmid pRN500 containing either no insert (vector), or the C-terminal domains of either Gal83 or Sip2 as indicated. Cells were spotted onto synthetic complete media lacking tryptophan with either glucose (Glu) or glycerol–ethanol (GE) as the carbon source.
The β-subunits are required for invertase derepression
Derepression of invertase in response to low glucose concentrations requires the Snf1 kinase activity (Carlson et al., 1984). We examined the role of the β-subunits of Snf1 kinase in invertase regulation by using our set of strains bearing complete deletions of one or more β-subunit genes (Figure 3A). When wild-type cells grown in 2% glucose are transferred to media containing 0.05% glucose, invertase expression was induced over 35-fold. Deletion of any one of the β-subunit genes had little, if any, effect on this regulation. A similar pattern of expression was also observed in cells that were deleted for any two of the β-subunit genes. Therefore, expression of any one of the β-subunits is sufficient for invertase regulation. However, expression of at least one β-subunit is essential since deletion of all three genes completely abolished invertase derepression. The complete lack of invertase expression in the sip1Δ sip2Δ gal83Δ strain is indistinguishable from that observed in the strain lacking the catalytic subunit of the Snf1 kinase (snf1Δ10).

Fig. 3. The β-subunits of Snf1 kinase are required for regulation of invertase. Quantitative invertase assays were performed on cells with the indicated mutations. Three independent colonies of each strain were assayed and the mean is plotted with the error bar representing one standard error. The strains used are the same as those used in Figure 1. (A) Invertase was assayed using cells grown under repressing (2% glucose) and derepressing conditions (0.05% glucose). (B) Cells grown under repressing conditions were transformed with high copy plasmids YEp352 (vector) or p6A5U (STD1).
Increased gene dosage of STD1 (MSN3) induces invertase expression under repressing conditions (Hubbard et al., 1994; Zhang et al., 1998). Std1 acts upstream of the Snf1 kinase since no Std1-mediated induction of invertase (Hubbard et al., 1994) or repression of HXT1 (Schmidt et al., 1999) is observed in a snf1Δ10 mutant. In contrast, Snf4, the γ-subunit of the Snf1 kinase, is not essential for Std1-mediated induction of invertase (Hubbard et al., 1994), suggesting that Std1 may affect the activation of the Snf1 kinase. We tested whether the β-subunits are required for Std1-mediated induction of invertase by transforming our set of strains with a high copy number plasmid encoding STD1 (Figure 3B). In wild-type cells, increased gene dosage of STD1 causes a 3.4-fold increase in invertase expression. Deletion of the SNF1 gene completely blocks the ability of Std1 to induce invertase while deletion of the γ-subunit of the Snf1 kinase (SNF4) reduces but does not eliminate the Std1-mediated induction. Std1-mediated induction of invertase was observed in all combinations of single and double β-subunit mutants, although the absolute level of Std1-mediated induction was variable, ranging from 2.5- to 13-fold. Cells expressing Sip1 as the only β-subunit showed the largest Std1-mediated induction of invertase. Deletion of all three β-subunit genes completely abrogated the ability of Std1 to induce invertase expression.
Mig1 phosphorylation in vivo requires the β-subunits
Phosphorylation of the Mig1 protein in vivo requires the Snf1 kinase (Treitel et al., 1998). We analyzed the requirement of the β-subunits for the phosphorylation of Mig1 in vivo in response to glucose limitation. Cells transformed with a single copy plasmid expressing epitope-tagged Mig1 protein were shifted to low glucose media. Protein extracts were prepared and the SDS gel mobility of Mig1, an indicator of phosphorylation, was examined by western blotting (Figure 4). When cells are shifted to low glucose media, the mobility of Mig1 protein is reduced. This reduction in gel mobility is due to phosphorylation since it requires Snf1 kinase activity (Figure 4, lane 4) and can be reversed by phosphatase treatment (Treitel and Carlson, 1995). Phosphorylation of Mig1 is not observed in the strain lacking all three β-subunits, demonstrating that the β-subunits are required for Snf1 kinase activity in vivo. All three double mutants show apparently normal phosphorylation of Mig1, indicating that each of the three β-subunits is individually sufficient for directing Mig1 phosphorylation in vivo.
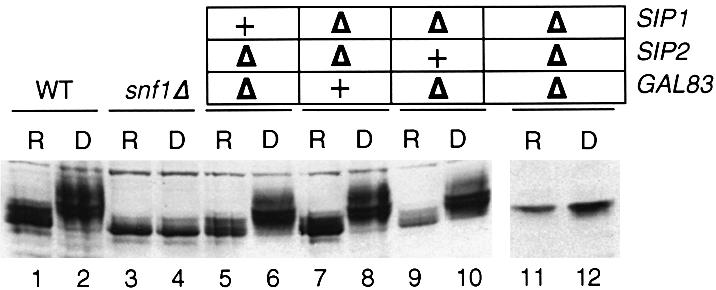
Fig. 4. The β-subunits of Snf1 kinase are required for Mig1 phos phorylation in vivo. Cells with the indicated mutations were grown under repressing (R) and derepressing (D) conditions. Protein extracts were prepared and the mobility of Mig1-HA was analyzed by western blotting.
β-subunits affect Sip4 phosphorylation in vivo
The glycerol–ethanol growth defect of the SIP1 sip2Δ gal83Δ strain suggested that the Sip1-form of the Snf1 kinase was unable to direct the phosphorylation of one or more downstream targets required for growth on non-fermentable carbon sources. One candidate for such a Snf1 substrate is the Sip4 protein, a zinc cluster transcription factor (Lesage et al., 1996) that binds to and activates expression of promoters that contain the carbon source-responsive element (Vincent and Carlson, 1998). Since Sip4 phosphorylation is Snf1 dependent (Lesage et al., 1996), we examined the requirement of the β-subunits for the phosphorylation of Sip4 protein. Tandem copies of the HA epitope tag were introduced to the C-terminus of the SIP4 reading frame in its chromosomal locus and the phosphorylation state of the Sip4 protein was inferred from its SDS gel mobility. Earlier studies have shown that a slower migrating Sip4 species appears in glucose-starved cells, is dependent on Snf1 kinase activity and is due to phosphorylation since the reduction in SDS gel mobility can be reversed by phosphatase treatment (Lesage et al., 1996). The accumulation and phosphorylation state of Sip4-HA was examined in cells bearing deletions in the genes encoding subunits of the Snf1 kinase (Figure 5). In wild-type cells grown in the presence of glucose, the Sip4-HA protein is barely detectable and the protein migrates at the faster mobility indicating that under repressing conditions, the protein is expressed at low levels and is unphosphorylated. When cells are shifted to glycerol–ethanol media for 6 h, the accumulation of Sip4-HA is greatly increased and a large fraction of the protein migrates at the reduced mobility of the phosphorylated species (Figure 5A, lane 2, and B, lane 1). In cells lacking all three β-subunit genes, very little increase in accumulation is observed and most if not all of the protein appears unphosphorylated. The same results are observed in snf1Δ10 cells (see below). We conclude that the β-subunits are required for Snf1-dependent induction and phosphorylation of Sip4-HA.

Fig. 5. Induction and phosphorylation of Sip4 protein requires specific β-subunits. Western blots of yeast cell extracts were analyzed for the accumulation and mobility of the Sip4-HA protein. The gel mobilities of the phosphorylated and unphosphorylated forms of Sip4-HA are indicated. (A) Extracts were prepared from wild-type (lanes 1 and 2) or sip1Δ sip2Δ gal83Δ cells (lanes 3 and 4) grown in glucose media (repressed; R) or after shifting to glycerol–ethanol media for 6 h (derepressed; D). (B) Cell extracts were prepared from derepressed cells of strains bearing the indicated gene deletions. Lane 1 shows a shorter exposure of lane 3.
The requirement of the individual β-subunits for Sip4 induction and phosphorylation was examined in derepressed cultures bearing deletions of Snf1 kinase subunit genes (Figure 5B). Deletion of SNF1 resulted in a complete lack of induction of Sip4 protein levels and lack of phosphorylation (lane 4). A similar result has been reported for HA-Sip4 expressed from a high copy number plasmid (Lesage et al., 1996). Deletion of the GAL83 gene greatly reduced the induction of Sip4-HA protein and the majority of the Sip4-HA migrated as unphosphorylated protein. Deletion of the SIP1 gene also reduced the induction of Sip4 protein levels but in contrast, the majority of the Sip4 protein migrated as phosphorylated protein. Cells lacking the Sip2 protein induced and phosphorylated Sip4-HA normally (lane 11). Thus deletion of the individual β-subunits had distinct effects on the expression level and the phosphorylation state of the Sip4-HA protein. Examination of Sip4-HA expression in the double β-subunit mutants showed that both the sip2Δ gal83Δ and the sip1Δ gal83Δ strains had reduced induction and phosphorylation of Sip4-HA, emphasizing the prominent role played by Gal83 in controlling Sip4-HA phosphorylation. In contrast, the sip1Δ sip2Δ strain showed near wild-type levels of Sip4-HA induction and phosphorylation.
Discussion
The Snf1 kinase and its mammalian homolog, AMPK, are heterotrimeric enzymes. The yeast α- and γ-subunits are each encoded by single genes (SNF1 and SNF4, respectively), while the β-subunit is encoded by three distinct but related genes. Genetic studies indicated that the α- and γ-subunits were essential for Snf1 kinase function in vivo (Celenza and Carlson, 1989; Celenza et al., 1989) but that the β-subunits were dispensable (Erickson and Johnston, 1993; Yang et al., 1994). This result was perplexing in light of the strong evidence from studies of the mammalian enzyme that showed that all three subunits were essential for reconstitution of enzymatic activity (Dyck et al., 1996). The data reported here solve this apparent contradiction. The β-subunits are in fact absolutely required for Snf1 kinase activity. We show that complete deletion of all three β-subunits completely inactivates the Snf1 kinase enzyme in vivo as judged by the following criteria. (i) The sip1Δ sip2Δ gal83Δ strain exhibits growth defects on media containing glycerol–ethanol, raffinose or galactose as the carbon sources (Figure 1), as well as on inositol-deficient media (not shown). (ii) The sip1Δ sip2Δ gal83Δ strain is completely unable to derepress invertase (Figure 3). (iii) The sip1Δ sip2Δ gal83Δ strain is unable to direct the phosphorylation of Mig1 and Sip4 in vivo (Figures 4 and 5). In all of these activities, the sip1Δ sip2Δ gal83Δ strain is indistinguishable from the snf1Δ10 strain, which lacks the catalytic subunit of the Snf1 kinase complex. Therefore, the β-subunits are essential for Snf1 kinase function in vivo.
If the β-subunits are required for Snf1 kinase function, why did earlier studies (Erickson and Johnston, 1993; Yang et al., 1994) fail to detect any phenotype in strains with disruptions in all three β-subunit genes? The simple answer to this question is that disruption alleles are not necessarily null alleles. We show here that the gal83::URA3 disruption allele is in fact a functional allele of GAL83. Despite the lack of regulatory sequences for transcription and translation, we hypothesize that the C-terminal 139 amino acids of Gal83 encoded by the gal83::URA3 disruption are expressed and functional. This idea is supported by deletion analysis of the Gal83 protein, which has shown that the C-terminal 219 amino acids are sufficient for interaction with the α- and γ-subunits (Jiang and Carlson, 1997). Recent studies have shown that Gal83 is required for the Snf1-dependent phosphorylation of Sip4 and that the C-terminal 83 amino acids of Gal83 are sufficient for interaction with Sip4 (Vincent and Carlson, 1999). Thus it is not unreasonable to think that the C-terminal domain of the Gal83 protein is sufficient for supplying some Gal83 functions. Furthermore, we have engineered plasmids to express the C-terminal domains of the Gal83 and Sip2 proteins, and have found that both were able to complement the glycerol–ethanol growth defect of the sip2Δ gal83Δ double mutant (Figure 2C). This finding demonstrates that the C-terminal domains of these proteins are functional and that this property is not unique to the Gal83 protein. Finally, it is worth noting that the SIP1 and SIP2 disruptions used in the earlier studies (Erickson and Johnston, 1993; Yang et al., 1994) also retain the C-terminal domains, and these alleles may contribute some β-subunit function. In particular, the C-terminal 297 amino acids of the Sip1 protein, containing the entire KIS and ASC domains are left intact in the sip1Δ1::URA3 disruption allele used in both earlier studies (Erickson and Johnston, 1993; Yang et al., 1994).
Now that true null alleles of the β-subunit genes have been constructed, it is worth reconsidering whether the three β-subunit proteins play distinct roles in vivo. The very existence of three genes suggests some specialization of function. Our studies provide two lines of evidence to support the hypothesis that each β-subunit directs the Snf1 kinase complex to a distinct subset of target proteins. First, a novel synthetic growth phenotype was uncovered in the SIP1 sip2Δ gal83Δ strain. This strain, expressing Sip1 protein as the only β-subunit, is able to grow normally on raffinose but is unable to grow on glycerol–ethanol media. The separation of these two growth phenotypes in the SIP1 sip2Δ gal83Δ strain is in marked contrast to cells lacking catalytic subunit (snf1Δ10) and unable to grow on either media. The simplest explanation for these data is that each β-subunit directs the Snf1 kinase to a subset of its targets in vivo. The targets of Snf1 phosphorylation required for growth on raffinose must be distinct from those required for growth on non-fermentable carbon sources. Mig1 is a Snf1 target that is important for growth on raffinose (Ostling et al., 1996; Treitel et al., 1998) and our studies show that Mig1 phosphorylation in vivo is apparently normal in all combinations of double β-subunit deletions. Likewise, invertase expression is required for growth on raffinose and its regulation appears normal in all three double mutants (Figure 3). Thus, any one of the β-subunits is sufficient in vivo for directing the phosphorylation of Mig1 protein, the regulation of invertase and growth on raffinose.
If the SIP1 sip2Δ gal83Δ is unable to grow on non-fermentable carbon sources, then our hypothesis predicts that a subset of Snf1 targets whose phosphorylation are critical for growth under these conditions are not properly modified. Two excellent candidates for the critical Snf1 targets that are required for growth on non-fermentable carbon sources are the Cat8 and Sip4 proteins (Randez-Gil et al., 1997; Vincent and Carlson, 1998). Indeed, the Snf1-dependent phosphorylation of Sip4 requires the Gal83 protein (Vincent and Carlson, 1999). We examined the induction and phosphorylation of the Sip4 protein in response to growth on glycerol–ethanol media. The deletion of the individual β-subunit genes produced distinct effects on the Sip4 protein. Consistent with earlier studies (Vincent and Carlson, 1999), we found that deletion of GAL83 had the largest effect on Sip4 phosphorylation. However, our studies detect a large effect of Snf1 kinase mutations on the accumulation of the Sip4 protein (Figure 5) that was not evident in studies using a high copy number plasmid to express epitope-tagged Sip4 (Vincent and Carlson, 1999). We found that deletion of either SIP1 or GAL83 greatly reduced accumulation of Sip4 protein. Although present at reduced levels, the Sip4 that was expressed in a sip1Δ strain was efficiently phosphorylated while the Sip4 present in a gal83Δ strain was not. These data, combined with the evidence for direct interaction between Sip4 and Gal83 proteins (Vincent and Carlson, 1999), lead us to propose that the Sip1 protein may exert its effect on Sip4 indirectly, perhaps through Cat8, while the Gal83 protein plays a more direct role in targeting the Snf1 kinase to phosphorylate the Sip4 protein. Taken together, the growth phenotypes and the varied effects on Sip4 accumulation and phosphorylation strongly suggest that the β-subunits play distinct roles in vivo and direct the Snf1 kinase to a subset of its targets.
Two possible mechanisms are proposed by which the β-subunits of the Snf1 kinase might direct the kinase to a subset of its targets (Figure 6). In the localization model, the β-subunits control the subcellular localization of the Snf1 kinase. While there is currently no direct evidence to support this mode of action, the β1-subunit of the mammalian AMPK has been shown to be modified by N-terminal myristoylation (Mitchelhill et al., 1997). Numerous regulatory proteins, including kinases, phosphatases and trimeric G proteins, are modified by N-terminal myristoylation and thereby targeted to the plasma membrane (Resh, 1999). One of the yeast β-subunits, Sip2, has the glycine residue at position 2 that is critical for N-terminal myristoylation, and a synthetic peptide derived from the N-terminal sequence of Sip2 is efficiently myristoylated in vitro by yeast Nmt1 (Ashrafi et al., 1998). While this evidence is circumstantial at best, the fact that both mammals and yeast express a β-subunit that can be myristoylated suggests that this β-subunit may target one form of this trimeric kinase to a specific subcellular localization. In the direct interaction model, the β-subunits specify Snf1 kinase targets through direct protein–protein interactions between substrate proteins and the β-subunits. Recent studies of the Snf1-dependent phosphorylation of Sip4 support this mechanism by demonstrating interaction between Gal83 protein and Sip4 by two hybrid assays and by immunoprecipitation (Vincent and Carlson, 1999). The studies reported support their conclusion that Gal83 plays a critical role in controlling Sip4 phosphorylation in vivo. Precedence for a regulatory subunit providing substrate specificity to the catalytic subunit of a protein kinase can be found in the cyclin-dependent protein kinases. For instance, direct interactions between the retinoblastoma protein (Rb) and the D-type cyclins are critical for the cdk4-dependent phosphorylation of Rb (Ewen et al., 1993; Kato et al., 1993). Lastly, it is important to note that these two models of substrate specification are not mutually exclusive and in fact, may be simultaneously acting to control Snf1 substrate selection.

Fig. 6. Models for β-subunit function. β-subunits may define Snf1 substrates by (A) controlling subcellular localization of the Snf1 kinase complexes or (B) by direct interactions with different substrate molecules, shown here as proteins X, Y and Z.
Our data support a model of Snf1 function in which the β-subunits are more than enzyme assembly factors holding the α- and γ-subunits in proximity. The β-subunits are essential subunits of the Snf1 kinase complex that direct the kinase to a subset of its targets.
Materials and methods
Yeast strains, media and genetic techniques
Saccharomyces cerevisiae strains utilized in this study are described in Table I. Except where indicated, growth of yeast utilized standard media at 30°C (Rose et al., 1990). For carbon sources, glucose, galactose or raffinose were present at 2% (g/100 ml) while the glycerol–ethanol mixture was present at 3% (v/v) and 2% (v/v), respectively. Antimycin A was included at 1 µg/ml in all raffinose media. Standard procedures were utilized for genetic crosses, sporulation and tetrad analysis (Rose et al., 1990). Transformations of yeast strains utilized the lithium acetate procedure (Gietz et al., 1995). Expression of the C-terminal domains of the Gal83 and Sip2 proteins was accomplished by inserting the PCR-amplified fragments encoding the C-terminal 139 and 138 amino acids of each protein into pRN500, a derivative of pRS414 containing the MET3 promoter (gift of R.Nash).
Table I. Saccharomyces cerevisiae strains used in this study.
| Strain | Genotype | Source or reference |
|---|---|---|
| MSY182 | MATa ura3-52, leu2Δ1, trp1Δ63, his3Δ200 | Ganster et al. (1998) |
| FY1193 | MATα ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 snf1-Δ10 | Fred Winston |
| FY454 | MATa ura3-52 leu2Δ1 his4-912δ lys2-128δ snf4Δ1, | Fred Winston |
| MSY528 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 | this study |
| MSY520 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip2Δ::HIS3 | this study |
| MSY522 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 gal83Δ::HIS3 | this study |
| MSY545 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 | this study |
| MSY552 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 gal83Δ::HIS3 | this study |
| MSY543 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip2Δ::HIS3 gal83Δ::HIS3 | this study |
| MSY558 | MATa ura3-52 leu2Δ1 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 gal83Δ::HIS3 | this study |
| MSY555 | MATα ura3-52 trp1Δ63 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 snf1Δ10 | this study |
| MSY548 | MATa ura3-52 leu2Δ1 his3Δ200 sip1Δ::HIS3 gal83Δ::HIS3 snf1Δ10 | this study |
| MSY562 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip2Δ::HIS3 gal83Δ::HIS3 snf1Δ10 | this study |
| MSY560 | MATa ura3-52 trp1Δ63 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 gal83Δ::HIS3 snf1Δ10 | this study |
| MSY566 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 SIP4-6HA::TRP1 | this study |
| MSY587 | MATα ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 snf1-Δ10 SIP4-6HA::TRP1 | this study |
| MSY580 | MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 gal83Δ::HIS3 SIP4-6HA::TRP1 | this study |
| MSY582 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 SIP4-6HA::TRP1 | this study |
| MSY583 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 gal83Δ::HIS3 SIP4-6HA::TRP1 | this study |
| MSY581 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip2Δ::HIS3 gal83Δ::HIS3 SIP4-6HA::TRP1 | this study |
| MSY586 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 SIP4-6HA::TRP1 | this study |
| MSY584 | MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip2Δ::HIS3 SIP4-6HA::TRP1 | this study |
| MSY585 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 gal83Δ::HIS3 SIP4-6HA::TRP1 | this study |
Gene deletions
Snf1 kinase β-subunit genes were deleted by homologous recombination using the method of PCR synthesis of marker cassettes (Wach, 1996). The HIS3 gene from plasmid pRS403 (Sikorski and Hieter, 1989) was amplified with the primers 5′-GTTGGCGGGTGTCGGGGCTGG and 5′-CATAAGAACACCTTTGGTGG, which were extended at the 5′ ends to contain 50 bases of sequence identical to the genomic sequences upstream of the ATG codons and downstream of the stop codons of the SIP1, SIP2 and GAL83 genes. Each gene was individually deleted from the wild-type strain MSY182 and confirmed by Southern blotting. Each deletion mutant was backcrossed to a wild-type strain to ensure only one HIS3 cassette had integrated and to obtain the deletion in strains of both mating types. In order to construct strains that carried multiple β-subunit deletions, deletion strains were crossed and diploids were induced to sporulate. Haploid segregants were analyzed by PCR using sets of three primers. For the SIP1 gene, the unique upstream primer (Sip1-T, 5′-GCA AAATGTACAGATGACTG) and the downstream primers (Sip1-B, 5′-GCTAGTATGACTGGCGCGTC; and His3-B, 5′-CTTCCCTGA CTAATGCCGTG) amplified a 514 bp product for the wild-type gene and a 350 bp product for the sip1Δ::HIS3 allele. A similar analysis for the SIP2 and GAL83 genes used the same His3-B primer and the following gene-specific primers: Sip2-T, 5′-ACGTTTACCTATACGAATTG; Sip2-B, 5′-ATCTCCCTCTTCATCATCGC; Gal83-T, 5′-CGTTAA AACACAGGCCACGC; Gal83-B, 5′-TCAGAATCAAAGGTCTCCCC. These primers amplified products of 538 and 338 bp for SIP2 and 514 and 375 bp for GAL83 for the wild-type and deletion alleles, respectively. Segregation of the snf1Δ10 allele was analyzed by three-primer PCR using the following primers that distinguish between the wild-type allele (567 bp product) and the deletion (402 bp product): Snf1-T, 5′-CTTACTGCGCATTCGTGTCC; Snf1-B1, 5-TTTGATGATGTG GGGGTGTC; Snf1-B2, 5′-TCATCCGAAGAAATAATGCC.
Invertase assays
Quantitative invertase assays were performed as described previously (Schmidt et al., 1999).
Epitope tagging
The Mig1 protein was epitope tagged by engineering a MunI site at the C-terminus of the MIG1 reading frame and inserting an oligonucleotide encoding three copies of the HA epitope. The MIG1-HA gene was introduced to cells on the CEN URA3 plasmid pRS316 (Sikorski and Hieter, 1989). The Sip4 protein was tagged with six tandem copies of the HA epitope using a PCR-based method (Knop et al., 1999). Strains MSY182, FY1193, MSY558, MSY545, MSY552, MSY543, MSY528, MSY520 and MSY522 were transformed to Trp+ using the recommended PCR product of plasmid pYM3 to generate the Sip4-HA tagged strains MSY566, MSY587, MSY580, MSY582, MSY583, MSY581, MSY586, MSY584 and MSY585, respectively.
Western blotting
Tagged proteins were detected by western blotting using the method described previously (Schmidt et al., 1999) except that mouse monoclonal antibody against the HA epitope was purchased from Santa Cruz Biotechnology.
Acknowledgments
Acknowledgements
We thank Mark Johnston for generously and expediently sharing plasmids and strains, Marian Carlson for suggesting experiments that strengthened our conclusions and Jana L.Patton-Vogt for testing the inositol phenotypes of our strains. This work was supported by grant GM46443 from the National Institutes of Health.
References
- Ashrafi K., Farazi,T.A. and Gordon,J.I. (1998) A role for Saccharomyces cerevisiae fatty acid activation protein 4 in regulating protein N-myristoylation during entry into stationary phase. J. Biol. Chem., 273, 25864–25874. [DOI] [PubMed] [Google Scholar]
- Carlson M., Osmond,B.C. and Botstein,D. (1981) Mutants of yeast defective in sucrose utilization. Genetics, 98, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M., Osmond,B.C., Neigeborn,L. and Botstein,D. (1984) A suppressor of SNF1 mutations causes constitutive high-level invertase synthesis in yeast. Genetics, 107, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celenza J.L. and Carlson,M. (1989) Mutational analysis of the Saccharomyces cerevisiae SNF1 protein kinase and evidence for functional interaction with the SNF4 protein. Mol. Cell. Biol., 9, 5034–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celenza J.L., Eng,F.J. and Carlson,M. (1989) Molecular analysis of the SNF4 gene of Saccharomyces cerevisiae: evidence for physical association of the SNF4 protein with the SNF1 protein kinase. Mol. Cell. Biol., 9, 5045–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriacy M. (1977) Isolation and characterization of yeast mutants defective in intermediary carbon metabolism and in carbon catabolite derepression. Mol. Gen. Genet., 154, 213–220. [DOI] [PubMed] [Google Scholar]
- Dale S., Wilson,W.A., Edelman,A.M. and Hardie,D.G. (1995) Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1 and mammalian calmodulin-dependent protein kinase I. FEBS Lett., 361, 191–195. [DOI] [PubMed] [Google Scholar]
- Dyck J.R.B., Gao,G., Widmer,J., Stapleton,D., Fernandez,C.S., Kemp,B.E. and Witters,L.A. (1996) Regulation of 5′-AMP-activated protein kinase activity by the noncatalytic β and γ subunits. J. Biol. Chem., 271, 17798–17803. [DOI] [PubMed] [Google Scholar]
- Erickson J.R. and Johnston,M. (1993) Genetic and molecular characterization of GAL83: its interaction and similarities with other genes involved in glucose repression in Saccharomyces cerevisiae. Genetics, 135, 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewen M.E., Sluss,H.K., Sherr,C.J., Matsushime,H., Kato,J. and Livingston,D.M. (1993) Functional interactions of the retino blastoma protein with mammalian D-type cyclins. Cell, 73, 487–497. [DOI] [PubMed] [Google Scholar]
- Ganster R.W., McCartney,R.R. and Schmidt,M.C. (1998) Identification of a calcineurin-independent pathway required for sodium ion stress response in Saccharomyces cerevisiae. Genetics, 150, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R.D., Schiestl,R.H., Willems,A.R. and Woods,R.A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure Yeast, 11, 355–360. [DOI] [PubMed] [Google Scholar]
- Hanks S.K. and Hunter,T. (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J., 9, 576–596. [PubMed] [Google Scholar]
- Hardie D.G. and Carling,D. (1997) The AMP-activated protein kinase—fuel gauge of the mammalian cell? Eur. J. Biochem., 246, 259–273. [DOI] [PubMed] [Google Scholar]
- Hardie D.G., Carling,D. and Carlson,M. (1998) The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem., 67, 821–855. [DOI] [PubMed] [Google Scholar]
- Hubbard E.J., Jiang,R. and Carlson,M. (1994) Dosage-dependent modulation of glucose repression by MSN3 (STD1) in Saccharomyces cerevisiae. Mol. Cell. Biol., 14, 1972–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R. and Carlson,M. (1996) Glucose regulates protein interactions within the yeast SNF1 protein kinase complex. Genes Dev., 10, 3105–3115. [DOI] [PubMed] [Google Scholar]
- Jiang R. and Carlson,M. (1997) The Snf1 protein kinase and its activating subunit, Snf4, interact with distinct domains of the Sip1/Sip2/Gal83 component in the kinase complex. Mol. Cell. Biol., 17, 2099–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J., Matsushime,H., Hiebert,S.W., Ewen,M.E. and Sherr,C.J. (1993) Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev., 7, 331–342. [DOI] [PubMed] [Google Scholar]
- Knop M., Siegers,K., Pereira,G., Zachariae,W., Winsor,B., Nasmyth,K. and Schiebel,E. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast, 15, 963–972. [DOI] [PubMed] [Google Scholar]
- Lesage P., Yang,X. and Carlson,M. (1996) Yeast SNF1 protein kinase interacts with SIP4, a C6 zinc cluster transcriptional activator: a new role for SNF1 in the glucose response. Mol. Cell. Biol., 16, 1921–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michell B.J., Stapleton,D., Mitchelhill,K.I., House,C.M., Katsis,F., Witters,L.A. and Kemp,B.E. (1996) Isoform-specific purification and substrate specificity of the 5′-AMP-activated protein kinase. J. Biol. Chem., 271, 28445–28450. [DOI] [PubMed] [Google Scholar]
- Mitchelhill K.I., Michell,B.J., House,C.M., Stapleton,D., Dyck,J., Gamble,J., Ullrich,C., Witters,L.A. and Kemp,B.E. (1997) Posttranslational modifications of the 5′-AMP-activated protein kinase β1 subunit. J. Biol. Chem., 272, 24475–24479. [DOI] [PubMed] [Google Scholar]
- Ostling J., Carlberg,M. and Ronne,H. (1996) Functional domains in the Mig1 repressor. Mol. Cell. Biol., 16, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randez-Gil F., Bojunga,N., Proft,M. and Entian,K.D. (1997) Glucose derepression of gluconeogenic enzymes in Saccharomyces cerevisiae correlates with phosphorylation of the gene activator Cat8p. Mol. Cell. Biol., 17, 2502–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh M.D. (1999) Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta, 1451, 1–16. [DOI] [PubMed] [Google Scholar]
- Rose M.D., Winston,F. and Hoeter,P. (1990) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Salt I., Celler,J.W., Hawley,S.A., Prescott,A., Woods,A., Carling,D. and Hardie,D.G. (1998) AMP-activated protein kinase: greater AMP dependence and preferential nuclear localization, of complexes containing the α2 isoform. Biochem. J., 334, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.C., McCartney,R.R., Zhang,X., Tillman,T.S., Solimeo,H., Wolfl,S., Almonte,C. and Watkins,S.C. (1999) Std1 and Mth1 proteins interact with the glucose sensors to control glucose-regulated gene expression in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 4561–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith F.C., Davies,S.P., Wilson,W.A., Carling,D. and Hardie,D.G. (1999) The SNF1 kinase complex from Saccharomyces cerevisiae phosphorylates the transcriptional repressor protein Mig1p in vitro at four sites within or near regulatory domain 1. FEBS Lett., 453, 219–223. [DOI] [PubMed] [Google Scholar]
- Treitel M.A. and Carlson,M. (1995) Repression by SSN6-TUP1 is directed by MIG1, a repressor/activator protein. Proc. Natl Acad. Sci. USA, 92, 3132–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treitel M.A., Kuchin,S. and Carlson,M. (1998) Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol. Cell. Biol., 18, 6273–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven A.J., Woods,A., Brennan,C.H., Hawley,S.A., Hardie,D.G., Scott,J., Beri,R.K. and Carling,D. (1995) The AMP-activated protein kinase gene is highly expressed in rat skeletal muscle. Alternative splicing and tissue distribution of the mRNA. Eur. J. Biochem., 228, 236–243. [PubMed] [Google Scholar]
- Vincent O. and Carlson,M. (1998) Sip4, a Snf1 kinase-dependent transcriptional activator, binds to the carbon source-responsive element of gluconeogenic genes. EMBO J., 17, 7002–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent O. and Carlson,M. (1999) Gal83 mediates the interaction of the Snf1 kinase complex with the transcription activator Sip4. EMBO J., 18, 6672–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A. (1996) PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast, 12, 259–265. [DOI] [PubMed] [Google Scholar]
- Wilson W.A., Hawley,S.A. and Hardie,D.G. (1996) Glucose repression/derepression in budding yeast: SNF1 protein kinase is activated by phosphorylation under derepressing conditions and this correlates with a high AMP:ATP ratio. Curr. Biol., 6, 1426–1434. [DOI] [PubMed] [Google Scholar]
- Woods A., Salt,I., Scott,J., Hardie,D.G. and Carling,D. (1996) The α1 and α2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett., 397, 347–351. [DOI] [PubMed] [Google Scholar]
- Yang X., Hubbard,E.J. and Carlson,M. (1992) A protein kinase substrate identified by the two-hybrid system. Science, 257, 680–682. [DOI] [PubMed] [Google Scholar]
- Yang X., Jiang,R. and Carlson,M. (1994) A family of proteins containing a conserved domain that mediates interaction with the yeast SNF1 protein kinase complex. EMBO J., 13, 5878–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Shen,W. and Schmidt,M.C. (1998) Amino acid residues in Std1 protein required for induction of SUC2 transcription are also required for suppression of TBPΔ57 growth defect in Saccharomyces cerevisiae. Gene, 215, 131–141. [DOI] [PubMed] [Google Scholar]
- Zimmermann F.K., Kaufmann,I., Rasenberger,H. and Haubetamann,P. (1977) Genetics of carbon catabolite repression in Saccharomyces cerevisiae: genes involved in the derepression process. Mol. Gen. Genet., 151, 95–103. [DOI] [PubMed] [Google Scholar]