

Abstract
Endometrial cancer is the most commonly diagnosed female genital tract malignancy. Krüppel-like factor 9 (KLF9), a member of the evolutionarily conserved Sp family of transcription factors, is expressed in uterine stroma and glandular epithelium, where it affects cellular proliferation, differentiation, and apoptosis. Deregulated expression of a number of Sp proteins has been associated with multiple types of human tumors, but a role for KLF9 in endometrial cancer development and/or progression is unknown. Here, we evaluated KLF9 expression in endometrial tumors and adjacent uninvolved endometrium of women with endometrial carcinoma. KLF9 mRNA and protein levels were lower in endometrial tumors coincident with decreased expression of family member KLF4 and growth-regulators FBJ murine osteosarcoma viral oncogene homolog (FOS) and myelocytomatosis viral oncogene homolog (MYC) and with increased expression of telomerase reverse transcriptase (TERT) and the chromatin-modifying enzymes DNA methyltransferase 1 (DNMT1) and histone deacetylase 3 (HDAC3). Expression of estrogen receptor alpha (ESR1) and the tumor-suppressor phosphatase and tensin homolog deleted in chromosome 10 (PTEN) did not differ between tumor and normal tissue. The functional relevance of attenuated KLF9 expression in endometrial carcinogenesis was further evaluated in the human endometrial carcinoma cell line Ishikawa by siRNA targeting. KLF9 depletion resulted in loss of normal cellular response to the proliferative effects of estrogen concomitant with reductions in KLF4 and MYC and with enhancement of TERT and ESR1 gene expression. Silencing of KLF4 did not mimic the effects of silencing KLF9 in Ishikawa cells. We suggest that KLF9 loss-of-expression accompanying endometrial carcinogenesis may predispose endometrial epithelial cells to mechanisms of escape from estrogen-mediated growth regulation, leading to progression of established neoplasms.
Keywords: endometrial carcinoma, endometrium, estradiol/estradiol receptor, Ishikawa cells, Krüppel-like factor 9, telomerase, uterine cancer, uterus
The loss of expression of transcription factor Krüppel-like factor 9 in human endometrial cells leads to aberrant growth regulation and underlies the progression of endometrial carcinoma.
INTRODUCTION
Endometrial carcinoma (EC) is the most common gynecologic malignancy of women in the United States, with approximately 40 000 new cases and 8000 deaths reported in 2009 [1]. EC consists of two major histological subtypes—namely, the typically low-grade endometrioid EC (type I), which is associated with a favorable outcome, and the nonendometrioid EC (type II), which is high grade and correlated with poor prognosis. The estrogen-dependent type I EC develops from endometrial glands and accounts for approximately 80% of all cases. Risk factors for type I include early menarche, late menopause, nulliparity, obesity, and estrogen-only hormone replacement therapy [2]. Despite the fact that EC is readily treatable when diagnosed early, the mean 5-yr survival rate decreases by 70% when metastasis occurs [1]. Detection of atypical hyperplasia in endometrial biopsies is associated with increased risk of subsequent carcinoma; by contrast, simple hyperplasia and complex hyperplasia have low risk of progression and the potential for overdiagnosis and overtreatment [2]. Given the physical, emotional, and increasing financial burdens of EC and its potential misdiagnosis, the current detection and treatment paradigms for this disease could benefit from a continuing search to find new biomarkers for risk prediction.
The uterus, predominantly under the influence of the ovarian steroid hormones 17β-estradiol (estrogen [E2]) and pregn-4-ene-3,20-dione (progesterone [P4]), undergoes cycles of proliferation, differentiation, and apoptosis necessary to support its key reproductive function—namely, the implantation of a viable embryo leading to successful pregnancy [3]. E2 and P4 exert their actions by binding to cognate-receptors estrogen receptor-α and -β (ESR1 and ESR2) and progesterone receptors A and B (PGR-A and PGR-B), respectively [4, 5]. The actions of E2 and P4 leading to transcriptional activation of specific genes that modify cellular phenotype and behavior have resulted in a generalized molecular scheme for ESR and PGR signaling [4, 6], but these pathways have become increasingly complex due to the participation of numerous nuclear coregulators, each of which demonstrates specific spatial and temporal expression and differentially affects ESR and PGR activities [7, 8].
Our laboratory has identified the transcription factor Krüppel-like factor (KLF) 9, an Sp-family member [9], as a transcriptional coregulator of ESR1 and PGR-B signaling in uterine cells [10–15]. KLF9 is expressed in uterine endometrial stroma and glandular, but not luminal, epithelium in mice [16], a finding recently confirmed for human endometrial cells [17]. Using mice null for KLF9 [16], we demonstrated the numerous contributions of KLF9 to uterine morphology; embryo implantation, and fertility; sensitivity to E2 and P4; and timing of parturition [12, 14, 15, 18–20]. These findings have raised the possibility of yet unidentified roles for KLF9 in the etiology of gynecological diseases such as endometriosis and EC, which are characterized by unopposed E2 signaling coincident with increased P4 resistance. Notably, these findings also suggest the intriguing likelihood that KLF9 and/or its gene targets, the latter regulated in concert with PGR and ESR1, may serve as prognostic predictors for these uterine-associated diseases.
Initial studies from several groups, including our own, have suggested that KLF9 expression is attenuated or lost in human and mouse uterine endometriotic and EC tissues, EC cell lines, and human uterine leiomyoma [21–23]. Despite these findings, it remains unclear whether diminished KLF9 expression is causal to the manifestation of these diseases. The present study was designed to test the hypothesis that altered expression status of growth-regulatory genes is associated with aberrant KLF9 expression in EC. Using human EC tissue and cell line, we demonstrate that loss of KLF9 expression may predispose to mechanisms of escape from estrogen-mediated growth regulation to promote the progression of EC.
MATERIALS AND METHODS
Materials
Reagents were obtained from the following commercial sources: Dulbecco modified Eagle medium (DMEM), antibiotic-antimycotic (ABAM), Lipofectamine 2000 transfection reagent, OPTI-MEM I reduced serum medium, and TRIzol reagent from Invitrogen/GIBCO; Eagle minimum essential medium (MEM), dimethyl sulfoxide (DMSO), and E2 from Sigma-Aldrich; fetal bovine serum (FBS) from Fisher Scientific; nontargeting (scrambled [Scr]), KLF9, and KLF4 siRNAs from Dharmacon, Inc.; MTT Cell Proliferation Assay Kit from American Type Culture Collection; and Caspase-Glo 3/7 Apoptosis Assay Kit from Promega Corp.
Biological Samples
Tissue samples were obtained from 13 patients recruited from a cohort of women diagnosed with EC and undergoing hysterectomy at the University of Arkansas for Medical Sciences (UAMS) between October 2009 and April 2010. Subject recruitment and related procedures for tissue harvest at surgery followed the protocol approved by the UAMS Institutional Review Board (IRB 11185). Inclusion criteria included age (25–75 yr) and no previous diagnosis of cancer. Risk-related information, such as age, parity, oral contraceptive pill use, body mass index (BMI), and in utero exposure to synthetic estrogens, was collected from each subject and considered in the data analyses. EC and surrounding nontumor (normal endometrium [NE]) tissues were excised by a surgical pathologist. Collected tissues were bisected and snap-frozen for subsequent molecular analyses. A portion of the EC for each subject was analyzed at the UAMS Pathology Core Laboratory as part of the routine cancer diagnosis, confirmation, grading, and staging.
Cell Culture and Treatments
The human EC cell line Ishikawa was maintained at 37°C in an atmosphere of 5% CO2/95% air in flasks containing DMEM supplemented with 10% (v/v) FBS and 1% (v/v) ABAM (DMEM-FBS) [24]. For siRNA targeting studies, Ishikawa cells were seeded in six-well plates at a density of 6 × 105 cells/well and incubated overnight in ABAM-free DMEM-FBS. Cells were treated for 6 h with Lipofectamine 2000/OPTI-MEM I containing human KLF4 or KLF9 siRNAs and, as control, Scr siRNAs (50 nM each). After transfection, cells were incubated overnight in MEM supplemented with 0.5% (v/v) charcoal-stripped FBS and 1% ABAM (5% CS-FBS-MEM). For proliferation and apoptosis studies, cells were seeded in 96-well plates at a density of 3 × 104 cells/well and processed following the siRNA transfection procedures. Treatment with medium (10% [v/v] CS-FBS-MEM) containing 0.1% (v/v) DMSO (vehicle) or 100 nM E2 in medium was performed for the indicated times as described for each experiment. Cell proliferation was measured as a function of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide oxidation (MTT Cell Proliferation Assay Kit), whereas apoptosis was measured as a function of caspase 3/7-facilitated luciferase activity (Caspase-Glo 3/7 Apoptosis Assay Kit), both following the manufacturer's instructions. Experiments were performed in quadruplicate (siRNA transfections) or quintuplicate (proliferation and apoptosis) on two or three separate occasions.
RNA Isolation, Quantitative RT-PCR, and Western Blot Analyses
Total RNA was isolated from uterine tissues or cells with TRIzol reagent following the manufacturer's protocol. RNA concentrations were determined using an ND-1000 spectrophotometer (Nanodrop). The cDNA was synthesized from 1 μg of total RNA using the Taqman Reverse Transcription Reagents Kit (Applied Biosystems) or the iScript cDNA Synthesis Kit (Bio-Rad), then analyzed by quantitative RT-PCR (QPCR) using SYBR Green (Bio-Rad) and the MyiQ optical module and iCycler thermal cycler (Bio-Rad) as previously described [24]. Intron-flanking primers (Table 1) were designed to eliminate the amplification of genomic DNA using Primer Express (Applied Biosystems) or PrimerQuest (Integrated DNA Technologies, Inc.) and were obtained from Integrated DNA Technologies, Inc. Western blot analyses were carried out as previously described [14, 20] using rabbit anti-rat KLF9 antibody [20] and antibodies against glyceraldehyde phosphate dehydrogenase (GAPDH; monoclonal anti-GAPDH; Sigma) and lamin A (Sigma) as loading controls.
TABLE 1.
Human primer sequences used for QPCR.

Telomere Length Measurement
Telomere measurement was performed as previously described [25]. Genomic DNA was isolated using the QIAamp DNA Mini Kit (Qiagen). DNA (15 ng) was used in a 30-μl reaction containing SYBR Green (Bio-Rad) and the following primers: Tel A (5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′) and Tel B (5′-GGCTTG CCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′). Primers A and B were used at a final concentration of 100 and 300 nM, respectively. Standards were generated by serial dilution of pooled DNA in the range of 15 to 0.469 ng.
Data Analysis
Statistical analyses were performed using SigmaStat (version 3.5; Systat Software). Data (mean ± SEM) were analyzed for statistical significance (P ≤ 0.05) by two-way ANOVA followed by all pairwise multiple-comparison procedures (Tukey test) or Student t-test.
RESULTS
Demographic Information for Study Subjects
Table 2 summarizes the demographic features of the women in the present study. The average age, age at menarche, and number of years of menstruation were 51 yr (range, 30–69 yr), 12 yr (range, 10–13 yr), and 35 yr (range, 19–47 yr), respectively. Only 1 of 13 women (7.7%) had a BMI within the normal range (BMI, 19–24), whereas the remainder (12/13, 92.3%) were classified as obese or extremely obese (BMI, >30). Nine of the 13 women had at least one successful delivery.
TABLE 2.
Demographic features of women enrolled in endocarcinoma study.a

Reduced KLF9 Expression in EC
Endometrial tumors and adjacent tissues collected from women in the study were processed for hematoxylin-and-eosin (H&E) staining, and tumors were scored by grade and stage (Table 2). Representative H&E-stained sections of normal and tumor tissues from two patients are shown in Figure 1A. Because of the small cohort size and most of the tumors (9/13) being grade 1, adjacent NE and EC tissues were collectively analyzed, irrespective of tumor stage and grade. Compared to NE tissues, EC tissues had decreased KLF9 transcript levels (by 50%; P = 0.001) (Fig. 1B). The reduction in transcript levels was confirmed at the protein level by Western blot analysis (Fig. 1C). Densitometric quantification of the anti-KLF9 immunoreactive bands indicated significantly lower KLF9 protein in EC compared to NE (Fig. 1C). Among family members, KLF4 transcript levels were down-regulated (P = 0.03), but those for KLF13 were up-regulated (P = 0.03), in EC relative to NE (Fig. 1B). In contrast, KLF5 and KLF6 transcript levels did not differ with tumor status.
FIG. 1.

Analyses of KLF9 expression in human EC tissues. A) Representative H&E-stained sections of adjacent NE and EC tissues from two patients undergoing hysterectomy after diagnosis with EC. GE, glandular epithelium; LE, luminal epithelium; ST, stroma. Original magnification ×10. B) Messenger RNA transcript levels of KLF9 and KLF family members were quantified by QPCR and normalized to those of TATA box-binding protein (TBP). Data (mean ± SEM) are expressed as the fold-change relative to corresponding NE (n = 13 samples/tissue type). C) Western blot analyses of nuclear proteins isolated from NE and EC tissues using rabbit anti-rat KLF9 and anti-GAPDH (loading control) antibodies. A representative blot (left) with the quantified data from densitometric scans of normalized immunoreactive bands from NE and EC samples pooled from individual patients (right) is shown. *P < 0.05 (Student t-test).
Changes in Expression of Growth-Associated Genes with Loss of KLF9 Expression in EC
To determine whether attenuated KLF9 expression in EC is associated with loss of growth control, the expression of a subset of growth-regulatory genes was evaluated in NE and EC by QPCR. We previously reported the aberrant expression of these genes in Klf9-null mice with deregulated ESR1 signaling due to early postnatal exposure to diethylstilbestrol (DES) [15]. Loss of KLF9 expression in EC was accompanied by decreased expression of FBJ murine osteosarcoma viral oncogene homolog (FOS; P = 0.03), a modest down-regulation of myelocytomatosis viral oncogene homolog (MYC; P = 0.08), and a dramatic increase in telomerase reverse transcriptase (TERT; by 20-fold; P = 0.02) transcript levels (Fig. 2A). ESR1 as well as phosphatase and tensin homolog deleted in chromosome 10 (PTEN) transcript levels were unchanged with carcinoma status. The increase in epithelial-specific cadherin (CDH1) transcript levels in EC relative to NE, which contrasts with the comparable transcript levels of stromal-specific vimentin (VIM) in EC and NE (Fig. 2A), is consistent with the higher glandular epithelial status associated with the transformation of NE to EC [2].
FIG. 2.
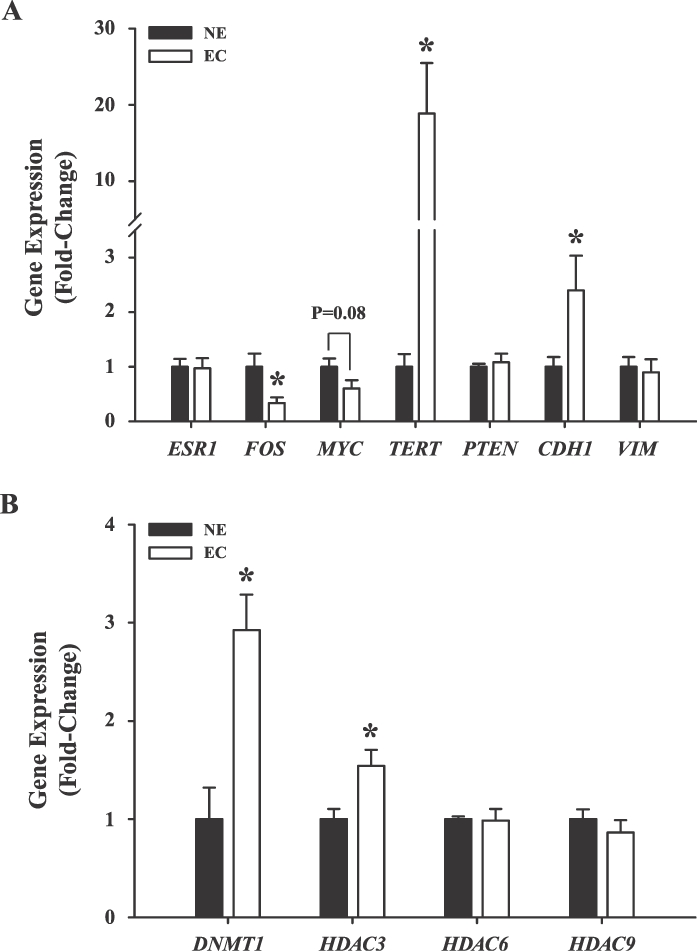
The expression of growth-associated genes in EC versus NE. The expression levels of ESR1, FOS, MYC, TERT, PTEN, CDH1, and VIM (A) and of DNMT1, HDAC3, HDAC6, and HDAC9 (B) were quantified in NE and EC tissues by QPCR. Data (mean ± SEM, n = 13 samples/tissue type) were normalized to TATA box-binding protein (TBP) and renormalized to values for NE. *P < 0.05 (Student t-test).
Because altered expression of tumor-suppressor genes and oncogenes in cancer has been attributed in part to epigenetic mechanisms leading to gene silencing or activation, the expression of a number of chromatin-modifying enzymes were compared in NE and EC. The transcript levels of DNA methyltransferase 1 (DNMT1) and histone deacetylase 3 (HDAC3) were similarly up-regulated in EC relative to NE, whereas those for HDAC6 and HDAC9 were not different between tumor and nontumor endometria (Fig. 2B).
The enhanced expression of TERT with EC (Fig. 2A) suggested elongated telomeres with EC status. However, no significant differences (P = 0.45) in the mean telomere length (copy number) were observed between NE (14.88 ± 2.99) and EC (11.99 ± 2.24) tissues.
Expression of Growth-Regulatory Genes with KLF9 Knockdown
To evaluate the causal relationship between attenuated KLF9 expression in EC and the deregulated expression of growth-regulatory genes that might underlie the genesis and/or progression of endometrial carcinogenesis, siRNA targeting of KLF9 expression in Ishikawa human EC cells was performed. Cells were transfected with control Scr siRNA or siRNA directed against human KLF9 for 6 h and then treated with vehicle containing 100 nM E2 for 72 h, the latter to mimic the high estrogenic environment of EC. Transcript levels for KLF9 and growth-regulatory genes (as described in Fig. 2A) were then measured by QPCR. Transfection of E2-treated cells with KLF9 siRNA reduced KLF9 transcript levels by 60% (P < 0.001); this was accompanied by a greater than 90% loss of KLF9 protein, as determined by Western blot analyses of nuclear proteins from Scr siRNA- and KLF9 siRNA-transfected cells (Fig. 3, inset). Attenuated KLF9 enhanced ESR1 (P = 0.005), FOS (P = 0.043), and TERT (P = 0.004) expression; reduced KLF4 (P < 0.001) and MYC (P < 0.001) expression; and had no effect on KLF13 expression (Fig. 3). The increase in ESR1 expression with KLF9 knockdown under conditions of high E2 recapitulated that found for Ishikawa cells in a previous study [13]. Moreover, increased TERT expression (by ∼2.5-fold) and decreased KLF4 and MYC expression with KLF9 siRNA targeting (Fig. 3) mimicked that observed in EC relative to NE (Fig. 2A). However, the 2-fold increase in FOS expression and the lack of change in KLF13 expression with KLF9 knockdown relative to control siRNA-transfected cells differed from that observed in EC versus NE (Fig. 2A).
FIG. 3.

Altered gene expression upon KLF9 gene silencing in human Ishikawa cells treated with E2. The transcript levels of KLF9 and other genes for which expression were altered in EC versus NE (see Fig. 2) were quantified in Ishikawa cells transfected with either control (Scr) siRNA or siRNA directed against KLF9 and treated with vehicle (0.1% DMSO in treatment medium; data not shown) or 100 nM E2 in vehicle for 72 h. Data (mean ± SEM) were normalized to TBP and are expressed as the fold-change relative to vehicle-treated, scRNA-transfected controls (n = 4 individual wells/transfection). Inset) KLF9 knockdown resulted in more than 90% loss of KLF9 protein levels. *P < 0.05 (Student t-test).
KLF9 Knockdown Effects on Cell Proliferation and Apoptosis
To directly link the loss of KLF9 regulation of growth-regulatory genes in endometrial glands with disruption of cellular proliferation and apoptosis occurring with EC status [26], control (vehicle alone) and E2-treated Ishikawa cells transfected with Scr or KLF9 siRNA were assayed for cell proliferation (MTT) and apoptosis (induction of caspase3/7 activity) after 72 h. As expected, E2 treatment increased cell proliferation in cells normally expressing KLF9 (P = 0.04) (Fig. 4A). KLF9 knockdown in non-E2-treated cells increased proliferation (P = 0.03) to the same extent as that found for E2-treated, KLF9-expressing cells. However, the number of epithelial cells upon E2-treatment was decreased with KLF9 knockdown, suggesting the loss of normal proliferative response to E2 of cells lacking KLF9 (Fig. 3B). Cellular apoptosis was disrupted by KLF9 knockdown but only in the background of high E2 (Fig. 4B). Notably, a modest but significant decrease in apoptosis (P < 0.05) was observed under estrogenic conditions in cells lacking KLF9, which was not observed in KLF9-expressing cells.
FIG. 4.
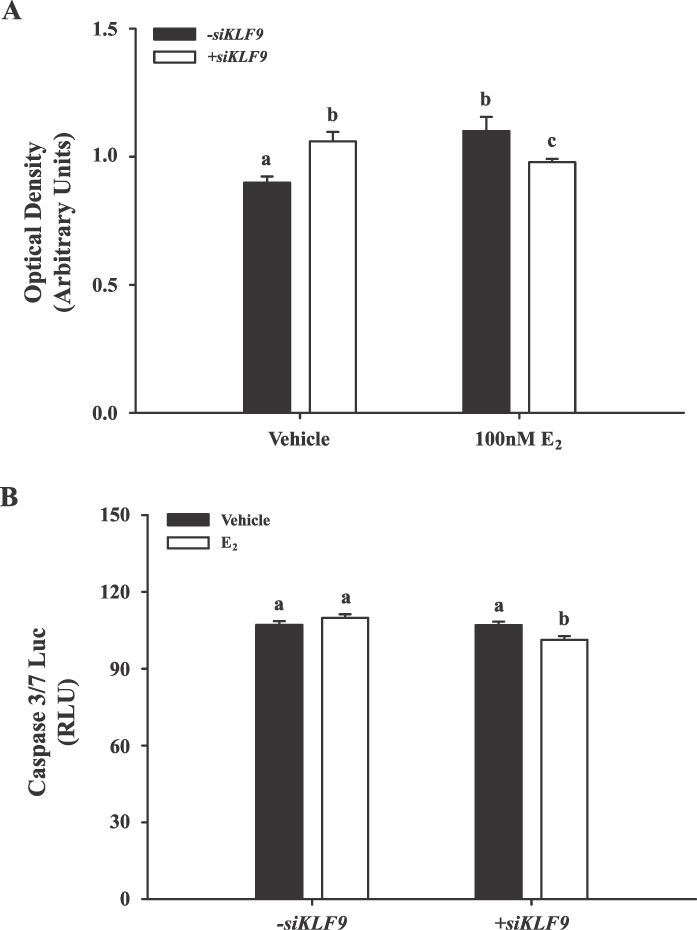
Proliferative and apoptotic responses of human Ishikawa cells to KLF9 gene silencing. A) Proliferation status of Ishikawa cells transfected with either Scr or KLF9 siRNAs and treated with vehicle (0.1% DMSO in treatment medium) or 100 nM E2 was measured using the MTT assay after 3 days. Data (mean ± SEM) are from five independent experiments. B) Apoptotic status of Ishikawa cells transfected with either Scr or KLF9 siRNAs and treated with vehicle (0.1% DMSO in treatment medium) or 100 nM E2 for 72 h was measured using the caspase 3/7-Luciferase assay kit and expressed as relative light units (RLUs). Data (mean ± SEM) are from five independent experiments. Means with different letters differed significantly (P < 0.05, two-way ANOVA followed by Tukey test).
KLF4 Effects on KLF9-Regulated Genes
Given the coincident loss of KLF4 and KLF9 expression in EC (Fig. 1B) and in Ishikawa cells under a background of high E2 (Fig. 3), we determined whether KLF4 mediates KLF9 regulation of MYC and TERT gene expression, which was decreased and increased, respectively, with loss of KLF9 expression (Fig. 2). Ishikawa cells were transfected with Scr siRNA or siRNA targeting KLF4 and then treated with E2 (100 nM). Transfection with KLF4 siRNA resulted in a significant reduction (by 60%; P < 0.001) of KLF4 expression (Fig. 5). However, both MYC and TERT expression were unaffected by KLF4 knockdown.
FIG. 5.

Analyses of gene expression as a function of KLF4 gene silencing in Ishikawa cells treated with E2. Expression of KLF4, MYC, and TERT in Ishikawa cells transfected with either Scr or KLF4 siRNAs and treated with vehicle (0.1% DMSO in treatment medium; data not shown) or 100 nM E2 for 72 h was quantified by QPCR. Data (mean ± SEM) were normalized to TBP and are expressed as the fold-change relative to vehicle-treated, scRNA-transfected controls (n = 4 individual transfections/treatment). *P < 0.05 (Student t-test).
DISCUSSION
Previous reports have shown KLF9 to influence the transcription of PGR- and ESR1-regulated genes [10, 11, 13, 15], to mediate cell-cycle component expression [27], and to control cell proliferation and apoptosis [18, 27]. Female mice mutant for Klf9 exhibited subfertility [12] and delayed onset of parturition [19], resulting in part from defective PGR signaling. Moreover, endometriosis, a P4-resistant condition, was associated with loss of KLF9 expression in eutopic endometrium of an experimental mouse model of endometriosis [22]. To further understand how disruptions in steroid hormone receptor coregulator functions may lead to uterine pathology, we examined whether KLF9 expression is deregulated in EC and, if so, whether this is linked to loss of normal growth regulation of endometrial epithelial cells. We found that KLF9 mRNA and protein levels were significantly attenuated in EC relative to NE. The loss of KLF9 expression likely reflects a true down-regulation rather than a change in the proportion of KLF9-expressing cells between EC and NE, given the higher glandular epithelial status associated with the transformation of NE to EC [2]. Notably, the loss of KLF9 expression was accompanied by increased expression of TERT, KLF13, DNMT1, and HDAC3 and decreased expression of FOS, KLF4, and MYC. By using the human EC cell line Ishikawa, we confirmed KLF9-mediated up-regulation of TERT and down-regulation of KLF4 and MYC expression by silencing KLF9 in glandular epithelial cells under a high-estrogenic environment, mimicking that found in EC. The lack of confirmatory responses in vitro of KLF13 (no change) and FOS (increased rather than decreased) gene expression and the dramatic induction in ESR1 expression with KLF9 silencing in Ishikawa cells likely reflect the distinct, cumulative effects of KLF9 loss-of-expression in whole endometrial tissue as opposed to isolated glandular epithelial cells. In this regard, whereas KLF13 is predominantly expressed in endometrial stromal cells [20], FOS and ESR1 are ubiquitously expressed in all endometrial cell types. Finally, we showed, to our knowledge for the first time, the existence in endometrial glandular epithelial cells of a regulatory link between KLF9 and KLF4 on one hand and between KLF9 and TERT on the other, which has important implications for growth control.
Our studies identified KLF4 and TERT as candidate KLF9 downstream targets by which KLF9 may contribute to the progression of endometrial carcinogenesis. To our knowledge, this is the first report of decreased KLF4 expression in EC. KLF4 functions as a prodifferentiation and antiproliferative protein [28] and is a downstream target [29] as well as a transcriptional repressor [30] of the tumor-suppressor p53. KLF4 has also been reported to inhibit oncogene WNT signaling by interacting with β-catenin, thus disrupting β-catenin-mediated recruitment of the coactivators p300/CBP [31]. KLF9 regulation of KLF4 expression aligns with our previous findings showing that forced overexpression of KLF9 in the human EC HEC-1A cells resulted in increased KLF4 transcript levels [21]. The coincident down-regulation of KLF4 and KLF9 has also been reported in human colorectal cancer [32, 33]. Together, these findings raise the intriguing possibility of interactions of KLF9, through its regulation of KLF4, with p53 (TP53) and WNT signaling pathways, both of which are deregulated in cancer, and may offer important clues for understanding the contribution of KLF9 to growth regulation.
Of particular interest is the negative correlation between KLF9 and TERT expression in EC and in E2-treated Ishikawa cells, consistent with our previous report of enhanced Tert expression in uteri of Klf9-null mice upon deregulation of uterine Esr1 signaling because of early postnatal exposure to DES [15]. TERT encodes the telomerase gene transcriptase, which constitutes the catalytic subunit of the telomerase holoenzyme [34]. Previous studies have shown that a strong association exists between TERT transcript levels and telomerase activity [34, 35], that TERT expression specifically correlates with cell proliferation [36], and that TERT activity is increased in human EC relative to the normal cycling endometrium [37–39]. Furthermore, an E2-dependent, ESR1-facilitated increase in TERT has been reported in various tissues [40–42]. The latter raises the possibility that the observed increase in ESR1 expression with KLF9 siRNA targeting in glandular epithelial cells [13] (present study) underlies enhanced TERT expression, providing a common pathway by which KLF9 and ESR1 mediate endometrial TERT expression. In addition, given the role for TERT in promoting progression of established neoplasms [43], our data also suggest a mechanism by which KLF9 loss-of-expression may promote a cellular context favoring tumorigenesis. The lack of the coincident increase in telomere length with increased telomerase activity in EC relative to NE is in keeping with mounting evidence for telomere length-independent pathways of growth stimulation by TERT [44, 45].
The coincident loss of KLF9 and KLF4 with increased TERT expression suggests a transcriptional cross-regulatory network between these KLFs and TERT, as reported in mouse embryonic fibroblasts wherein Tert overexpression resulted in the coincident down-regulation of Klf4 and Klf9 expression [36]. However, KLF4 knockdown in Ishikawa cells failed to elicit TERT up-regulation, identical to that found with KLF9 knockdown, indicating distinct cell type-specific programs of gene regulation. Furthermore, attenuated KLF4 expression had no effect on KLF9 expression (data not shown). Thus, in the hierarchy of KLF family cross-regulation, KLF9 may occupy a higher “seat,” given that it also influences the expression of KLF13 [20] as well as KLF5 and KLF6 (data not shown), albeit under different endometrial cell contexts.
Several other findings from the present study are novel and worth noting. First, KLF9 knockdown in E2-treated Ishikawa cells resulted in the loss of normal epithelial response to E2 (i.e., increased proliferation). The decrease in glandular cell numbers observed by blocking KLF9 expression is in agreement with our previous finding of lower gland numbers, but increased gland sizes, in DES-exposed uteri from Klf9-null mice compared to those of KLF9-expressing (wild-type) mice [15]. In keeping with the association of resistance to apoptosis with tumorigenicity, we found a modest, albeit significant, decrease in apoptotic status of E2-treated cells upon KLF9 knockdown. Second, despite reports that PTEN is one of the most frequently altered genes in EC [46, 47], we did not find a correlation between PTEN expression and EC status in the present study. Whereas the latter may be related to the small sample size and the use of adjacent (nontumor) tissues as control for EC tissues in the present study, our results are consistent with those in a recent report suggesting loss of PTEN expression as an unreliable marker of progression to EC [48]. Nevertheless, because forced expression of PTEN in PTEN-null Ishikawa cells was found to inhibit TERT expression [49], early loss of endometrial PTEN expression may facilitate neoplastic transformation in part by enhancing TERT expression, similar to the effect elicited by KLF loss. Third, the increased expression of the chromatin-modifying enzymes DNMT1 and HDAC3 in EC is consistent with the reported epigenetic deregulation in EC [50]. Indeed, aberrant promoter methylation of PGR [51] and of the WNT-inhibitor adenomatosis polyposis coli [52] in EC had been proposed as possible mechanisms for alterations in gene expression noted with this disease. Finally, the positive association of obesity and incidence of EC, a well-established phenomenon based on epidemiological studies [53] in our patient cohort with loss of endometrial KLF9 expression, is intriguing given the recent report implicating KLF9 in adipogenesis [54]. Our collective data raise the possibility that KLF9-promoter silencing, consequent to the deregulated expression of a number of chromatin-modifying enzymes, may facilitate the progression of EC. Future studies at the level of the KLF9 promoter will address this predicted mechanism.
In summary, using a subset of previously identified growth-associated genes for which expression was altered in uteri of Klf9 mice with dysregulated Esr1 signaling [15] to query EC and NE tissues, we have provided support for the functional relevance of KLF9 to human EC. We suggest that KLF9 as well as its downstream targets KLF4 and TERT may function as novel predictors/diagnostic markers of EC status. Future work utilizing a larger cohort of patients as well as further exploration of the role of KLF9 in glandular epithelial biology will expand our current understanding of the regulatory grid mediated by KLF9 in the pathogenesis of EC and, perhaps, other reproductive diseases.
ACKNOWLEDGMENTS
We thank members of our laboratories for their insights and helpful discussions during the course of these studies and Margie Brackeen, Arkansas Children's Hospital Research Institute, for her invaluable assistance during the preparation of IRB protocols.
Footnotes
Supported by National Institutes of Health grant HD21961 (R.C.M.S.), Arkansas Children's Hospital Research Institute (R.C.M.S., C.D.S.), the University of Arkansas for Medical Sciences Committee for Allocating Graduate Student Research Funds Award (C.D.S.), and the Southern Regional Education Board State Doctoral Scholars Program (C.D.S.).
REFERENCES
- American Cancer Society. Cancer Facts & Figures 2009. Atlanta: American Cancer Society; 2009. [Google Scholar]
- Sherman ME. Theories of endometrial carcinogenesis: a multidisciplinary approach. Mod Pathol 2000; 13: 295 308. [DOI] [PubMed] [Google Scholar]
- Dey SK, Lim H, Das SK, Reese J, Paria BC, Daikoku T, Wang H. Molecular cues to implantation. Endocr Rev 2004; 25: 341 373. [DOI] [PubMed] [Google Scholar]
- Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 1993; 63: 451 486. [DOI] [PubMed] [Google Scholar]
- Franco HL, Jeong JW, Tsai SY, Lydon JP, DeMayo FJ. In vivo analysis of progesterone receptor action in the uterus during embryo implantation. Semin Cell Dev Biol 2008; 19: 178 186. [DOI] [PubMed] [Google Scholar]
- Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem 2001; 276: 36869 36872. [DOI] [PubMed] [Google Scholar]
- Lonard DM, O'Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell 2006; 125: 411 414. [DOI] [PubMed] [Google Scholar]
- Green KA, Carroll JS. Estrogen-receptor-mediated transcription and the influence of cofactors and chromatin state. Nat Rev Cancer 2007; 7: 713 722. [DOI] [PubMed] [Google Scholar]
- Suske G, Bruford E, Philipsen S. Mammalian SP/KLF transcription factors: bring in the family. Genomics 2005; 85: 551 556. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhang X-L, Michel FJ, Blum JL, Simmen FA, Simmen RCM. Direct interaction of the Krüppel-like family (KLF) member BTEB1 and progesterone receptor mediates progesterone-responsive gene expression in endometrial epithelial cells. Endocrinology 2002; 141: 62 73. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Zhang D, Michel FJ, Blum JL, Simmen FA, Simmen RCM. Selective interactions of Krüppel-like factor 9/basic transcription element-binding protein with progesterone receptor isoforms A and B determine transcriptional activity of progesterone-responsive genes in endometrial epithelial cells. J Biol Chem 2003; 278: 21474 21482. [DOI] [PubMed] [Google Scholar]
- Simmen RCM, Eason RR, McQuown JR, Linz AL, Kang T-J, Chatman L, Till SR, Fujii-Kuriyama Y, Simmen FA, Oh SP. Subfertility, uterine hypoplasia, and partial progesterone resistance in mice lacking the Krüppel-like factor 9/basic transcription element-binding protein-1 (Bteb1) gene. J Biol Chem 2004; 279: 29286 29294. [DOI] [PubMed] [Google Scholar]
- Velarde MC, Zeng Z, McQuon JR, Simmen FA, Simmen RCM. Krüppel-like factor 9 is a negative regulator of ligand-dependent estrogen receptor alpha signaling in Ishikawa endometrial adenocarcinoma cells. Mol Endocrinol 2007; 21: 2988 3001. [DOI] [PubMed] [Google Scholar]
- Pabona JMP, Velarde MC, Zeng Z, Simmen FA, Simmen RCM. Nuclear receptor coregulator Krüppel-like factor 9 and prohibitin 2 expression in estrogen-induced epithelial cell proliferation in the mouse uterus. J Endocrinol 2009; 200: 63 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons C, Pabona J, Zeng Z, Velarde M, Gaddy D, Simmen F, Simmen R. Response of adult mouse uterus to early disruption of estrogen receptor-α signaling is influenced by Krüppel-like factor 9. J Endocrinol 2010; 205: 147 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Kobayashi A, Yamashita T, Shimanuki T, Nakajima O, Takahasi S, Ikegami S, Inokuchi K, Yamashita K, Yamamoto M. Functional analysis of basic transcription element binding protein by gene targeting technology. Mol Cell Biol 2003; 23: 2489 2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Sarno J, Taylor HS. HOXA10 inhibits Kruppel-like factor 9 expression in the human endometrial epithelium. Biol Reprod 2010; 83: 205 211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde MC, Geng Y, Eason RR, Simmen FA, Simmen RC. Null mutation of Kruppel-like factor 9/basic transcription element binding protein-1 alters peri-implantation uterine development in mice. Biol Reprod 2005; 73: 472 481. [DOI] [PubMed] [Google Scholar]
- Zeng Z, Velarde MC, Simmen FA, Simmen RCM. Delayed parturition and altered myometrial progesterone receptor isoform A expression in mice null for Krüppel-like factor 9. Biol Reprod 2008; 78: 1029 1037. [DOI] [PubMed] [Google Scholar]
- Pabona JM, Zeng Z, Simmen FA, Simmen RC. Functional differentiation of uterine stromal cells involves cross-regulation between bone morphogenetic protein 2 and Krüppel-like factor (KLF) family members KLF9 and KLF13. Endocrinology 2010; 151: 3396 3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen FA, Su Y, Xiao R, Zeng Z, Simmen RC. The Krüppel-like factor 9 (KLF9) network in HEC-1-A endometrial carcinoma cells suggests the carcinogenic potential of dys-regulated KLF9 expression. Reprod Biol Endocrinol 2008; 6: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Du H, Taylor HS. Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium. Biol Reprod 2009; 80: 79 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackow BW, Taylor HS. Submucosal uterine leiomyomas have a global effect on molecular determinants of endometrial receptivity. Fertil Steril 2010; 93: 2027 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde MC, Iruthayanathan M, Eason RR, Zhang D, Simmen FA, Simmen RCM. Progesterone receptor transactivation of the secretory leukocyte protease inhibitor gene in Ishikawa endometrial epithelial cells involves recruitment of Krüppel-like factor 9/basic transcription element binding protein-1. Endocrinology 2006; 147: 1969 1978. [DOI] [PubMed] [Google Scholar]
- Hapangama DK, Turner MA, Drury JA, Martin-Ruiz C, Von Zglinicki T, Farquharson RG, Quenby S. Endometrial telomerase shows specific expression patterns in different types of reproductive failure. Reprod Biomed Online 2008; 17: 416 424. [DOI] [PubMed] [Google Scholar]
- Hecht JL, Mutter GL. Molecular and pathologic aspects of endometrial carcinogenesis. J Clin Oncol 2006; 24: 4783 4791. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Simmen FA, Michel FJ, Simmen RC. Increased expression of the Zn-finger transcription factor BTEB1 in human endometrial cells is correlated with distinct cell phenotype, gene expression patterns, and proliferative responsiveness to serum and TGF-beta1. Mol Cell Endocrinol 2001; 181: 81 96. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Ghaleb AM, Nandan MO, Yang VW. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays 2007; 29: 549 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, Biggs JR, Kraft AS, Yang VW. The gut-enriched Krüppel-like factor (Krüppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem 2000; 275: 18391 18398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland BD, Bernards R, Peeper DS. The KLF4 tumor suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol 2005; 7: 1074 1082. [DOI] [PubMed] [Google Scholar]
- Evans PM, Chen X, Zhang W, Liu C. KLF4 interacts with beta-catenin/TCF4 and blocks p300/CBP recruitment by beta-catenin. Mol Cell Biol 2010; 30: 372 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lu B, Xu F, Gu H, Fang Y, Huang Q, Lai M. Dynamic down-regulation of Krüppel-like factor 4 in colorectal adenoma-carcinoma sequence. J Cancer Res Clin Oncol 2008; 134: 891 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Lu B, Xu J, Hu H, Lai M. Down-regulation of Krüppel-like factor 9 in human colorectal cancer. Pathol Int 2008; 58: 334 338. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997; 277: 955 959. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y, Takahashi Y, Morishita S, Hashimoto M, Niwa K, Tamaya T. Telomerase activity in the human endometrium throughout the menstrual cycle. Mol Hum Reprod 1998; 4: 173 177. [DOI] [PubMed] [Google Scholar]
- Geserick C, Tejera A, González-Suárez E, Klatt P, Blasco MA. Expression of mTert in primary murine cells links the growth-promoting effects of telomerase to transforming growth factor-beta signaling. Oncogene 2006; 25: 4310 4319. [DOI] [PubMed] [Google Scholar]
- Yan P, Benhattar J, Seelentag W, Stehle JC, Bosman FT. Immunohistochemical localization of hTERT protein in human tissues. Histochem Cell Biol 2004; 121: 391 397. [DOI] [PubMed] [Google Scholar]
- Oshita T, Nagai N, Ohama K. Expression of telomerase reverse transcriptase mRNA and its quantitative analysis in human endometrial cancer. Int J Oncol 2000; 17: 1225 1230. [DOI] [PubMed] [Google Scholar]
- Lehner R, Enomoto T, McGregor JA, Shroyer AL, Haugen BR, Pugazhenthi U, Shroyer KR. Quantitative analysis of telomerase hTERT mRNA and telomerase activity in endometrioid adenocarcinomas and in normal endometrium. Gynecol Oncol 2001; 84: 120 125. [DOI] [PubMed] [Google Scholar]
- Misiti S, Nanni S, Fontemaggi G, Cong YS, Wen J, Hirte HW, Piaggio G, Sacchi A, Pontecorvi A, Bachetti S, Farsetti A. Induction of hTERT expression and telomerase activity by estrogens in human ovary epithelium cells. Mol Cell Biol 2000; 20: 3764 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha Y, Kwon SJ, Seol W, Park KS. Estrogen receptor-alpha mediates the effects of estradiol on telomerase activity in human mesenchymal stem cells. Mol Cells 2008; 26: 454 458. [PubMed] [Google Scholar]
- Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, Young NS. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood 2009; 114: 2236 2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis 2010; 31: 9 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA. Telomerase beyond telomeres. Nat Rev Cancer 2002; 2: 627 633. [DOI] [PubMed] [Google Scholar]
- Flores I, Cayuela ML, Blasco MA. Effects of telomerase and telomere length on epidermal stem cells behavior. Science 2005; 309: 1253 1256. [DOI] [PubMed] [Google Scholar]
- Risinger JI, Hayes AK, Berchuck A, Barrett JC. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res 1997; 57: 4736 4738. [PubMed] [Google Scholar]
- Mutter GL, Baak JPA, Fitzgerald JT, Gray R, Neuberg D, Kust G, Gentleman R, Gullans S, Wei L, Wilcox M. Global expression changes of constitutive and hormonally regulated genes during endometrial neoplastic transformation. Gynecol Oncol 2001; 83: 177 185. [DOI] [PubMed] [Google Scholar]
- Lacey JV, Jr, Mutter GL, Ronnett BM, Ioffe OB, Duggan MA, Rush BB, Glass AG, Richesson DA, Chatterjee N, Langholz B, Sherman ME. PTEN expression in endometrial biopsies as a marker of progression to endometrial carcinoma. Cancer Res 2008; 68: 6014 6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Bae-Jump VL, Whang YE, Gehrig PA, Boggess JF. The PTEN tumor suppressor inhibits telomerase activity in endometrial cancer cells by decreasing hTERT mRNA levels. Gynecol Oncol 2006; 101: 305 310. [DOI] [PubMed] [Google Scholar]
- Seeber LM, Zweemer RP, Marchioni L, Massuger LF, Smit VT, van Baal WM, Verheijen RH, van Diest PJ. Methylation profiles of endometrioid and serous endometrial cancers. Endocr Relat Cancer 2010; 17: 663 673. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Kaneuchi M, Fujimoto S, Tanaka Y, Dahiya R. Hypermethylation can selectively silence multiple promoters of steroid receptors in cancers. Mol Cell Endocrinol 2003; 202: 201 207. [DOI] [PubMed] [Google Scholar]
- Ignatov A, Bischoff J, Ignatov T, Schwarzenau C, Krebs T, Kuester D, Costa SD, Roessner A, Semczuk A, Schneider-Stock R. APC promoter hypermethylation is an early event in endometrial tumorigenesis. Cancer Sci 2010; 101: 321 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader AN, Arriba LN, Frasure HE, von Gruenigen VE. Endometrial cancer and obesity: epidemiology, biomarkers, prevention and survivorship. Gynecol Oncol 2009; 114: 121 127. [DOI] [PubMed] [Google Scholar]
- Pei H, Yao Y, Yang Y, Liao K, Wu JR. Krüppel-like factor KLF9 regulates PPARγ transactivation at the middle stage of adipogenesis. Cell Death Differ 2011; 18: 315 327. [DOI] [PMC free article] [PubMed] [Google Scholar]