

Abstract
It is often argued that the efficacy of herbal medicines is a result of the combined action of multiple constituents that work synergistically or additively. Determining the bioactive constituents in these mixtures poses a significant challenge. We have developed an approach to address this challenge, synergy directed fractionation, which combines comprehensive mass spectrometry profiling with synergy assays and natural products isolation. The applicability of synergy directed fractionation was demonstrated using the botanical medicine goldenseal (Hydrastis canadensis) as a case study. Three synergists from goldenseal were identified, sideroxylin (1), 8-desmethyl-sideroxylin (2), and 6-desmethyl-sideroxylin (3). These flavonoids synergistically enhance the antimicrobial activity of the alkaloid berberine (also a constituent of H. canadensis) against Staphylococcus aureus by inhibition of the NorA multidrug resistance pump. The flavonoids possess no inherent antimicrobial activity against S. aureus; therefore, they could have been missed using traditional bioactivity directed fractionation. The flavonoid synergists are present at higher concentration in extracts from H. canadensis leaves, while the antimicrobial alkaloid berberine is present at higher levels in H. canadensis roots. Thus, it may be possible to produce an extract with optimal activity against S. aureus using a combination of goldenseal roots and leaves.
Investigators who study botanical medicines face a formidable challenge. The effects of such preparations can result from the interaction of an array of chemically diverse components, whose identity and concentration may vary from one preparation to the next. Proponents of the medicinal use of botanicals often argue that their components interact synergistically, such that the combined effect is greater than the sum of the parts. Indeed, there are many examples in the literature whereby constituents are more effective in the phytochemical matrix than when isolated.1–2 However, the specific components responsible for these effects, and the fundamental mechanisms by which they interact, are rarely known. Methods are needed to unravel the complexity of botanical medicines and identify their key bioactive constituents. Such information would in turn enable better quality control and more meaningful and reproducible clinical trials.
A common approach for studying complex plant preparations is bioactivity guided fractionation. With this technique, extracts are screened in pursuit of those that contain biologically active compounds. The active extracts are then partitioned and purified, with the ultimate goal of identifying single active compounds. A limitation of bioactivity guided fractionation for studying botanical medicines is that it may not facilitate the identification of synergists. One type of synergy, potentiation, occurs when a compound possesses no activity on its own, but enhances the activity of another active compound.3 It is possible with bioactivity guided fractionation for potentiators to end up in inactive fractions throughout the isolation process, and thereby to be overlooked. With the studies presented here, we sought to develop a new method, synergy directed fractionation, which would facilitate identification of an array of biologically active compounds, including synergists, in a complex botanical medicine.
The botanical medicine goldenseal, Hydrastis canadensis L. (Ranunculaceae), is presented as a case study for the development of the methods presented herein. A recent survey ranked goldenseal among the 20 most popular herbal supplements used worldwide,4 and it is reported as the 6th most commonly used herbal preparation by children under 18 years of age.5 Goldenseal is recommended as a treatment for a number of ailments, including viral and bacterial infections and as an antidiarrheal.6 This use dates back to traditional application by Native Americans; the Cherokee employed the plant topically for eye and skin disease, and it was used by the Iroquois for gastrointestinal issues.7 There is some justification in the scientific literature for the effectiveness of goldenseal and its constituents. Crude goldenseal root8–12 and leaf10 extracts, and their most abundant alkaloid berberine,13–15 have been shown to possess antimicrobial activity in vitro. Berberine has also demonstrated efficacy in vivo against severe diarrhea16 and eye infections caused by Chlamydia trachomatis.17
While the antibacterial activity of goldenseal is generally attributed to alkaloids, several investigators have suggested a role for additional compounds that synergistically or additively enhance the effectiveness of the alkaloids.8–9,18 Most recently, we reported that extracts from the leaves of goldenseal contain synergists that increase the antimicrobial activity of the alkaloid berberine against Staphylococcus aureus.19 Berberine alone has only weak antimicrobial activity (MIC ~150 μg/mL).20 This low activity can be attributed, at least in part, to berberine being a substrate for bacterial multidrug resistant (MDR) efflux pumps, which function to extrude the compound from bacterial cells, rendering it inactive at low concentrations.20 We have determined that goldenseal leaf extracts contain unidentified constituents that appear to act as inhibitors of NorA, the major chromosomal multidrug resistance efflux pump expressed by Staphylococcus aureus.19 These findings are significant given that a major mechanism by which bacteria become resistant to treatment is by the over-expression of efflux pumps.21 Thus, it may be possible, using extracts that contain both efflux pump inhibitors and antimicrobial agents, to achieve efficacy against drug resistant bacteria and to limit the development of resistance. A major goal of these studies was to employ synergy directed fractionation to identify the compounds in H. canadensis leaf extracts that synergize the antimicrobial activity of berberine. In so doing, we sought to demonstrate the applicability of synergy directed fractionation as a method to study botanical medicines. Additional goals were to investigate efflux pump inhibition as the likely mode of action for synergists from H. canadensis, and to compare the abundance of active compounds in extracts from the roots and leaves of this plant.
RESULTS AND DISCUSSION
The approach for synergy directed fractionation developed as a result of these studies is outlined in Figure 1. Crude extracts are subjected to synergy testing to identify those likely to contain synergists. Active extracts are fractionated, and each fraction is profiled using liquid chromatography-mass spectrometry (LC-MS) and again subjected to a synergy assay. LC-MS profiles are compared to bioactivity data, and statistical correlations are used to identify potential bioactive compounds (which could include both synergists and those responsible for direct activity). The process of fractionation, synergy testing, and analysis with LC-MS is repeated iteratively until sufficient pure material is obtained for structure elucidation with NMR and MS.
Figure 1.

Synergy directed fractionation. Synergy testing is used to identify extracts and fractions likely to contain combinations of compounds that work together. These extracts are then fractionated, and LC-MS profiles are compared to bioassay data to identify potential bioactive compounds. The process is repeated iteratively as necessary to obtain pure compounds (synergists), the structures of which are elucidated using NMR and other spectroscopic techniques.
The synergy directed fractionation approach is perhaps best illustrated by example. In the following section, we demonstrate its application to identify synergists from the botanical medicine goldenseal (Hydrastis canadensis). Leaf material was chosen for this study because previous studies showed greater synergistic antimicrobial activity of goldenseal leaf extracts than goldenseal root extracts.19 The first step in the process of identifying the compounds responsible for this effect was to select an appropriate method to assay synergy. The checkerboard assay,22 which has been widely applied to study antimicrobial synergists, was chosen. To conduct the checkerboard assay, a crude H. canadensis leaf extract was tested at a range of concentrations in combination with the known antimicrobial agent berberine (also a component of H. canadensis). Minimum inhibitory concentrations (MIC) were measured for each combination of berberine/goldenseal extract concentrations, and an isobologram was plotted (Figure 2). Wagner has presented an excellent review of the use and interpretation of isobolograms for evaluation of synergy among phytochemical compounds.2 Briefly, an isobologram is a plot where each x,y data pair represents a combination of concentrations at which a desired activity is obtained (i.e. growth of bacteria is completely inhibited). The shape of the isobologram is indicative of either synergistic, additive, or antagonistic interactions among the compounds or extracts tested. As a quantitative measure of synergy, fractional inhibitory concentrations (FIC) can also be calculated using the same data on which the isobologram is based, as described elsewhere.19 An FIC cutoff of <0.5 is usually applied to indicate synergy,23 while an antagonistic effect is characterized by FIC >4, and FIC values between 0.5 and 4 indicate no interaction. 22 In this study, the crude H. canadensis extract demonstrated synergism when combined with berberine, as indicated by the convex shape of the isobologram (Figure 2) and the FIC value of 0.19 (Table 1).
Figure 2.

Isobolograms for berberine, the tannin free Hydrastis canadensis extract (after liquid-liquid extraction, see Supporting Information, Figure S1), the most active fraction from the first stage of the separation (fraction 4) and the most active fraction from the second stage of the separation (sub-fraction 2). All were tested in combination with berberine. The crude extract and its two fractions synergistically enhanced the antimicrobial activity of berberine, as demonstrated by the convex shape of the isobolograms, and the FIC values reported in Table 1.
Table 1.
MIC and FIC values (indicative of synergy) for a Hydrastis canadensis extract and fractions against wild type Staphylococcus aureus (NCTC8325-4).
| Treatment | MIC (μg/mL) | FIC Index |
|---|---|---|
| berberine | 150 | 1.00 |
| Hydrastis canadensis extracta | 100 | 0.19 |
| fraction 4b | >50 | 0.13 |
| sub-fraction 2c | >50 | 0.03 |
The tannin free Hydrastis canadensis extract after liquid-liquid partitioning (see Supporting Information, Figure S1) was tested.
Fraction 4 was the most active of the fractions collected from the first stage separation of the tannin free Hydrastis canadensis extract. Due to limited solubility, the maximum concentration tested was 50 μg/mL. No antimicrobial activity was observed at this concentration.
Sub-fraction 2 was the most active of the fractions collected from the second stage separation (using flash chromatography over silica gel with fraction 4 as starting material).
Once synergy had been demonstrated for the crude H. canadensis extract, the next step was to identify the compounds responsible for this activity. Toward this goal, the extract was fractionated (see Figure S1 provided as Supporting Information), and the activity of each fraction in combination with berberine was evaluated. Because of limited time and materials, rather than construct a full isobologram, the activity of each fraction was measured at a single concentration (75 μg/mL) in combination with a range of concentrations of berberine. The MIC of berberine in the presence of the fraction was then compared to the MIC of berberine alone, and fractions were deemed “active” if they enhanced the activity of the berberine (decreased its MIC). Note that this activity does not necessarily imply synergy; a fraction could enhance the activity of berberine either due to potentiation or to an additive antimicrobial effect. Several of the fractions collected from the first stage fractionation did, indeed, decrease the MIC of berberine (Figure 3A). The most active of these was fraction 4, which decreased the MIC of berberine 16-fold, from 75 μg/mL (berberine alone) to 4.7 μg/mL (berberine in combination with fraction 4).
Figure 3.

Panel A shows minimum inhibitory concentration (MIC) of starting material (sm), berberine alone, and berberine in combination with 11 fractions (each at a fixed concentration of 75 μg/mL) from the first stage of separation (flash chromatography over silica gel with CHCl3:MeOH gradient) of the Hydrastis candensis leaf extract. The MIC of berberine (75 μg/mL) was reduced significantly by combining with the starting material and with several of the fractions, with the most active fraction being fraction 4. Panels B and C show the distribution of flavonoids (measured with negative-ion LC-MS), and alkaloids (measured with positive-ion LC-MS), respectively in the fractions. Fraction 4 contains the highest levels of the 3 flavonoids. There is a moderate multiple correlation between MIC and flavonoid peak area (R = 0.77, p = 0.016).
A limitation of the simplified approach employed for screening fractions (constant concentration of fraction in combination with a range of concentrations of berberine) is that it does not enable additive effects to be distinguished from synergistic effects. Thus, the observed activity of fraction 4 was tested further with an in-depth synergy assay. A checkerboard assay identical to that employed for the crude extract was conducted using a range of combinations of fraction 4 with berberine. The resulting isobologram (Figure 2) and the FIC value of 0.13 (Table 1) were indicative of synergy.
Once it had been determined that fraction 4 did, indeed, contain synergist(s), LC-MS profiles were compared to the bioactivity data (Figure 3) to determine whether activity could be attributed to specific known or unknown compounds. LC-MS analysis revealed the presence of several ions that were not known constituents of H. canadensis in fraction 4. These ions were identified initially based on their m/z values (M - H− at 311 and 297). NMR analysis of pure isolated compounds eventually enabled their structures to be elucidated as a group of three flavonoids (1, 2, and 3), as described below.
It was possible, using LC-MS, to verify that the activity of fraction 4 was not due to the presence of the three major known alkaloids in H. canadensis, berberine (4), hydrastine and canadine. From Figure 3C, it is apparent that fractions 9, 10, and 11 contained the highest levels of berberine. The weak activity of these fractions (2-fold decrease in MIC) was attributed to this compound, and they were not investigated further. Hydrastine and canadine were present at higher concentrations in fraction 3 than in fraction 4, but fraction 4 was significantly more active. This suggested that other compounds were responsible for the activity of fraction 4, a finding that was not surprising given that hydrastine and canadine have been shown to be inactive against S. aureus, both alone and in combination with berberine.19
Visual inspection of the data in Figure 3 revealed separate correlations with the presence of compounds at m/z 297 and 311. The strength of this correlation was evaluated jointly using a statistical method. A multiple correlation analysis was conducted to examine the extent of linear association between peak areas of ions 297 and 311, and the MIC of the fractions obtained from the first stage separation. A moderate correlation was observed (R = 0.77, p = 0.02), which suggests that the joint presence of ions 297 and 311 explains approximately 60% (0.772), but does not completely explain the activities observed for the fractions. Presumably, other compounds that play a role in the overall activity of the crude extract are also present. Indeed, visual inspection of the data could also have led to this conclusion, given that fractions 5 and 6 demonstrated pronounced activity (8-fold decrease in MIC of berberine) even without significant levels of berberine or of the ions with m/z 297 and 311.
Fraction 4 was subjected to a second stage of separation using flash chromatography with a hexane/ethyl acetate gradient (see fractionation scheme, Supporting Information Figure S1). Mass spectrometry profiles were compared to MIC data (Figure 4), and again the compounds with m/z 297 and 311 were present at highest concentration in the most active fraction (sub-fraction 2). Sub-fraction 2 was also subjected to the synergy assay (Figure 2), and an even more pronounced synergistic enhancement of the activity of berberine was observed (FIC = 0.03). The second stage of purification yielded multiple active fractions (Figure 4), which explains the weak multiple correlation observed between MIC and peak area of compounds with m/z 297 and 311 for the second stage of separation (R = 0.55, p = 0.37). This weak correlation implies that the compounds with m/z 297 and 311 do not fully account for the activity of fraction 4, and that, again, additional active compounds were present in this fraction.
Figure 4.

Comparison between MIC (A) and distribution of flavonoids (B) and alkaloids (C) after the second stage of the separation (flash chromatography over silica gel with hexane:EtOAc gradient) of the Hydrastis canadensis leaf extract. Sub-fraction 2 contained the highest levels of the three flavonoids, and was one of the most active fractions. Note, however, that other compounds appear to play a role in the activity of the starting material (sm); several fractions with little to no flavonoid content have activity on the same order as sub-fraction 2. The presence of additional active compounds would also explain the weak multiple correlation between activity and flavonoid content after this second stage of separation (R = 0.55, p = 0.37).
Two rounds of preparative HPLC starting with sub-fraction 2 ultimately lead to the isolation of the flavonoids sideroxylin (1), 8-desmethyl-sideroxylin (2), and 6-desmethly sideroxylin (3). These flavonoids were the same compounds observed in the LC-MS spectra at m/z 311 (sideroxylin) and 297 (isomeric 8-desmethyl-sideroxylin and 6-desmethyl-sideroxylin). The flavonoids have been reported previously as constituents of Eucalyptus spp. (1 and 2)24–26 and Dracaena cochinchinensis (3, referred to in the reference by the name 4′,5-dihydroxy-7-methoxy-8-methylflavone).27 Ours is the first publication to report these flavonoids as constituents of H. canadensis.28 Other flavonoids have, however, been identified in H. canadensis9, and, as previously mentioned, it is well known that the plant contains the alkaloid berberine (4). Both 1H and 13C NMR chemical shift data (Supporting Information, S2) were in excellent agreement with those reported for the flavonoids,24–27 and high resolution mass spectrometry measurements confirmed the molecular formulae of C18H16O5 for sideroxylin and C17H14O5 for the isomers 8-desmethyl-sideroxylin and 6-desmethyl-sideroxylin.
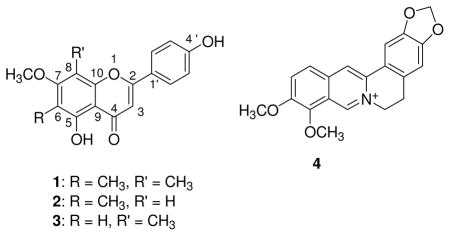
Although the crude extract and early stage fractions were quite soluble in the antimicrobial assay medium (Müeller-Hinton broth with 2% DMSO), the isolated flavonoids demonstrated very poor solubility (<13 μg/mL) in this same medium. As such, it was not possible to observe an influence of the pure flavonoids on the MIC of berberine against wild type S. aureus. However, activity was observed for 8-desmethyl-sideroxylin against a NorA efflux pump overexpressing strain of S. aureus (S. aureus K2378, norA++, Table 2), which is more sensitive to inhibitors. Note that although the reported assay concentration of 8-desmethyl-sideroxylin was 75 μg/mL (Table 2), the actual concentration was likely much lower, due to the aforementioned poor flavonoid solubility. The flavonoid demonstrated no antimicrobial activity alone (MIC >300 μg/mL) against the norA++ S. aureus, but decreased the MIC of berberine 2-fold. Thus, it can be said that 8-desmethyl-sideroxylin (2) synergistically enhances, or potentiates, the antimicrobial activity of berberine against norA++ S. aureus.
Table 2.
MIC of berberine alone and in combination with 8-desmethyl-sideroxylin (2) against S. aureus K2378 (a norA++ strain). Reserpine served as a positive control.
| Treatment | MIC (μg/mL) |
|---|---|
| berberine (4) | 300 |
| 8-desmethyl-sideroxylin (2) | >300 |
| berberine (4) + 8-desmethyl-sideroxylin (2) (75 μg/mL) | 150 |
| berberine (4) + reserpine (75 μg/mL) | 75 |
Previous literature has shown flavonoids to act as efflux pump inhibitors,20,29–30 and our group has demonstrated efflux pump inhibitory activity for goldenseal leaf extracts.19 Thus, we hypothesized efflux pump inhibition as a likely mode of action for the H. canadensis flavonoids. To test this hypothesis, the activity of each of the three individual flavonoids was evaluated with an ethidium bromide efflux assay (Figure 5). Ethidium bromide is a substrate of the S. aureus efflux pump NorA, and fluoresces strongly (due to intercalation with DNA) inside bacterial cells.20 With the ethidium bromide efflux assay, cells are loaded with ethidium bromide and fluorescence is monitored over time. A decrease in fluorescence indicates efflux of ethidium bromide from the cells, and in the presence of an efflux pump inhibitor, fluorescence should decrease more slowly. The ethidium bromide efflux assay has the advantages of being more sensitive than the checkerboard synergy assay, and also having a greater tolerance for DMSO (10% DMSO was used in this assay, as compared to 2% DMSO in the checkerboard assays). These features enabled the solubility problems with the pure flavonoids to be circumvented when using the efflux assay.
Figure 5.

The three flavonoids from Hydrastis canadensis inhibit the NorA efflux pump of Staphylococcus aureus. Decrease in fluorescence over time is due to efflux of ethidium bromide, which is blocked by the positive control (CCCP) and all three flavonoids, sideroxylin (1), 8-desmethyl-sideroxylin (2), and 6-desmethyl-sideroxylin (3). Sideoxylin is the least active of the three flavonoids. The effect is observed with wild type Staphylococcus aureus (NCTC8325-4) (A) but not a NorA deletion mutant (K1758) (B). All of the flavonoids and controls were tested at a final concentration of 100 μM in Müeller-Hinton broth containing 10% DMSO (vehicle). Each point is an average % fluorescence from three independent measurements, with error bars equal to ± standard error (SE).
Figure 5A shows the results of the ethidium bromide efflux assay with wild type S. aureus (NCTC 8325-4). The positive control, carbonyl cyanide m-chlorophenylhydrazone (CCCP), and compounds 1, 2, and 3 inhibited efflux of ethidium bromide. Significant differences between vehicle control (10% DMSO) and treatment with sideroxylin (1) (p = 0.02), 8-desmethyl-sideroxylin (2) (p = 0.0002), 6-desmethyl-sideroxylin (3) (p = 0.002), and CCCP (p = 0.0002) were observed at t = 300 s. When the same experiment was repeated using the norA deletion mutant S. aureus (K1758), neither CCCP nor any of the flavonoids significantly inhibited efflux of ethidium bromide (Figure 5B). The results demonstrate that all three flavonoids from S. aureus inhibit the NorA efflux pump of S. aureus.
Although all three flavonoids demonstrated some efflux pump inhibitory activity, they differed in potency (Figure 5A). Sideroxylin (1), the dimethyl analog, was a less effective inhibitor than either of the monomethyl analogs, 8-desmethyl-sideroxylin (2) and 6-desmethyl-sideroxylin (3). It appears that substitution on both the 6 and 8 positions decreases efflux pump inhibitory activity.
A relevant question to the quality control of dietary supplements from H. canadensis aerial portions is whether the flavonoids and alkaloids are present at detectable levels in such preparations. To address this question, ethanolic extracts were analyzed that had been prepared using a method based on that employed in the manufacture of dietary supplements.31 Both flavonoids and alkaloids were detected in the ethanolic extracts. Retention times and CID fragmentation patterns for these compounds (see Supporting Information, Table S2) matched those of standards. In agreement with previous literature, the alkaloids were present at higher levels in extracts prepared from goldenseal roots than leaves.18 Conversely, the flavonoids were detected only at very low levels in the root/rhizome extracts, but at much higher levels in the leaf extracts (Table 3). Goldenseal leaf extracts contained an average of 1.9 ± 0.4 mM 8-desmethyl sideroxylin/6-desmethyl-sideroxylin (quantified as a mixture) and 73.3 ± 0.9 μM sideroxylin. Levels in the root extracts were over 50 times lower. Notably, the same leaf extracts for which data is presented in Table 3 have been shown previously to have efflux pump inhibitory activity,19 which is likely attributable, at least in part, to the presence of flavonoids 1, 2, and 3.
Table 3.
Concentrations (± SE, N = 6) of alkaloids (berberine (4), hydrastine, canadine) and flavonoids [sideroxylin (1), 8-demethy-sideroxylin (2), and 6-desmethyl-sideroxylin (3)] in extracts from the roots/rhizomes and leaves of Hydrastis canadensis. Data are averages of measurements made from five separate leaf extracts and five separate root/rhizome extracts (each from a single plant).
| Berberine (mM) ± SE | Hydrastine (mM) ± SE | Canadine (mM) ± SE | Flavonoids 2/3a (mM) ± SE | Flavonoid 1 (mM) ± SE | |
|---|---|---|---|---|---|
| goldenseal (H. canadensis) root/rhizome | 36.4 ± 6.4 | 3.14 ± 0.50 | 0.0687 ± 0.0035 | 0.0378 ± 0.011 | 0.00084 ± 0.00033 |
| goldenseal (H. canadensis) leaf | 7.78 ± 0.60 | 1.12 ± 0.12 | 0.4500 ± 0.0040 | 1.85 ± 0.41 | 0.0733 ± 0.0089 |
Flavonoids 2 and 3 were quantified as a mixture because the two isomers were not resolved using reversed phase HPLC-MS with a C-18 stationary phase.
The finding that goldenseal leaf extracts have higher levels of synergists while root extracts contain higher levels of alkaloids suggests the potential benefit of using a mixture of root and leaf material in the production of dietary supplements from goldenseal. Further studies would, however, be needed to evaluate the safety and efficacy of goldenseal leaf extracts in vivo. If goldenseal leaf material were shown to be efficacious, this could add value to cultivated goldenseal crops. In addition, the use of goldenseal leaf material has the added advantage of reducing impact on wild goldenseal populations. Goldenseal leaves can be harvested in the fall, after the berries have dropped, without killing the plants. Sustainable production of goldenseal would be particularly desirable given that it is listed as threatened by CITES in much of its native habitat.32
In terms of the broader relevance of this study, it has been shown that synergy directed fractionation can be employed to identify both compounds with direct antimicrobial activity (berberine) and efflux pump inhibitors that synergistically enhance berberine’s activity (the three flavonoids) from a complex botanical extract. The results highlight three important elements that distinguish synergy directed fractionation from bioactivity guided fractionation. First, the application of synergy testing to the crude extract made it possible to determine that it contained synergists even prior to fractionation. Second, integration of synergy testing with the fractionation process allowed synergists to be tracked as they were purified. This process ultimately facilitated the isolation of synergists that possess no inherent antimicrobial activity, compounds that may have been missed in inactive fractions with traditional bioactivity guided fractionation. Third, comparison of LC-MS data and biological activity data after each stage of separation allowed potential active compounds to be identified and tracked throughout the isolation process. This establishes an important linkage between biological activity of the crude extract and the presence of specific active compounds. In the example presented here, it was possible based on LC-MS profiles to identify ions corresponding to the active flavonoids even after only one stage of separation. Importantly, it was not necessary to have prior knowledge of the identities of the compounds detected in the active fractions to correlate their presence with biological activity. These compounds were initially identified based solely on characteristic m/z values, and it was only after their likely role in the biological activity of the extract had been established that they were isolated and their identities determined via NMR.
Visual inspection and statistical analyses of the data after the first and second stages of separation suggests the presence of multiple active compounds, including some that have not yet been identified. By no means does this finding negate the importance of the flavonoids and alkaloids in the activity of H. canadensis. Rather, the results suggest that the antimicrobial activity of the crude H. canadensis extract is due to multiple compounds, including antimicrobial alkaloids, flavonoids that synergistically enhance the antimicrobial activity of the alkaloids, and several additional compounds whose identities and modes of action have yet to be determined. This finding is consistent with the claims often made for botanical medicines; that their activity results from the combined (and perhaps synergistic) action of an array of chemically diverse constituents.
EXPERIMENTAL SECTION
General Experimental Procedures
NMR spectra were acquired with a JEOL ECA-500 spectrometer (500 MHz for 1H and 125 MHz for 13C) using DMSO-d6. LC-MS measurements were made using an LCQ Advantage (Thermo) coupled to an Agilent HP1100 HPLC with a Prevail C18 column (3 μm; 110 Å, 50 × 2.1 mm). HRESIMS data were acquired using an LTQ-Orbitrap (Thermo). Flash chromatography separations were accomplished using an automated CombiFlash RF system (Teledyne-Isco). HPLC separations were performed on a Varian HPLC system (ProStar 210 pumps, ProStar 710 fraction collector, ProStar 335 photodiode array detector) with Galaxie Chromatography Workstation software (version 1.9.3.2). Preparative scale HPLC separations employed a C-18 YMC-Pack ODS-A column (5 μm, 120Å; 250 × 20 mm, Waters) or a Luna PFP(2) column (pentafluorophenyl propyl, 5 μm, 100Å; 250 × 21.2 mm, Phenomenex). Analytical scale HPLC separations were accomplished with YMC ODS-A (5 μm, 120Å; 150 × 4.6 mm, Waters) and Luna PFP(2) (5 μm, 100Å; 150 × 4.6 mm, Phenomenex) columns. For antimicrobial assays, OD600 was read using a POLARstar Optima microplate reader (BMG Labtech, Inc). Fluorescence was monitored in efflux assays using a Spex FluoroMax-2 spectrofluorometer (Instruments S. A., Inc). Müeller-Hinton broth, carbonyl cyanide m-chlorophenylhydrazone (CCCP), berberine (4) (purity >98% by HPLC), (1R,9S)-(−)-β-hydrastine (purity >98% by HPLC) and reserpine (purity >98% by TLC) were purchased from Sigma Aldrich and canadine (tetrahydroberberine, purity >98% by HPLC, stereochemistry unconfirmed) was obtained from Chromadex. Other reagents were purchased from Fisher Chemical, except EtOH (95%), and HPLC grade MeOH, which were obtained from Pharmco-AAPER. Nanopure water was prepared with a nanodiamond water purification system (Barnstead).
Plant Material
Individual goldenseal (Hydrastis canadensis) plants and bulk leaf material were collected in July 2010 from William Burch in Hendersonville, North Carolina (NC, N 35° 24.277′, W 082° 20.993′, 702.4 m elevation). The plants were cultivated in their native environment, a hardwood forest understory, and were at least 5 years old at time of harvest. A voucher specimen was deposited at the Herbarium of the University of North Carolina at Chapel Hill (NCU583414) and authenticated by Dr. Alan S. Weakly. Samples were air dried at 37 °C before extraction. This temperature was chosen because it was sufficient to dry plant material without causing discoloration of the leaves.
Extraction
Ethanolic H. canadensis extracts (50:50 EtOH:H2O) were prepared via a procedure used for the preparation of botanical dietary supplements, as described previously.19 Extracts were prepared separately for the root and leaf material from five individual goldenseal plants (for a total of ten extracts, five leaf and five root). An additional larger scale extraction (2.3 kg leaf material) was performed to facilitate isolation. Leaves were homogenized and percolated in MeOH overnight, and the MeOH extract was concentrated in vacuo and subjected to liquid-liquid extraction, as described previously.33 Briefly, the extract was defatted by partitioning between hexane and 10% aqueous MeOH (1:1). The dried aqueous MeOH fraction was partitioned further between 4:1:5 CHCl3:MeOH:H2O, and the organic extract was washed with 1% saline solution to remove hydrosoluble tannins.34
Isolation
The isolation scheme is provided as supporting information (Figure S1). The first stage of normal phase flash chromatography (330 g silica gel column) was conducted at a 60 mL/min flow rate with a 110 min hexane:CHCl3:MeOH gradient. The most active fraction from the first stage separation (fraction 4) was subjected to a second stage of normal phase flash chromatography (120 g silica gel column), which employed an 85 mL/min flow rate and a 50 min. hexane:EtOAc:MeOH gradient. The most active fraction from the second stage separation (sub-fraction 2) was purified using reversed phase preparative HPLC with a YMC ODS-A column at a 8.0 mL/min flow rate. A MeOH:H2O gradient was employed, which progressed linearly from 70:30 to 85:15 over 60 min. Compound 1 eluted at 44.85 min (22.2 mg, 98.6 % purity, 0.00096% yield) and compounds 2 and 3 eluted as a mixture (53.4 mg) between 36.8 and 37.3 min. The mixture of 2 and 3 was subjected to an additional stage of preparative HPLC using a Luna PFP(2) (pentafluorophenyl propyl) stationary phase with a 21.2 mL/min flow rate. An CH3CN:H2O gradient was used, initiating at 45:55 (isocratic for 12 min), increasing linearly to 58:42 from 12.1 to 15.0 min, and from 58:42 to 60:40 from 15.1 to 20.0 minutes. Compound 2 eluted at 17.4 min (14.8 mg, 99% purity, 0.00064 % yield) and compound 3 eluted at 15.21 min (2.40 mg, 98.9% purity, 0.0001 % yield)
Sideroxylin (1): white solid; HRESIMS m/z 311.0922 [M - H−] (calcd for C18H15O5−, 311.0925); 1H NMR (500 MHz DMSO-d6) and 13C NMR (125 MHz DMSO-d6) chemical shifts were in agreement with literature values24–26 and are provided as Supporting Information (Table S1 and Figure S2).
8-Desmethyl-sideroxylin (2): white solid; HRESIMS m/z 297.0765 [M - H−] (calcd for C17H13O5−, 297.0768); 1H NMR (500 MHz DMSO-d6) and 13C NMR (125 MHz DMSO-d6) were in agreement with literature values,24–26 as indicated by chemical shift data (Table S1) and NMR spectra (Figure S2) provided as Supporting Information.
6-Desmethyl-sideroxylin (3): white solid; HRESIMS m/z 297.0761 [M - H−] (calcd for C17H13O5−, 297.0768). 1H NMR (500 MHz DMSO-d6) and 13C NMR (125 MHz DMSO-d6)7 data were in agreement with literature27 and are provided for reference as Supporting Information (Table S1 and Figure S2).
LC-MS
Alkaloids and flavonoids were quantified and identified using LC-MS with an ion trap mass spectrometer. Reversed phase HPLC was used for the separation with a Prevail C18 column and a gradient employing 1% aqueous acetic acid in acetonitrile. The gradient was as follows (ratios represent CH3CN:H2O): 0 to 5 min, 0:100 to 32:68; 5.1 to 20 min, 32:68 to 100:0; 20 to 25 min, isocratic 100:0; 25 to 25.10 min, 100:0 to 0:100. Extracts were analyzed both in the positive ion mode (for detection and quantification of alkaloids) and the negative ion mode (for detection and quantification of flavonoids). Standards were analyzed over a concentration range of 7.8 × 10−7 M to 1.0 × 10−4 M (flavonoids) and 5.0 × 10−7 M to 1.0 × 10−4 M (alkaloids). Calibration curves were plotted as the area of the relevant selected ion trace (as detected by the LC-MS) vs. concentration. Extracts were diluted so that constituents fell within the linear range of the calibration curves. The alkaloids berberine, hydrastine, and canadine were identified by comparison of retention time, m/z, and CID fragmentation patterns with those of commercial standards (see Table S2 provided as Supplementary Information), as described previously.19 The flavonoids were identified in the same fashion using compounds 1, 2, and 3, isolated as part of this study, as standards.
Antimicrobial Assays
Two strains of S. aureus were employed, wild type (NCTC 8325-4)35 and NorA overexpressing (K2378, norA++).36 Minimum inhibitory concentration (MIC) was evaluated according to Clinical Laboratory Standards Institute (CLSI) guidelines.37 A single colony inoculum of S. aureus was grown to log phase in Müeller-Hinton broth and was adjusted to a final assay dilution of 1.0 × 105 CFU/mL based on absorbance at 600 nm (OD600). The negative control consisted of 2% DMSO in broth (vehicle), and the known efflux pump inhibitor reserpine38 served as the positive control. Triplicate wells were prepared with all treatments and controls. Additional wells were included containing the samples without bacteria, for the purpose of background subtraction. OD600 was read after incubation for 20 hrs at 37 °C. MIC was defined as the concentration at which no statistically significant difference was observed (via ANOVA) between the negative control and treated samples.
Synergy Testing
Broth microdilution antimicrobial checkerboard assays19,22 were performed to evaluate synergy for the crude extracts. Extracts were tested in combination with berberine over a concentration range of 5 to 300 μg/mL. The antibiotic norfloxacin (positive control) demonstrated an MIC value of 2 μg/mL, consistent with the literature.39 The vehicle control consisted of 2% DMSO in broth. To evaluate synergistic interactions, isobolograms were plotted and FIC indices calculated, as described previously.19 After each round of fractionation, a simplified version of the synergy assay was used to enable rapid screening of the large number of fractions generated. This involved testing berberine at a range of concentrations (5 to 300 μg/mL) in combination with a constant concentration of each of the extract fractions (75 μg/mL). The known efflux pump inhibitor reserpine38, also at 75 μg/mL, was employed as a positive control for these experiments. Fractions were deemed active if they enhanced the activity of berberine (reducing its MIC against S. aureus by at least 2-fold), and were advanced to the next stage of separation. Using the simplified synergy assay, it is not possible to distinguish synergistic effects from additive effects, so the most active fractions were also subjected to the synergy checkerboard assay as described for the crude extracts.
Efflux Pump Inhibition Assay
The efflux pump inhibition assay was conducted using a previously published method.19 Wild type S. aureus (NCTC 8325-4)35 and an isogenic NorA deletion mutant (K1758 norA−)40 were grown to OD600 = 0.74 in Müeller-Hinton broth. Ethidium bromide (25 μM) and carbonyl cyanide m-chlorophenylhydrazone (CCCP, 100 μM) were added, and the bacteria were incubated at 20 °C with agitation (300 RPM) for 20 min. This solution was diluted to OD600 = 0.40 with broth containing ethidium bromide and CCCP at 25 μM and 100 μM, respectively. Aliquots (1 mL) of this solution were centrifuged at 20 °C for 5 min at 13,000 × g in a Spectrafuge 24D centrifuge (Labnet). The pellets were stored on ice for <1 hr. Prior to measurement, the pellets were thawed for 5 min and 1 mL of fresh broth containing DMSO (10% final assay concentration) and treatments (CCCP or flavonoid) was added. Fluorescence of these solutions was measured every second for 300 s with λex = 530 nm, λemiss = 600 nm, and slit widths of 5 mm.
Statistics
Statistical significance of means vs. control was evaluated using two factor ANOVA with p < 0.05 considered significant. Multiple (canonical) correlations were computed to assess the degree of association of MIC jointly with the peak area of several ions (flavonoid 1, m/z 311, and the flavonoid 2/3 mixture, m/z 297). The peak areas appeared to be non-normally distributed, and sample sizes were small, so p-values for assessing the statistical significance of the multiple correlations were calculated using permutation tests.
Supplementary Material
Acknowledgments
This research was supported by Grant Number 1 R15 AT005005 from the National Center for Complementary and Alternative Medicine (NCCAM), a component of the National Institutes of Health (NIH). Thanks are owed to W. Burch for providing goldenseal plant material, B. Ehrmann for technical assistance, and G. Kaatz, J. Falkinham, and R. Cech for helpful advice.
Footnotes
Supporting Information. 13C and 1 H NMR data, a fractionation scheme, and LC-MS fragmentation data, and retention times for alkaloids and flavonoids. This information is available free-of-charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.Spelman K, Duke JA, Bogenschutz-Godwin MJ. In: Natural Products from Plants. 2. Cseke L, Kirakosyan A, Kaufman PB, Warber SL, Duke JA, Brielmann HL, editors. CRC Taylor and Francis; Boca Raton: 2006. pp. 475–501. [Google Scholar]
- 2.Wagner H, Ulrich-Merzenich G. Phytomed. 2009;16:97–110. doi: 10.1016/j.phymed.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Berenbaum M. Pharmacol Rev. 1989;41:93–141. [PubMed] [Google Scholar]
- 4.Herbal Supplements, Prescription Drugs, Rx-to-OTC Products. BCC Research; Wellesley: 2006. BCC Research Report; p. 202. [Google Scholar]
- 5.Barnes P, Powell-Griner E, McFann K, Nahin R. CDC Advance Data Report. 2004;343 [PubMed] [Google Scholar]
- 6.Upton R. Goldenseal root Hydrastis canadensis: Standards of Analysis, Quality Control, and Therapeutics. American Herbal Pharmacopoeia; Santa Cruz: 2001. p. 36. [Google Scholar]
- 7.Blumenthal M, Hall T, Goldberg A, Kunz T, Dinda K, Brinckmann J, Wollschlaeger B. The ABC Clinical Guide to Herbs. American Botanical Council; Austin, Texas: 2003. p. 512. [Google Scholar]
- 8.Gentry EJ, Jampani HB, Keshavarz-Shokri A, Morton MD, Velde DV, Telikepalli H, Mitscher LA, Shawar R, Humble D, Baker W. J Nat Prod. 1998;61:1187–1193. doi: 10.1021/np9701889. [DOI] [PubMed] [Google Scholar]
- 9.Hwang BY, Roberts SK, Chadwick L, Wu CD, Kinghorn DA. Planta Med. 2003;69:623–627. doi: 10.1055/s-2003-41115. [DOI] [PubMed] [Google Scholar]
- 10.Knight SE. Bios. 1999;70:3–10. [Google Scholar]
- 11.Mahady GB, Pendland SL, Stoia A, Chadwick L. Phytother Res. 2003;17:217–221. doi: 10.1002/ptr.1108. [DOI] [PubMed] [Google Scholar]
- 12.Scazzocchio F, Cometa MF, Tomassini L, Palmery M. Planta Med. 2001;67:561–564. doi: 10.1055/s-2001-16493. [DOI] [PubMed] [Google Scholar]
- 13.Amin AH, Subbaiah TV, Abbasi KM. Can J Microbiol. 1969;15:1067–76. doi: 10.1139/m69-190. [DOI] [PubMed] [Google Scholar]
- 14.Hyeon-Hee Y, Kang-Ju K, Jeong-Dan C, Hae-Kyong K, Young-Eun L, Na-Young C, Yong-Ouk Y. J Med Food. 2005;8:454–461. [Google Scholar]
- 15.Noskin GA, Rubin RJ, Schentag JJ, Kluytmans J, Hedblom EC, Smulders J, Lapetina E, Gemmen E. Arch Intern Med. 2005;165:1756–1761. doi: 10.1001/archinte.165.15.1756. [DOI] [PubMed] [Google Scholar]
- 16.Purohit KR, Rao KS. Antiseptic. 1969;66:855–863. [Google Scholar]
- 17.Khosla PK, Neeraj VI, Gupta SK, Satpathy G. Rev Int Trach Pathol Ocul Trop Subtrop Sante Publique. 1992;69:147–165. [PubMed] [Google Scholar]
- 18.McNamara CE, Perry NB, Follett JM, Parmenter GA, Douglas JA. J Nat Prod. 2004;67:1818–1822. doi: 10.1021/np049868j. [DOI] [PubMed] [Google Scholar]
- 19.Ettefagh KA, Burns JT, Junio HA, Kaatz GW, Cech NB. Planta Med. 2010 doi: 10.1055/s-0030–1250606. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stermitz FR, Lorenz P, Tawara JN, Zenewicz LA, Lewis K. Proc Nat Acad Sci. 2000;97:1433–1437. doi: 10.1073/pnas.030540597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webber MA, Piddock LJV. J Antimicrob Chemother. 2003;51:9–11. doi: 10.1093/jac/dkg050. [DOI] [PubMed] [Google Scholar]
- 22.Eliopolous GM, Moellering RC. In: Antibiotics in Laboratory Medicine. 3. Iorian V, editor. Williams and Wilkins; Baltimore, MD: 1996. pp. 330–396. [Google Scholar]
- 23.Odds FC. J Antimicrob Chemother. 2003;52:1–1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 24.Wollenweber E, Kohorst G. Z Naturforsch. 1981;36:913–915. [Google Scholar]
- 25.Lamberton JA. Aust J Chem. 1964;17:692–696. [Google Scholar]
- 26.Sarkera SD, Bartholomew B, Nash RJ, Simmonds MSJ. Biochem Syst Ecol. 2001;29:759–762. doi: 10.1016/s0305-1978(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 27.Tu P-F, Tao J, Hu Y-Q, Zhao M-B, Zhongguo TY. Chin J Nat Med. 2003;1:27–29. [Google Scholar]
- 28.Chin Y-W, Parekh H, Yildiz S, Beavers R, Kinghorn AD, Wu CD. No peer reviewed publications. A conference abstract mentions flavonoids 1 and 2 (but not 3) as constituents of H. canadensis. IADR/AADR/CADR 85th General Session and Exhibition; March 21–24, 2007; New Orleans, LA. [Google Scholar]
- 29.Belofsky G, Carreno R, Lewis K, Ball A, Casadei G, Tegos GP. J Nat Prod. 2006;69:261–264. doi: 10.1021/np058057s. [DOI] [PubMed] [Google Scholar]
- 30.Stermitz FR, Scriven LN, Tegos G, Lewis K. Planta Med. 2002;68:1140–1141. doi: 10.1055/s-2002-36347. [DOI] [PubMed] [Google Scholar]
- 31.Cech RA. Making Plant Medicine. Horizon Herbs: Williams; 2000. p. 276. [Google Scholar]
- 32.McGraw JB, Sanders SM, Voort MVD. J Torrey Bot Soc. 2003;130:62–69. [Google Scholar]
- 33.Gu J-Q, Graf TN, Lee D, Chai H-B, Mi Q, Kardono LBS, Setyowati FM, Ismail R, Riswan S, Farnsworth NR, Cordell GA, Pezzuto JM, Swanson SM, Kroll DJ, Falkinham JO, Wall ME, Wani MC, Kinghorn AD, Oberlies NH. J Nat Prod. 2004;67:1156–1161. doi: 10.1021/np040027m. [DOI] [PubMed] [Google Scholar]
- 34.Wall ME, Wani MC, Brown DM, Fullas F, Olwald JB, Josephson FF, Thorton NM, Pezzuto JM, Beecher WW, Farnsworth NR, Cordell GA, Kinghorn AD. Phytomed. 1996;3:281–285. doi: 10.1016/S0944-7113(96)80067-5. [DOI] [PubMed] [Google Scholar]
- 35.Novick R. Virology. 1967;33:155–166. doi: 10.1016/0042-6822(67)90105-5. [DOI] [PubMed] [Google Scholar]
- 36.Tomkiewicz D, Casadei G, Larkins-Ford J, Moy TI, Garner J, Bremner JB, Ausubel FM, Lewis K, Kelso MJ. Antimicrob Agents Chemother. 2010;54:3219–3224. doi: 10.1128/AAC.01715-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anonymous. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard. 8. Clinical and Laboratory Standards Institute; 2009. p. 65. M07-A8. [Google Scholar]
- 38.Markham PN. Antimicrob Agents Chemother. 1999;43:988–989. doi: 10.1128/aac.43.4.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barry AL, Jones RN, Thornsberry C, Ayers LW, Gerlach EH, Sommers HM. Antimicrob Agents Chemother. 1984;25:633–637. doi: 10.1128/aac.25.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price CTD, Kaatz GW, Gustafson JE. Int J Antimicrob Agents. 2002;20:206–213. doi: 10.1016/s0924-8579(02)00162-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.