

Abstract
We report on 2 relatives with a segmental duplication of 5q11.2 → 13.3. The phenotype is surprisingly limited for the degree of chromosome imbalance, the propositus presenting with schizophrenia. Using RFLP markers, we have shown that the gene for HEXB lies within the duplicated region. We suggest this region as a candidate region for the location of a single major gene which predisposes to schizophrenia and which may be assessed by linkage analysis.
Keywords: segmental duplication 5q, HEXB, gene mapping
INTRODUCTION
Autosomal abnormalities are most often seen in patients with mental retardation, multiple minor malformations, and pre- and postnatal growth retardation [Schinzel, 1983]. These general, nonspecific phenotypic effects may be attributed to a general effect of genomic imbalance on embryogenesis. Psychiatric problems are less commonly described among individuals with specific chromosome abnormalities except for sex chromosome anomalies.
In general, duplications have a less deleterious effect than deletions of the same segment. Increasing the size of a duplication leads to an increasing number and severity of phenotypic effects [Epstein, 1986]. Within this general context of disordered growth and development, particular syndromes can be recognized clinically by particular constellations of manifestations associated with particular aneuploidies.
Phenotypic mapping of specific anomalies (the result of numerous interactions during cellular development) to particular chromosome regions is unlikely to illuminate the location or effect of underlying single gene loci [Epstein, 1986]. On the other hand, the level of expression of many single gene loci shows dosage effects. The association of a particular phenotype with a specific aneuploidy may lead to the definition of a single gene responsible for that phenotype. The association of trisomy 21 with Alzheimer disease and the subsequent mapping of a single major gene to chromosome 21 presents such a paradigm [St. George-Hyslop et al., 1987].
We now report a duplication of 5q11.2 → q13.3. This aneuploidy has surprisingly limited phenotypic effects in view of the degree of chromosome imbalance. The particular characteristics of schizophrenia and renal anomalies suggest that single major genes associated with these phenotypes may lie within this aneuploid region. A preliminary report of this family has been published [Bassett et al., 1988].
CLINICAL REPORTS
The patients reported here are the propositus and his maternal uncle.
Patient 1
The propositus came to our attention following admission to hospital with acute psychiatric problems. At that time he was noted to have an unusual appearance and a family history of schizophrenia [Bassett et al., 1988].
He was born in 1965, at term, after an unremarkable pregnancy. His birth weight was 3,730 g. Ptosis on the left side was noted at birth. He had normal psychomotor development. His scholastic performance was below average, but he did not repeat any grades. He was investigated at age 7 years for school problems. At that assessment his intelligence was normal (WISC verbal score 97, performance score 92), but poor coordination and minimal neurological abnormalities were noted. He had surgery at age 8 for the ptosis and was investigated at age 12 for obesity and hypogonadism. He was found to have low testosterone levels, but normal response to luteinizing hormone releasing hormone (LHRH) stimulation. Testosterone levels increased after human chorionic gonadotropin (CG) injection.
At age 16, he was noted to have depression and withdrawal requiring psychiatric consultation. At age 20, he was hospitalized with typical symptoms of acute schizophrenia, including auditory hallucinations, paranoid delusions without mood change, and a gradual decline in school performance and social relationships. A standard battery of neuropsychiatric tests, including a WAIS-R, documented low average verbal intelligence. The psychotic symptoms responded well to low-dose haloperidol, but residual symptoms of low motivation, blunted emotion, and poverty of speech remained.
On clinical examination the propositus had macrocephaly (occipital frontal circumference 58.5 cm) with frontal bossing and a round face. Height was below the 3rd centile, while weight was on the 75th centile. He also had hypertelorism, over folding of the ears, a small phallus (6 cm), and borderline small testes (Table I). There was mild shortness of the 4th and 5th metacarpals, and more pronounced shortness of the 4th and 5th metatarsals bilaterally. Dermatoglyphics demonstrated 6 arches, unlike either parent.
TABLE I.
Clinical Findings Present in Patients With Duplication 5q11.2 → q13.3 Trisomy
| Manifestation | Propositus | Uncle |
|---|---|---|
| Bossed forehead | + | + |
| Flat occiput | + | + |
| Hypertelorism | + | + |
| Short stature | + | + |
| Obesity | + | + |
| Short 4th and 5th metacarpals | + | + |
| Scoliosis | − | + |
| Renal abnormalities | + | + |
| Small phallus | + | + |
| Low average intelligence | + | + |
| Schizophrenia | + | + |
Patient 2
The maternal uncle of the propositus was, by history, similar in appearance to the propositus and had psychiatric problems. He was born in 1934 and had had educational problems. By age 20 years he had withdrawn socially, and exhibited paranoid delusions and auditory hallucinations without prominent mood changes. He was diagnosed as schizophrenic and managed successfully with low dose neuroleptics from 1971. His only relapses of psychotic symptoms occurred following withdrawal of medication. Both affected individuals met the DSM-III-R criteria for schizophrenia.
On clinical examination, the uncle was similar in appearance to the propositus with obesity, short stature, and a round face. He had hypertelorism, overfolded protuberant ears, and a flat occiput. The phallus was small with normal size of testes. Mild shortness of the 4th and 5th metacarpals and 4th metatarsals was noted.
Cytogenetic investigation
Differential diagnosis of these 2 patients included pseudopseudohypoparathyroidism, Aarskog syndrome, and fragile X syndrome. Additional indications for cytogenetic analysis were the inferior intellectual performance of these patients within the family context, and their congruent constellation of minor facial anomalies.
Both men were found to have a derivative chromosome 1 (1q +) which, following cytogenetic examination of the propositus’ mother, was interpreted as arising due to an inverted insertion of material from 5q [karyotype 46, XY −1, +der(1) inv ins (1;5)(q32.3;q13.3q11.2)]. Thus, both men have a duplication of the proximal long arm segment of chromosome 5q11.2 → 5q13.3. The propositus’ mother was shown to have a balanced inverted insertion [karyotype 46, XX, inv ins (1;5)(q32.3;q13.3q11.2)]. The father and brother of the propositus have normal chromosomes.
Other investigations
Farther investigations of both men and their sibs have included renal scans. The propositus lacks the left kidney, while the uncle is suspected to have a partially duplicated and inferiorly displaced left kidney. Both parents, and the propositus’ brother, have normal renal scans. CT scans of the head show both affected males have a mild degree of ventricular dilatation with the uncle having a cavum septi pellucidi. CT scans of both parents were normal.
Molecular genetic investigation
As a preliminary step toward correlating the duplicated region with the physical and linkage map [Leppert et al., 1987] of chromosome 5 we chose to test this family with a DNA probe for the hexosaminidase B (HEXB) gene. This gene has been mapped to 5q13 [George and Francke, 1978; Dana and Wasmuth, 1982; Fox et al., 1984], so it was of interest to determine whether this gene lay within the aneuploid region.
The results of conventional Southern blot analysis [Wood et al., 1988] are shown in Figure 1. The restriction fragments at 1.0 kb and 1.6 kb are allelic [O’Dowd et al., 1987] while the 1.2 kb fragment is a constant band. The mother (lane 4) is heterozygous for this RFLP and transmits the 1.6 kb allele to her affected son but not to her chromosomally normal, unaffected son who inherits the maternal 1.0 kb allele. Thus, the 1.0 kb allele segregates with the intact maternal chromosome 5 and the 1.6 kb allele segregates with the derivative chromosome 1 containing the insertion of chromsome 5. This 1.6 kb allele does not segregate with the maternal deletion chromosome 5 since neither son inherits this chromosome. Thus, the derivative chromosome 1 carries a 1.6 kb allele at the HEXB locus and this allele is present, as expected in DNA from the maternal uncle (lane 5). This indicates that the HEXB locus lies within the duplicated region of chromosome 5.
Fig. 1.
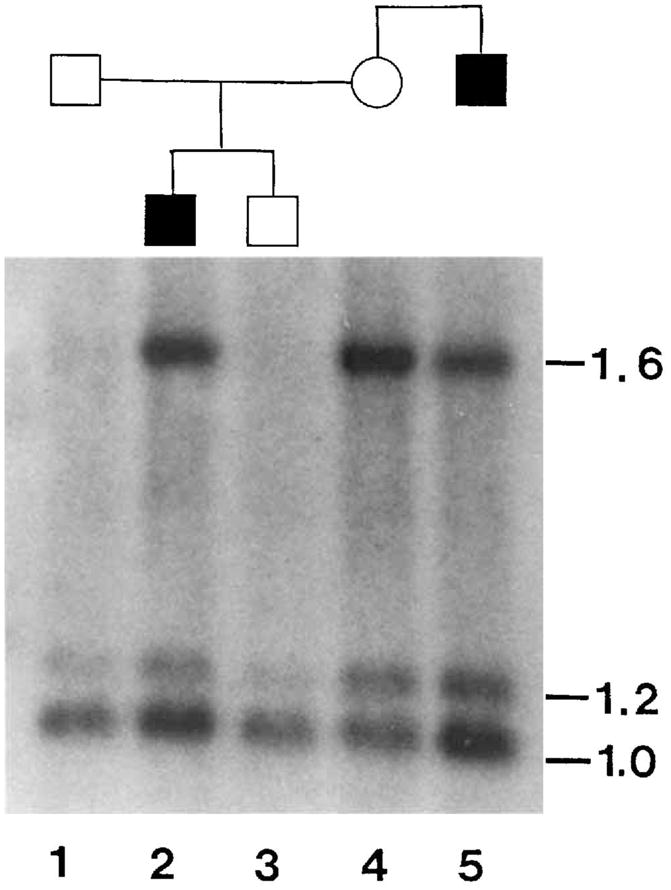
Segregation of HEXB alleles in relatives. The solid symbols indicate males with duplication 5q11.2 → 13.3. Lane 1: Father. Lane 2: Proband. Lane 3: Brother. Lane 4: Mother. Lane 5: Uncle. DNA was digested with PstI and Southern blots Drobed with a 1.0 kb PstI genomic HEXB fragment [O’Dowd et al., 1987]. Bands at 1.0 kb and 1.6 kb are allelic with a constant band at 1.2 kb.
The alternative interpretation, that one these HEXB alleles lies on the maternal deleted chromosome 5 and that one of her sons is recombinant, is unlikely in view of the reported physical location of this locus and is not supported by dosage analysis of the blot. Inspection of Figure 1 suggests that the intensity of the 1.0 kb band is greater than expected in the affected males for a single copy allele. In particular, the intensities of the 1.6 kb bands in the carrier mother and her affected son (lanes 2 and 4) are quite similar while the 1.0 kb band is more intense in the affected son. This indicates that the 1.6 kb band is present in single copy and the 1.0 kb band is present in 2 copies in the affected males. A Bio-Rad 620 video densitometer was used to measure the intensities of these bands. When the 1.6 kb bands are normalized to 1 with respect to the female carrier the relative intensities of the 1.0 kb bands in the affected son and uncle are calculated to be 1.83 and 2.56, respectively. This is in accordance with both affected males having 2 copies of this 1.0 kb fragment (one on each normal chromosome 5) whereas the mother has only one copy of this fragment.
DISCUSSION
This family was referred for genetic consultation because a young man and his maternal uncle had both been noted to have a similar psychiatric illness and clinical appearance. Both tested in the normal range for intelligence, but they were not as bright as expected for their family. Initially, X-linked inheritance was considered, although the clinical findings did not clearly fit a recognized X-linked syndrome.
Fra(X) mental retardation, Aarskog syndrome, pseudopseudohypoparathyroidism, and Robinow syndrome were included in the differential diagnosis. Chromosome studies were requested to exclude fra(X) syndrome and because both men had minor anomalies and were slow for their family.
Cytogenetic analysis showed both of the affected males to have a duplication of the proximal long arm of chromosome 5 (5q11.2 → q13.3) due to an inverted insertion of this segment into chromosome 1. The mother was balanced, having both the insertion 1 chromosome and the corresponding deletion 5 chromosome. Neither of the mother’s parents were living so the origin of the insertion could not be investigated further.
The psychiatric illness that was segregating with the chromosome imbalance may be a manifestation of this aneuploidy or may represent a severe manifestation of a familial psychiatric disorder. The possibility of a familial personality disorder was evaluated by extensive psychiatric testing of both parents, the uncle and both brothers, with the examiner being blind with regard to the medical history and chromosome findings. Only the 2 aneuploid males were judged to have schizophrenia, while the other relatives were classified as normal. The mother, who shares the der(1) chromosome with her affected son and brother, was normal. Thus, the psychiatric illness seems to be specifically related to the chromosome imbalance. This raises the possibility that a single major gene present within the duplicated region, and therefore present in 3 copies, may be the primary cause of schizophrenia in these males. The DNA of these affected individuals will have a triplex representation of the 5q11.2 → q13.3 region and can be used to map genes into this region.
The gene for HEXB has been localized to 5q13 using somatic cell hybrids, constructed from human cell lines carrying translocation chromosomes. Electrophoretic analysis of cellular hexosaminidase activity enabled George and Francke [1978] to localize the HEXB gene to 5cen → 5q13 and Fox et al. [1984] to localize HEXB to 5q13 → 5qter, giving 5q13 as the shortest region of overlap. Analysis of spontaneous segregants from cell hybrids allowed Dana and Wasmuth [1982] to localize HEXB to the middle of the 5q13 band. Our finding that the affected son in this family, who inherits both a normal chromosome 5 and a der(1) (containing 5q11.2 → q13.3) from his balanced carrier mother, also inherits both of her HEXB alleles is consistent with this localization for HEXB. This places a molecular marker within the duplicated region and provides a starting point for linkage and molecular studies.
This family also illustrates how offspring of balanced translocation carriers may provide gene mapping data. The mother transmits an intact chromosome 5 to both her offspring, a derivative chromosome 1 with chromosome 5 material to her affected son and a deletion chromosome 5 to neither son. Thus, for a marker locus that is heterozygous in the mother, the affected son will receive both alleles if the marker is located within the duplicated region as is the case for the HEXB locus. Furthermore, if the sons inherit single alternative alleles, then that locus must be present on the normal and deletion maternal chromosomes 5 and one son must be recombinant.
These studies demonstrate that the HEXB gene lies within the 5q11.2 → q13.3 region of chromosome 5 that is duplicated in both affected males. Multifactorial determination and single major gene models with limited penetrance are both consistent with the patterns of familial occurrence observed in schizophrenia [Gottesman and Shields, 1967; McGue et al., 1983; Risch and Baron, 1984]. No significant linkage has been reported. Neither have chromosome abnormalities been associated with schizophrenia, although systematic studies of affected individuals have not been reported. A putative major gene in the duplicated region could have an overexpressing allele that predisposes to schizophrenia in chromosomally normal individuals.
A gene for manic depressive illness has been mapped to chromosome 11 by focusing on the genetically isolated Old Order Amish population of Pennsylvania [Egeland et al., 1987]. This demonstrates that an affective disorder can have a single locus cause and illustrates the value of focusing the linkage search. Thus, lacking similar isolates with schizophrenia, it may be possible to map a gene for schizophrenia by focusing on the particular region of the genome that is duplicated in this family. If significant positive lod scores are obtained between schizophrenia and marker loci within this region of chromosome 5 it will be important to evaluate the extent of possible genetic heterogeneity.
Acknowledgments
We thank Dr. R.A. Gravel, Toronto Hospital for Sick Children, for providing the HEXB probe. This work was supported by the National Institute of Mental Health grant 1R03 MH 43730.
References
- Bassett AS, McGillivray BC, Jones BD, Pantzar JT. Partial trisomy chromosome 5 cosegregating with schizophrenia. Lancet. 1988;1:799–801. doi: 10.1016/s0140-6736(88)91660-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana S, Wasmuth JJ. Selective linkage disruption in human-Chinese hamster cell hybrids: Deletion mapping of the leuS, hexB, emtB, and chr genes on human chromosome 5. Mol Cell Biol. 1982;2:1220–1228. doi: 10.1128/mcb.2.10.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeland JA, Gerhard DS, Pauls DL, Sussex JN, Kidd KK, Allen CR, Hostetter AM, Houseman DE. Bipolar affective disorders linked to DNA markers on chromosome 11. Nature. 1987;325:783–787. doi: 10.1038/325783a0. [DOI] [PubMed] [Google Scholar]
- Epstein CJ. The Consequences of Chromosome Imbalance. New York: Cambridge University Press; 1986. [Google Scholar]
- Fox MF, DuToit DL, Warnich L, Retief AE. Regional localization of α-galactosidase (GLA) to Xpter →q22, hexosaminidase B (HEXB) to 5q13 → qter, and arylsulfatase B (ARSB) to 5pter → q13. Cytogenet Cell Genet. 1984;38:45–49. doi: 10.1159/000132028. [DOI] [PubMed] [Google Scholar]
- George DL, Francke U. Evidence for localization of the gene for hexosaminidase B to the cen → q13 region of human chromosome 5 using mouse X human hybrid cells. Cytogenet Cell Genet. 1978;22:408–411. doi: 10.1159/000130984. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Shields J. A polygenic theory of schizophrenia. Proc Natl Acad Sci USA. 1967;58:199–205. doi: 10.1073/pnas.58.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppert M, Dobbs M, Scambler P, O’Connell P, Nakamura Y, Stauffer D, Woodward S, Burt R, Hughes J, Gardner E, Lathrop M, Wasmuth J, Lalouel JM, White R. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science. 1987;238:1411–1413. doi: 10.1126/science.3479843. [DOI] [PubMed] [Google Scholar]
- McGue M, Gottesman II, Rao DC. The transmission of schizophrenia under a multifactorial threshold model. Am J Hum Genet. 1983;35:1161–1178. [PMC free article] [PubMed] [Google Scholar]
- O’Dowd BF, Neote K, Munroe DLG, Gravel RA, Mahuran D, Willard HF. PstI RFLP in the human hexosaminidase gene (HEXB) on chromosome 5. Nucleic Acids Res. 1987;15:3194. doi: 10.1093/nar/15.7.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Baron M. Segregation analysis of schizophrenia and related disorders. Am J Hum Genet. 1984;36:1039–1059. [PMC free article] [PubMed] [Google Scholar]
- Schinzel A. Catalog of Unbalanced Chromosome Aberrations in Man. New York: de Gruyter; 1983. [Google Scholar]
- St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, Myers RH, Feldman RG, Pollen D, Drachman D, Growdon J, Bruni A, Foncin JF, Salmon D, Frommelt P, Amaducci L, Sorbi S, Piacenti S, Stewart GD, Hobbs WJ, Conneally PM, Gusella JF. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science. 1987;235:885–890. doi: 10.1126/science.2880399. [DOI] [PubMed] [Google Scholar]
- Wood S, Shukin FJ, McGillivray BC, Ray PN, Worton RG. A grandpaternally derived de novo deletion within Xp21 initially presenting in carrier females diagnosed as Kugelberg-Welander syndrome. Am J Med Genet. 1988;29:419–423. doi: 10.1002/ajmg.1320290225. [DOI] [PubMed] [Google Scholar]