

Abstract
HSF1 is a transcription factor that plays a key role in the response to heat stress and was previously shown by us to also be essential for importation of p53 into the nucleus. Here we extend these studies and show that loss of HSF1 renders cells more sensitive to killing by ionizing radiation. Cells that lack a functional HSF1 were unable to arrest in G2 after exposure to ionizing radiation, suggesting that HSF1 activity was essential for activation of this cell cycle checkpoint. In addition, cells with no HSF1 showed a reduced capacity to repair radiation-induced double-stranded DNA breaks. We found that in these cells 53BP1 did not accumulate at sites of DNA damage, suggesting that HSF1 was also essential for the functioning of this DNA damage mediator. Taken together our results indicate that HSF1 plays an important role in checkpoint activation and DNA repair and suggest that there is overlap between the heat stress response pathway and the pathway that responds to ionizing radiation.
INTRODUCTION
The p53 tumor suppressor functions as a transcription factor that induces the expression of a variety of genes involved in cell cycle control, apoptosis and DNA repair (1). It is through its ability to activate gene expression that p53 exerts its tumor suppressor activity. Ionizing radiation and other DNA-damaging agents lead to rapid phosphorylation of the p53 protein, which accumulates in the nucleus, activates gene expression, and brings progression through the cell cycle to a halt and facilitates the repair of damaged DNA or inducing apoptosis in cells where the damage is unrepairable (2). The importance of p53’s transactivation capacity to its function as a tumor suppressor is exemplified by the observation that mutations in the p53 gene found in human tumors fall primarily within the DNA-binding domain of the protein and that these mutations inactivate p53’s ability to stimulate gene expression (3). Such mutations are found in approximately 50% of all human tumors.
It has been suggested that inactivation of p53 function occurs in nearly all tumors (4). However, since mutations in the p53 gene account for only half of all tumors, other components of the p53 pathway are also thought to be targets for inactivation during tumorigenesis. One of these is nuclear localization of the p53 protein. As with any transcription factor, entry into the nucleus is critical for function, and exclusion of p53 from the nucleus would be expected to negate its tumor suppressor activity. Consistent with this, cytoplasmic sequestration of p53 has been reported in inflammatory breast carcinoma (5), neuroblastoma (6) and ovarian cancer (7) and was associated with poor survival of patients with colorectal adenocarcinoma (8). Hence the process that transports p53 into the nucleus is critical for its function and is also inactivated in human tumors.
Previously we characterized the nuclear localization of p53 through derivation of a set of cell lines known as ALTR cell lines in which nuclear accumulation of p53 was defective (9). Subsequently we showed that in one of those lines, ALTR12, exclusion of p53 from the nucleus was due to loss of the heat-shock factor 1 (HSF1) (10). HSF1 is known to be activated by diverse environmental and physiological stresses through a multistep process that involves trimerization, nuclear accumulation and extensive post-translational modifications, leading to the expression of heat-shock protein (hsp) genes (11). Our observation that cells in which nuclear localization of p53 was defective also showed an absence of HSF1 demonstrated that HSF1 is an important component of the mechanism of nuclear localization p53 (10). Here we extend these studies and show that loss of HSF1 also compromised the cell’s ability to arrest cell cycle progression, repair damaged DNA, and activate the 53BP1 protein after exposure to ionizing radiation.
MATERIALS AND METHODS
Cell Culture and Reagents
A1-5 is a rat fibroblast cell line transfected with the temperature-sensitive murine p53val135 gene and has been described previously (12). ALTR12 is an A1-5 Low Temperature Resistant (ALTR) cell line generated from A1-5 cells by chemical mutagenesis in which p53 is sequestered in the cytoplasm and lacks HSF1 protein (10). ALTR12/ACT HSF1 cells were derived from ALTR12 cells by the introduction of an expression vector that drives expression of a constitutively active HSF1 (10). HCT116 is a human colon cancer cell line and was obtained from ATCC (American Type Culture Collection, Manassas, VA). All cells were maintained in complete DMEM consisting of 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco BRL, Gaithersburg, MD). HCT116 and A1-5 cells were incubated at 37°C and in an atmosphere containing 95% air/5% CO2. Cells of the ALTR12 cell lines were maintained under the same conditions as A1-5 cells except that they were normally incubated at 32°C.
Anti-53BP1, anti-histone H1, anti-α-tubulin and anti-β-actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-HSF1 (SPA-950) antibodies were purchased from Assay Designs/StressGen (Ann Arbor, MI).
Ionizing Radiation/Heat-Shock Survival Assay by Crystal Violet Staining
Cells grown on 10-cm plates to 80% confluence were trypsinized and diluted to 104/ml in complete DMEM that had been preheated to 42°C. After incubation at 42°C for 30 min, 1-ml aliquots were diluted into 9 ml PBS at room temperature. Five hundred-microliter (500 cells) aliquots of diluted cells were then put into a 60-mm plate with DMEM. After incubation at 37°C for 10 h, cells were exposed to ionizing radiation using a 60Co source at an average dose rate of 47 cGy/min. Colonies were stained with crystal violet and read on the colony counter after incubation at 37°C for ~10 days.
Comet Assay
Cells at 70–80% confluence were exposed to 10 Gy of ionizing radiation and then continuously incubated at 37°C. Cells were then collected at various times after irradiation for comet assays using a Trevigen CometAssay™ Kit (Trevigen, Gaithersburg, MD) according to the manufacturer’s protocol.
Mitotic Index
Cells at 70–80% confluence were exposed to different doses of ionizing radiation and then exposed to 0.3 μg/ml nocodazole for 20 h. The cells were harvested using trypsin-EDTA and washed with cold 50% PBS twice. After fixing in cold mitotic index fixer [50% PBS + 2% EtOH/acetic acid (3:1)] for 30 min on ice, the cells were centrifuged at 1000 rpm and the cell pellet was resuspended in EtOH/acetic acid (3:1) solution for 10 min on ice. The cell suspension was dropped onto slides, air-dried and stained with Giemsa solution for 2 min. Five hundred cells were counted by light microscopy after the slides were rinsed twice in water for 30 s. The mitotic index was calculated by dividing the number of mitotic cells by the total number of cells counted and multiplying by 100%.
53BP1 Focus Formation Assay
To assay for 53BP1 focus formation induced by ionizing radiation, cells plated on cover slips were either exposed to 10 Gy ionizing radiation or left untreated. After incubation at 37°C for 2 h, the cells were fixed and stained for 53BP1 by indirect immunofluorescence as described previously (13).
siRNA Transfection
A1-5 cells were grown in antibiotic-free DMEM supplemented with 5% fetal bovine serum to ~60% confluence. Cells were transfected with 20 nM of rat HSF1 siRNA pool (D-081010-00, Dharmacon), negative control siControl RISC free siRNA and siControl Non-targeting siRNA#1 (Dharmacon), respectively, using Dharma-FECT1 following the manufacturer’s protocol (Dharmacon, Lafayette, CO). After incubation at 37°C for 24 h, cells were again transfected with the same siRNAs. For Hct116 cells, human HSF1 siRNA pool (L-012109-00, Dharmacon) or 53BP1 siRNA pool (M-003548-01, Dharmacon) was transfected twice using the same protocol as above. Forty-eight hours after the last transfection, cells were analyzed for 53BP1 focus formation, mitotic index, comet assay and Western blotting.
Preparation of Cell Extracts and Western Blotting
For total cell extracts, cells were collected and lysed on ice for 30 min in RIPA buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% NP40, 0.25% Na-deoxycholate, 1 mM PMSF, 2 mM EDTA, and 1× protease inhibitor cocktail), then centrifuged at 4°C for 10 min at 16,000g. Nuclear and cytoplasmic proteins were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents according to the manufacturer’s instructions (Pierce Biotechnology, Rockford, IL). Western blot analysis was performed according to standard procedures using PVDF membranes (Millipore, Bedford, MA). Signals were detected by the chemiluminescence method using Super Signal West Pico chemiluminescence substrate (Pierce, Rockford, IL).
RNA Extraction and Quantitative RT-PCR
Cells grown on 10-cm plates to 80% confluence were either exposed to 5 Gy ionizing radiation or left untreated. After incubation at 37°C for 2 h, total RNA was isolated with the TRIzol reagent method using the Total RNA Isolation Reagent (Invitrogen, Carlsbad, CA). RNA concentration was calculated from the optical density at 260 nm, and purity was determined by 260 nm/280 nm absorbance.
Total RNA (1 μg) from each sample was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) for 30 min at 42°C. Quantitative real-time PCR was performed using Platinum Quantitative PCR SuperMix-UDG (Invitrogen), 50 μmol/μl gene-specific probe, forward and reverse primers, and 1 μl cDNA. The reaction conditions were as follows: 50°C for 2 min hold (UDG incubation), 95°C for 2 min hold, and 40 cycles of 95°C for 15 s, 60°C for 30 s. Reactions were conducted in the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Fluorescence was monitored during every PCR cycle at the annealing step. Three replicates for each experimental sample were performed, and differences were assessed with the 2−ΔΔCT method (14). Using the 2−ΔΔCT method, the data are presented as the relative change in 53BP1 gene transcripts normalized to the housekeeping gene GAPDH and compared to that of untreated A1-5 cells. The sequences of the specific primers are: GAPDH forward primer: TGGAGTCTACTGGCGTCTT; GAPDH reverse primer: TGTCATATTTCTCGTGGTTCA; GAPDH probe: [DFAM]CTGAAGGGTGGGGCCAAAAG[DTAM]; 53BP1 forward primer: GGGAAACAAGATGCCACTGT; 53BP1 reverse primer: CTCGAATCGTTCTCGTGTGA; 53BP1 probe: [DFAM]GCTCCTGTGGATGACACAGA[DTAM].
RESULTS
Heat Shock Induces Resistance to Ionizing Radiation in A1-5 Cells
Previously we characterized a set of mutant cell lines that are defective for nuclear importation of the temperature-sensitive p53val135 protein and found that one line, ALTR12 line, lacked expression of the heat-shock factor HSF1 (10). In addition to being required for the induction of Hsp70, we showed that HSF1 was also required for nuclear importation of p53 induced by ionizing radiation. This observation prompted us to test whether HSF1 might also be involved in the cellular response to radiation. To explore this, we first determined whether heat shock could affect the survival of cells exposed to radiation. We examined three cell lines. A1-5 cells are the parental cells from which the ALTR cells were derived (9). ALTR12 cells lack expression of HSF1 and are defective for nuclear importation of p53 (10). ALTR12/ACT HSF1 cells were derived from ALTR12 cells but express an exogenously introduced constitutively active HSF1. Cells of all three cell lines were exposed to increasing doses of radiation and then assayed for the fraction of surviving cells. Duplicate sets of cultures of A1-5 and ALTR12 cells were subjected to a heat shock at 42°C for 30 min prior to exposure to ionizing radiation (Fig. 1). Graphing the results showed that parental A1-5 cells were relatively resistant to radiation and that subjecting A1-5 cells to heat stress resulted in a small but reproducible increase in survival compared to cells not pretreated with heat. This increase in heat stress-induced survival was absent in ALTR12 cells. Indeed, pretreating ALTR12 cells with heat resulted in a slight decrease in survival. Moreover, ALTR12 cells were markedly more sensitive to radiation and showed an approximately fivefold decrease in survival at 6 Gy relative to A1-5 cells. The surviving fraction of ALTR12/ACT HSF1 cells was identical to that of A1-5 cells at 6 Gy, although they showed reduced survival compared to A1-5 cells at higher doses. Overall, however, ALTR12/ACT HSF1 cells were more resistant to radiation than were ALTR12 cells. Taken together, these results suggest that the heat-stress response mediated by HSF1 promotes resistance to ionizing radiation.
FIG. 1.

Radioresistance induced by heat shock is reduced in cells that lack a functional HSF1. A1-5 cells, ALTR12 cells and ALTR12 cells that constitutively express an activated HSF1 (ALTR12/ACT HSF1) were plated as described in the Materials and Methods, and the cells were exposed to increasing doses of ionizing radiation. In a parallel experiment, A1-5 and ALTR12 cells were subjected to a heat shock (42°C for 30 min) and then irradiated. All plates were incubated at 37°C for ~10 days. Surviving colonies were stained with crystal violet and quantified on a colony counter. The graph shows the average surviving fractions ± SE from three independent experiments.
HSF1 is Essential for the DNA Damage Response in A1-5 Cells
Since cell cycle arrest and repair of damaged DNA are key processes in the cellular response to radiation, we examined these two processes in our three cell lines. We first tested for irregularities in the radiation-induced G2 checkpoint. All three cell lines were exposed to increasing doses of radiation, and then progression through mitosis was blocked with nocodazole (Fig. 2A). As expected, A1-5 cells showed a progressive decrease in mitotic cells after irradiation, indicating that the cells delayed progression at G2 and that this checkpoint was functional. In contrast, in ALTR12 cells, the number of mitotic cells remained the same after irradiation, indicating that these cells progressed through G2. Hence the G2 checkpoint was not functional in the ALTR12 cell line. The G2 checkpoint was restored in the ALTR12/ACT HSF1 cells, indicating that HSF1 activity was required for activation of the G2 checkpoint in response to radiation.
FIG. 2.

Lack of HSF1 results in a defective G2 checkpoint and DNA repair in response to ionizing radiation. Panel A: A1-5, ALTR12 and ALTR12/ACT HSF1 cells were exposed to increasing doses of ionizing radiation as indicated. The cells were then incubated with nocodazole at 37°C for ~10 h and the fractions of mitotic cells were determined. The graph shows the average values normalized to the mitotic index for unirradiated cells ± SE determined from at least 500 individual cells. Panel B: A1-5, ALTR12 and ALTR12/ACT HSF1 cells were exposed to 10 Gy of ionizing radiation, incubated at 37°C, and then prepared for comet assay at various times after irradiation. The graph shows the average Olive tail moment, which was normalized to time zero. The values are averages ± SE from 100 individual cells. Panel C: A1-5, ALTR12 and ALTR12/ACT HSF1 cells were grown on cover slips at 37°C to 80% confluence and then exposed to 10 Gy or left untreated. Incubation was continued at 37°C for 2 h before fixing and staining for 53BP1 localization. The scale bars represent 5 μm. Panel D: The fraction of cells in panel C that exhibited 53BP1 foci was quantified and graphed. The values are averages ± SE from three independent experiments.
We next examined the rate of repair of radiation-induced double-stranded DNA breaks. Cells of all three cell lines were irradiated, harvested at regular intervals, and then prepared for the comet assay (Fig. 2B). As expected, in A1-5 cells most DNA damage was repaired by 1 h after irradiation. However, DNA damage persisted in ALTR12 cells for an extended period. In fact, some DNA damage remained 4 h after irradiation. Expression of the constitutively active HSF1 in ALTR12s (ALTR12/ACT HSF1) restored the DNA repair activity to parental A1-5 cell levels, suggesting that HSF1 is also essential for the repair of radiation-induced DNA damage.
Since our original characterization of ALTR cells focused on the activity of p53, we next examined the function of a p53 binding protein, 53BP1, also known to be involved in p53 function, G2/M checkpoint arrest and repair of double-stranded breaks in DNA (15, 16). Activation of DNA repair occurs at discrete sites within the nucleus that are visualized as foci after irradiation. Hence we examined the three cell lines for the formation of foci that contained 53BP1 (Fig. 2C). 53BP1-containing foci were readily visible in irradiated A1-5 and ALTR12/ACT HSF1 cells but were rarely observed in ALTR12 cells. Quantifying the data from Fig. 2C revealed a dramatic reduction (~75%) in the number of cells exhibiting 53BP1 foci compared to parental A1-5 cells (Fig. 2D). The number of cells exhibiting 53BP1 foci in the irradiated ALTR12/ACT HSF1 cells was similar to that in the parental A1-5 cells. Taken together, our results suggest that loss of HSF1 interferes with the cellular response to radiation and causes defects in radiation-induced cell cycle checkpoint activation and double-strand break repair, including the formation of 53BP1 foci.
Knockdown of HSF1 Expression Led to a Decrease in Radiation-Induced 53BP1 Foci
We sought to confirm the above results by suppressing HSF1 expression in A1-5 cells and human colorectal cancer-derived HCT116 cells using siRNA. First we confirmed that the anti-HSF1 siRNA was effective in suppressing HSF1 expression (Fig. 3A). Next we tested anti-HSF1 siRNA-transfected cells for formation of 53BP1 foci after irradiation (Fig. 3B). Our results demonstrate that suppression of HSF1 using siRNA also effectively diminished the formation of radiation-induced 53BP1 foci. Quantification of the number of cells with 53BP1 foci (Fig. 3C) indicated that transfection with HSF1 siRNA in A1-5 cells resulted in a ~50% reduction in cells with radiation-induced 53BP1 foci.
FIG. 3.

Suppression of HSF1 inhibits radiation-induced 53BP1 focus formation in A1-5 cells. Panel A: A1-5 cells grown on 30-mm dishes were transfected with the indicated siRNAs as described in the Materials and Methods. Total cell extracts were prepared 48 h after the last transfection. Then 50 μg of total protein mixed with loading buffer was applied to a 10% SDS-polyacrylamide gel for analysis of HSF1 protein levels by Western blotting using an anti-HSF1 antibody. The filter was probed for β-actin to control for loading variations. Panel B: In a parallel experiment, A1-5 cells were grown on cover slips and transfected with the indicated siRNAs. Then the cells were either exposed to 10 Gy ionizing radiation or left untreated. After incubation at 37°C for an additional 2 h, cells were fixed and stained for 53BP1 by indirect immunofluorescence using an anti-53BP1 antibody. A typical result is shown. The scale bars represent 5 μm. Panel C: The fractions of cells in panel B that exhibited 53BP1 focus formation were quantified and graphed. The values are averages ± SE from three independent experiments.
To ensure that our results were not cell line-specific, we conducted similar anti-HSF1 siRNA experiments in HCT116 cells (17). As can be seen in Fig. 4, suppression of HSF1 using siRNA again resulted in decreased HSF1 expression (Fig. 4A) that was accompanied by a ~50% reduction in the number of cells with radiation-induced 53BP1 foci (Fig. 4B, C). Moreover, transfection of HCT116 cells with HSF1 siRNA also resulted in failure of the G2 checkpoint (Fig. 4D) and a reduced capacity to repair DNA double-stranded breaks (Fig. 4E). These results indicated that our observations relating to the involvement of HSF1 in the repair of radiation-induced DNA damage were not restricted to A1-5 cells or related cell lines.
FIG. 4.

Suppression of HSF1 in Hct116 cells inhibits DNA repair and G2 checkpoint activation in response to ionizing radiation. Panel A: HCT116 cells grown on 30-mm dishes were transfected with the indicated siRNAs as described in the Materials and Methods. Total cell extracts were prepared 48 h after the last transfection. Then 50 μg of total protein mixed with loading buffer was applied to a 10% SDS-polyacrylamide gel for analysis of HSF1 protein levels by Western blotting using an anti-HSF1 antibody. The filter was probed for β-actin to control for variations in loading. Panel B: Hct116 cells were grown on cover slips and transfected with siRNAs as indicated and either not treated further or exposed to 10 Gy ionizing radiation. After incubation at 37°C for an additional 2 h, the cells were fixed and stained for 53BP1 by indirect immunofluorescence using an anti-53BP1 antibody. The scale bars represent 5 μm. Panel C: The fractions of cells in panel B exhibiting 53BP1 foci were quantified. The graphs show the averages ± SE from three independent experiments. Panel D: HCT116 cells were transfected with siRNAs as indicated and then exposed to increasing doses of ionizing radiation. The cells were blocked with nocodazole, and the fraction of mitotic cells was determined. The graph shows the average fraction of mitotic cells from at least 100 individual cells normalized to the fraction of mitotic cells in unirradiated samples ± SE. Panel E: HCT116 cells were transfected with the siRNAs as indicated and then exposed to 10 Gy ionizing radiation. The cells were harvested at regular intervals after irradiation and prepared for the comet assay. The graph shows the average relative Olive tail moment ± SE for at least 100 individual cells.
ALTR12 Cells Show Decreased 53BP1 Protein Levels
The results described above suggest that the loss of HSF1 activity has a profound impact on the functioning of 53BP1. Because our characterization of ALTR12 cells established that nuclear localization of p53 was defective, we reasoned that loss of HSF1 activity might also result in aberrant subcellular localization of 53BP1. To test this hypothesis, we fractionated A1-5, ALTR12 and ALTR12/ACT HSF1 cells into nuclear and cytoplasmic fractions and tested for the presence of 53BP1 by immunoblotting (Fig. 5A). Although we found 53BP1 exclusively in the nuclear fractions, indicating that 53BP1 was in fact localized to the appropriate subcellular compartment, we also found that the levels of 53BP1 were markedly reduced relative to parental A1-5 cells. In ALTR12/ACT HSF1 cells, the levels of 53BP1 protein were similar to the levels seen in the A1-5 cells, suggesting that in addition to affecting the activation of 53BP1, HSF1 may also play a role in controlling its protein levels. Since HSF1 is a transcription factor that has been shown to regulate gene expression, we wondered whether HSF1 might also be involved in regulating 53BP1 gene expression. To test this hypothesis, quantitative PCR was performed to analyze 53BP1 mRNA levels in A1-5, ALTR12 and ALTR12/ACT HSF1 cells (Fig. 5B). 53BP1 mRNA was found to be at similar levels in ALTR12, ALTR12/ACT HSF1 and parental A1-5 cells (Fig. 5C), indicating that HSF1 is not necessary for 53BP1 gene expression.
FIG. 5.
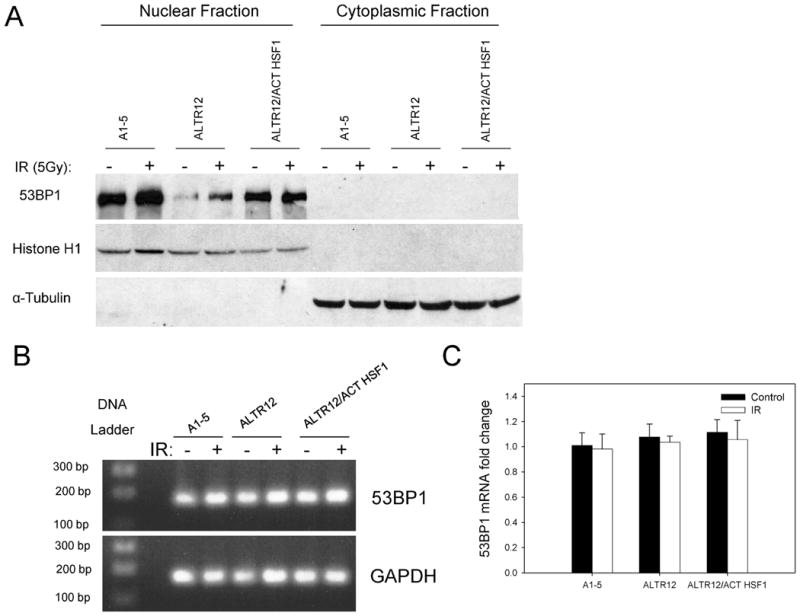
The quantity of 53BP1 protein is decreased in cells that lack a functional HSF1. Panel A: A1-5, ALTR12 and ALTR12/ActHSF1 cells were grown to 60% confluence and then either left untreated or exposed to 5 Gy of ionizing radiation (IR), and incubation continued at 37°C for 2 h. The cells were harvested and fractionated into nuclear and cytoplasmic fractions. Then 50 μg of total protein mixed with loading buffer was applied to a 10% SDS-polyacrylamide gel for analysis of 53BP1 protein by Western blotting using an anti-53BP1 antibody. The blots were also probed for histone H1 (a nuclear protein) or α-tubulin (a cytoplasmic protein) to assess the purity of the nuclear/cytoplasmic fractions. Panel B: A1-5, ALTR12 and ALTR12/ActHSF1 cells were grown as described in panel A and were either left untreated or exposed to 5 Gy of ionizing radiation and incubation was continued at 37°C for 2 h. Total RNA was extracted and 53BP1 mRNA measured using quantitative RT-PCR. The PCR products of 53BP1 and GAPDH separated on a 1% agarose electrophoresis and stained with ethidium bromide are shown. Panel C: The PCR products in panel B were analyzed using the 2−ΔΔCT quantitative PCR method. No apparent differences were seen in the 53BP1 gene expression among cell lines. Error bars represent SD for triplicate reactions. Fold change = relative change.
DISCUSSION
Previously, we determined that in ALTR12 cells the failure to transport p53 into the nucleus was the consequence of loss of HSF1, indicating that HSF1 played an essential role in a stress-activated nuclear importation mechanism. Here we show that HSF1’s influence on the cell is considerably more extensive than we anticipated. Unexpectedly, our data suggest that HSF1 is involved in activation of DNA repair and the G2 checkpoint. In support of this, we showed that ALTR12 cells, which lack HSF1 expression, showed a reduced ability to repair DNA double-stranded breaks as measured by the comet assay. We confirmed that this result was not a phenomenon restricted to ALTR12 cells by using an anti-HSF1 siRNA to suppress HSF1 expression in HCT116 cells and found that this resulted in a similar reduction in DNA repair capability after irradiation. Further support for HSF1 involvement in the DNA repair process was provided by our observation that the function of 53BP1 is compromised in cells lacking HSF1. 53BP1 is a key mediator in the DNA damage repair process and activation of 53BP1 leads to its accumulation at the sites of DNA strand breaks, forming discrete nuclear foci (15). 53BP1 is incapable of forming nuclear foci in cells lacking HSF1, suggesting that HSF1 activity is critical for 53BP1 function. Based on the original observation that in ALTR12 cells p53 was unable to localize to the nucleus, we reasoned that 53BP1 might also be inactive because it could not be transported to the nucleus. However, we found that 53BP1 protein did localize to the nucleus of cells lacking HSF1 but that the levels of protein were reduced. Hence it seems that HSF1 is important for maintenance of normal levels of 53BP1 protein rather than for its localization. However, the reduced protein levels are not the result of changes in 53BP1 gene expression, because quantitative PCR results showed that the 53BP1 mRNA levels were unchanged between the three cell lines (Fig. 5C). Since previously we showed that loss of HSF1 in ALTR12 resulted in abolishment in both heat-induced expression of HSP70 and the ability to reactivate heat-inactivated luciferase (10), we speculate that HSF1 may play a role in regulating the protein folding of 53BP1 and potentially its stability. However, we cannot rule out the possibility that the protein might also lack a critical post-translational modification. Taken together, our results suggest that HSF1 plays a critical role in activation of the DNA damage response pathway and is capable of regulating the function of mediators of the DNA repair process such as 53BP1. This could explain why a heat shock administered prior to irradiation can enhance survival.
HSF1 also appears to play a role in G2 checkpoint activation. ALTR12 cells continue to progress through G2 and into mitosis after irradiation, suggesting that HSF1 is critical for radiation-induced G2 checkpoint activation. Consistent with this, we showed that knocking down HSF1 expression with siRNA in parental A1-5 cells and in the unrelated HCT116 cells recapitulated the phenotype we observed in ALTR12 cells. Conversely, we showed that reintroduction of HSF1 into ALTR12 cells restored checkpoint activity to near normal. This is compelling evidence in favor of a role for HSF1 in regulation of checkpoint activation in response to ionizing radiation. However, it is not immediately obvious how HSF1 participates in this. To our knowledge, there is no direct connection between HSF1 and cell cycle regulators, although one report indicates that HSF1 can be phosphorylated by cdc2 in Arabidopsis (18). It seems likely that the link between HSF1 and cell cycle regulation is indirect. One possibility is that HSF1’s regulation of cell cycle checkpoints is manifested through its involvement with a mediator(s) of DNA damage response such as 53BP1 that is required for maintenance of the G2/M checkpoint (15). Indeed, suppression of 53BP1 by siRNA did result in a reduction in radiation-induced G2 checkpoint activation in HCT116 cells (data not shown); however, this reduction was less than what we observed in ALTR12 cells and HSF1-knockdown HCT116 cells (Fig. 4D), suggesting that, in addition to 53BP1, HSF1 is also likely to affect the function of other factors in the DNA damage response.
In conclusion, our work shows novel activities for HSF1 in regulation of DNA repair and the cell cycle and suggests that the heat-shock response is part of a global cellular response network that senses and responds to multiple stressful stimuli. This also implies that stress-related proteins previously thought to be responders to specific stimuli may in fact have overlapping functions and have roles in the response mounted to multiple stimuli.
Acknowledgments
These studies were supported by a grant from the National Cancer Institute (CA090776) to JM.
References
- 1.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 2.Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644–7655. doi: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 3.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 4.Lane DP, Cheok CF, Lain S. p53-based cancer therapy. Cold Spring Harb Perspect Biol. 2010;2:a001222. doi: 10.1101/cshperspect.a001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci USA. 1992;89:7262–7266. doi: 10.1073/pnas.89.15.7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman SC, Chen CY, Lansing TJ, Gilmer TM, Kastan MB. The p53 signal transduction pathway is intact in human neuroblastoma despite cytoplasmic localization. Am J Pathol. 1996;148:1381–1385. [PMC free article] [PubMed] [Google Scholar]
- 7.Runnebaum IB, Kieback DG, Mobus VJ, Tong XW, Kreienberg R. Subcellular localization of accumulated p53 in ovarian cancer cells. Gynecol Oncol. 1996;61:266–271. doi: 10.1006/gyno.1996.0137. [DOI] [PubMed] [Google Scholar]
- 8.Bosari S, Viale G, Bossi P, Maggioni M, Coggi G, Murray JJ, Lee AK. Cytoplasmic accumulation of p53 protein: an independent prognostic indicator in colorectal adenocarcinomas. J Natl Cancer Inst. 1994;86:681–687. doi: 10.1093/jnci/86.9.681. [DOI] [PubMed] [Google Scholar]
- 9.Li Q, Falsey RR, Gaitonde S, Sotello V, Kislin K, Martinez JD. Genetic analysis of p53 nuclear importation. Oncogene. 2007;26:7885–7893. doi: 10.1038/sj.onc.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Q, Feldman RA, Radhakrishnan VM, Carey S, Martinez JD. Hsf1 is required for the nuclear translocation of p53 tumor suppressor. Neoplasia. 2008;10:1138–1145. doi: 10.1593/neo.08430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anckar J, Sistonen L. Heat shock factor 1 as a coordinator of stress and developmental pathways. Adv Exp Med Biol. 2007;594:78–88. doi: 10.1007/978-0-387-39975-1_8. [DOI] [PubMed] [Google Scholar]
- 12.Martinez J, Georgoff I, Martinez J, Levine AJ. Cellular localization and cell cycle regulation by a temperature-sensitive p53 protein. Genes Dev. 1991;5:151–159. doi: 10.1101/gad.5.2.151. [DOI] [PubMed] [Google Scholar]
- 13.Gaitonde SV, Riley JR, Qiao D, Martinez JD. Conformational phenotype of p53 is linked to nuclear translocation. Oncogene. 2000;19:4042–4049. doi: 10.1038/sj.onc.1203756. [DOI] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 16.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brattain MG, Fine WD, Khaled FM, Thompson J, Brattain DE. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res. 1981;41:1751–1756. [PubMed] [Google Scholar]
- 18.Reindl A, Schoffl F, Schell J, Koncz C, Bako L. Phosphorylation by a cyclin-dependent kinase modulates DNA binding of the Arabidopsis heat-shock transcription factor HSF1 in vitro. Plant Physiol. 1997;115:93–100. doi: 10.1104/pp.115.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]