

Abstract
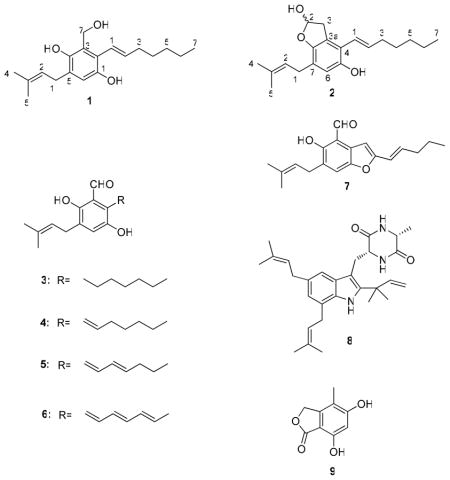
Bioassay-guided fractionation of the fungus Eurotium repens resulted in the isolation of two new benzyl derivatives, (E)-2-(hept-1-enyl)-3-(hydroxymethyl)-5-(3-methylbut-2-enyl)benzene-1,4-diol (1) and (E)-4-(hept-1-enyl)-7-(3-methylbut-2-enyl)-2,3-dihydrobenzofuran-2,5-diol (2) along with seven known compounds (3–9) including five benzaldehyde compounds, flavoglaucin (3), tetrahydroauroglaucin (4), dihydroauroglaucin (5), auroglaucin (6) and 2-(2′,3-epoxy-1′,3′-heptadienyl)- 6-hydroxy-5-(3-methyl-2-butenyl)benzaldehyde (7), one diketopiperazine alkaloid echinulin (8), and 5,7-dihydroxy-4-methylphthalide (9). The chemical structures of these compounds were established on the basis of extensive 1D, 2D NMR and HRMS data. Compounds 1–4 and 6 showed good binding affinity for human opioid or cannabinoid receptors. These findings have important implications for psychoactive studies with this class of compounds.
Central nervous system (CNS) disorders are worldwide common problems and annually about one-fourth of adult Americans suffer from a diagnosable psychotic disorder.1 Neuropathic pain is defined as a type of pain that is caused by a lesion or dysfunction of the nervous system. Worldwide as much as 7% to 8% of the population is affected by, while in the United States more than two million people suffer from, neuropathic pain.2 The treatment of neuropathic pain is challenging because the common causes are complex and may include diabetic neuropathy, nerve compression syndromes, postherpetic or trigeminal neuralgia, stroke, shingles, multiple sclerosis, spinal cord injury, cancer and/or HIV infection.3 The opioid and cannabinoid receptors are G-protein coupled receptors that have been classified into subtypes. The opioid receptor system includes three subtypes, δ, κ and μ, and the cannabinoid receptor system comprises at least two major subtypes, CB1 and CB2.4,5 Studies show that ligands of opioid or cannabinoid receptors have long been known to modulate pain.6 Furthermore, scientists found that components of neuropathic pain are affected significantly by the administration of opiates such as morphine and exogenous cannabinoids such as Δ9-tetrahydrocannabinol (compounds that have analgesic and addictive properties).7 These observations suggest that the opioid and the cannabinoid receptor systems are altered during neuropathic pain. Opioid and cannabinoid receptor agonists are potent analgesics and remain the more promising treatments for patients with neuropathic pains.8
Opioid and cannabinoid receptors are distributed in the regions associated with pain modulation. Agonists of opioid and cannabinoid receptors have been shown to activate pain inhibitory pathways in the central nervous system.6 So far, the majority of clinically available opioid analgesics are μ-agonists and includes morphine and its derivatives.7 However, morphine and its derivatives have many side effects such as tolerance and dependency.8 In order to meet the need for an efficacious analgesic without side effects, attention has focused on other opioid and cannabinoid receptors. Currently there are only a few agents that target κ and CB2 receptors, and none that target δ and CB1 receptors. Our findings provide interesting insights on new chemical scaffolds biosynthesized by nature that can lead the discovery of new selective ligands for opioid or cannabinoid receptors
In a high-throughput screening utilizing a receptor binding assay to find natural products with selective affinity for specific opioid and cannabinoid receptors, which could provide novel drug leads for neuropathic pain, we found that the ethyl acetate extract from the fungus Eurotium repens showed more than 40 % binding affinity for the opioid and cannabinoid receptors. We report herein the isolation, structure elucidation, and human opioid and cannabinoid receptors binding affinity of two new benzyl derivatives, (E)-2-(hept-1-enyl)-3-(hydroxymethyl)-5-(3-methylbut-2-enyl)benzene-1,4-diol (1) and (E)-4-(hept-1-enyl)-7-(3-methylbut-2-enyl)-2,3-dihydrobenzofuran-2,5-diol (2). In addition, seven known compounds (3–9) were also isolated, identified and their activity on these receptors are reported.
Compound 1 was obtained as a yellow solid. Its negative HRESIMS showed a molecular formula of C19H28O3, which is consistent with 6 degrees of unsaturation. The 13C NMR spectroscopic data of 1 (Table 1) showed the aromatic benzene signals at δC 145.9 (C-1), 121.2 (C-2), 123.0 (C-3), 147.7 (C-4), 128.5 (C-5) and 115.5 (C-6). The 1H NMR spectrum (Table 1) displayed signals due to the presence of two tertiary methyl groups at δH 1.76 and 1.78, a primary methyl group at δH 0.94, an oxygenated methylene at δH 4.83, five methylene groups at δH 1.36, 1.35, 1.52, 2.25 and 3.33, and three olefinic protons at δH 5.33, 5.90, and 6.24. Furthermore, the 1H NMR spectrum (Table 1) showed an aromatic singlet at δH 6.72 (H-6), which indicated a penta- substituted benzene. The first substituent in the benzene ring was determined to be a 3-methyl-2-butenyl group using 1H NMR, 1H-1H COSY and 1H-13C HMBC spectroscopic experiments. The proton triplet at δH 5.33, assigned to a methine proton at C-2″, showed connectivity to the proton doublet at δH 3.33, assigned to a methylene proton H-1″, and two tertiary methyl groups at δH 1.78 and 1.76, attributable to H-4″ and H-5″. The second substituent in the benzene ring was defined to be a heptadienyl group by the 1H-1H COSY correlations of H-2′ with H-1′ and H-3′, H-4′ with H-3′ and H-5′ and H-6′ with H-5′ and H-7′. The third substituent was determined to be a hydroxymethylene group (δH 4.83 (s), δC 60.9). The 13C NMR spectroscopic data (Table 1) also showed the presence of two quaternary oxyaryl carbons at δC 145.9 and 147.7 indicating the hydroxylation at C-1 and C-4 of the benzene ring. The HMBC correlations of H2-7 with C-3, C-2 and C-4, H-1″ with C-4, C-5 and C-6, H-2′ with C-2, and H-1′ with C-1 and C-2 confirmed the positions of different substituents in the benzene ring. The configuration of the C-1′ and C-2′ double bond was determined as E based on the large coupling constant value (J1′,2′ = 16.0 Hz). On the basis of the above spectral evidence, 1 was assigned as (E)-2-(hept-1-enyl)-3-(hydroxymethyl)-5-(3-methylbut-2-enyl) benzene-1,4-diol.
Table 1.
1H (500 MHz) and 13C (125 MHz) Spectroscopic Data for Compound 1 in CDCl3.
| Positions | δC, mult | δH (J in Hz) | HMBC (H→C) | COSY |
|---|---|---|---|---|
| 1 | 145.9, C | - | - | - |
| 2 | 121.2, C | - | - | - |
| 3 | 123.0, C | - | - | - |
| 4 | 147.7, C | - | - | - |
| 5 | 128.5, C | - | - | - |
| 6 | 115.5, CH | 6.72, s | C-1″, 1, 2, 4 | - |
| 7 | 60.9, CH2 | 4.83, s | C-2, 3, 4 | - |
| 1′ | 122.6, CH | 6.24, d (16.0) | C-1, 3′, 2 | H-2′ |
| 2′ | 139.7, CH | 5.90, dt (16.0, 7.0) | C-2, 3′, 4′ | H-1′, H-3′ |
| 3′ | 33.3, CH2 | 2.25, q (7.1) | C-1′, 2′,4′, 5′ | H-2′, H-4′ |
| 4′ | 28.9, CH2 | 1.52, m | C-6′, 3′, 5′ | H-3′, H-5′ |
| 5′ | 31.5, CH2 | 1.35, m | C-6′ | H-4′, H-6′ |
| 6′ | 22.5, CH2 | 1.36, m | C-5′ | H-5′, H-7′ |
| 7′ | 14.0, CH3 | 0.94, t (6.6) | C-5′, 6′ | H-6′ |
| 1″ | 28.9, CH2 | 3.33, d (7.1) | C-4, 5, 6, 2″, 3″, | H-2″ |
| 2″ | 121.9, CH | 5.33, t (7.3) | C-4″, 5″ | H-1″ |
| 3″ | 133.9, C | - | - | - |
| 4″ | 25.8, CH3 | 1.78, s | C-2″, 3″, 5″ | - |
| 5″ | 17.8, CH3 | 1.76, s | C-2″, 3″, 4″ | - |
Compound 2 was obtained as a yellow oil. The molecular formula of 2 was C20H28O3 on the basis of its negative HRESIMS data. Both the 1H and 13C NMR (Table 2) spectral data of 2 were similar to those of 1, but differences were observed including the carbinol group at C-7 of 1 being replaced with a hemiacetal group (δH 6.08, δC 100.5) in 2. Thus, the structure of (E)-4-(hept-1-enyl)-7-(3-methylbut-2-enyl)-2,3-dihydrobenzofuran-2,5-diol was assigned to 2, which was confirmed by DEPT, COSY, HSQC, and HMBC. Because 2 did not show any detectable optical rotation, it was defined as an enantiomer.
Table 2.
1H (500 MHz) and 13C (125 MHz) Spectroscopic Data for Compound 2 in CDCl3.
| Positions | δC, mult | δH (J in Hz) | HMBC | COSY |
|---|---|---|---|---|
| 1 | - | - | - | - |
| 2 | 100.5, CH | 6.08, m | - | H-3 |
| 3 | 38.7, CH2 | 3.40, dd (16.8, 6.4) 3.07, d (16.8) |
C-3a, C-7a C-2, C-3a |
H-2 H-2 |
| 3a | 122.7, C | - | - | - |
| 4 | 119.6, C | - | - | - |
| 5 | 147.5, C | - | - | - |
| 6 | 114.9, CH | 6.52, s | C-4, C-5, C-7a, C-1″ | - |
| 7 | 122.5, C | - | - | - |
| 7a | 149.4, C | - | - | - |
| 1′ | 123.1, CH | 6.37, d (16.2) | C-3a, C-4, C-5, C6, C-3″ | H-2′ |
| 2′ | 136.0, CH | 6.12, dt (16.2, 7.0) | C-4, C-3′, C-4′ | H-1′, H-3′ |
| 3′ | 33.8, CH2 | 2.25, m | C-1′, C-2′, C-4′, C-5′ | H-2′, H-4′ |
| 4′ | 29.1, CH2 | 1.50, m | C-2′, C-3′, C-5′, C-6′ | H-3′, H-5′ |
| 5′ | 31.4, CH2 | 1.36, m | C-6′ | H-4′, H-6′ |
| 6′ | 22.5, CH2 | 1.37, m | C-5′ | H-5′, H-7′ |
| 7′ | 14.0, CH3 | 0.93, t (6.7) | C-5′, C-6′ | H-6′ |
| 1″ | 27.9, CH2 | 3.27, d (6.5) | C-6, C-7a, C-2″, C-3″ | H-2″ |
| 2″ | 121.6, CH | 5.31, t (7.4) | C-4″, C-5″ | H-1″ |
| 3″ | 133.2, C | - | - | - |
| 4″ | 25.8, CH2 | 1.76, s | C-2″, C-3″, C-5″ | - |
| 5″ | 17.8, CH2 | 1.72, s | C-2″, C-3″, C-4″ | - |
The following known compounds were also identified; flavoglaucin (3),9 tetrahydroauroglaucin (4),10 dihydroauroglaucin (5),11 auroglaucin (6),11 and 2-(2′,3-epoxy-1′,3′-heptadienyl)- 6-hydroxy-5-(3-methyl-2-butenyl)benzaldehyde (7),9 echinulin (8),12 and 5,7-dihydroxy-4-methylphthalide (9),13 and this was confirmed by comparison of physical and spectroscopic data (UV, 1H and 13C NMR, and MS) with corresponding authentic samples or literature values.
Compounds 1–9 were evaluated at a concentration of 10 μM for their affinity to bind with opioid and cannabinoid receptors following the methods described previously.14,15 The biological data in Tables 3 and 4 is the first report that this class of compounds has good affinity for human opioid and cannabinoid receptors.
Table 3.
Binding Affinity of compounds (1–6) for Human Opioid (subtype δ, κ and μ) and Cannabinoid (subtype CB1 and CB2) Receptors.
| Compound a | Opioid Receptors (%) | Cannabinoid Receptors (%) | |||
|---|---|---|---|---|---|
| δ | κ | μ | CB1 | CB2 | |
| 1 | 62.2 | nab | na | na | na |
| 2 | na | 51.4 | na | ntc | nt |
| 3 | 52.5 | 48.0 | 67.1 | na | na |
| 4 | na | na | 59.1 | na | na |
| 5 | na | na | na | na | na |
| 6 | na | na | na | 62.6 | 43.1 |
| 7 | na | na | na | na | na |
| 8 | na | na | na | na | na |
| 9 | na | na | na | na | na |
| Naloxoned | 106.4 | 101.6 | 97.0 | nt | nt |
| CP 55,940e | nt | nt | nt | 104.3 | 102.6 |
All compounds were tested at the concentration of 10 μM.
Not active (< 40%).
Not tested.
For opioid receptor binding affinity assay, the opioid receptor antagonist naloxone was used as positive control.
For cannabinoid receptor binding affinity assay, the cannabinoid receptor agonist (CP 55,940) was used as positive control.
Table 4.
IC50 valuesf (μM) of compound 1, 2, 4, and 6
| Compound | Opioid Receptors (μM) | Cannabinoid Receptors (μM) | |||
|---|---|---|---|---|---|
| δ | κ | μ | CB1 | CB2 | |
| 1 | 5.4 | - | - | - | - |
| 2 | - | 32.4 | - | - | - |
| 3 | ntg | nt | nt | - | - |
| 4 | - | - | 7.2 | - | - |
| 6 | - | - | - | 15.2 | 19.9 |
IC50 is the concentration required for 50% inhibition 3H labelled ligand.
Not tested due to lack of material.
Experimental Section
General Experimental Procedures
Optical rotations were measured using a Rudolph Research Analytical Autopol V polarimeter. UV spectra were recorded on a Perkin-Elmer Lambda 3B UV/visspectrophotomer. NMR spectra were obtained on a Bruker model AMX 500 NMR spectrometer with standard pulse sequences, operating at 500 MHz in 1H and 125 MHz in 13C; CDCl3 was used as solvent and TMS was used as an internal standard. High-resolution mass spectra (HRESIMS) were recorded on a Micromass Q-Tof Micro mass spectrometer with a lock spray source. Column chromatography was carried out on silica gel (70–230 mesh, Merck) and Sephadex LH-20 (Mitsubishi Kagaku, Tokyo, Japan). Fractions obtained from column chromatography were monitored by TLC (silica gel 60 F254). Preparative TLC was carried out on silica gel 60 PF254+366 plates (20 × 20 cm, 1 mm thick). HPLC was performed on an ODS column (Phenomenex Luna C18, 10 × 250 mm, 5 μm) and the elution was monitored at 254 nm. All chemicals used were from Sigma-Aldrich (Poole, Dorset, U.K.) with the following exceptions. For the binding experiments, [3H]CP 55940, (CB1/CB2 agonist)-(174.8 Ci/mmol), [3H]DAMGO, (highly selective peptide agonist for the μ opioid receptor)-(53.4 Ci/mmol) [3H]U-69,593, (kappa agonist) (42.7 Ci/mmol), [3H]enkephalin (DPDPE; prototypical selective δ-opioid receptor agonist peptide) (45 Ci/mmol), were obtained from Perkin-Elmer Life Sciences Inc. (Boston, MA, U.S.A.). Non-tritium labelled CP 55,940, DAMGO, DPDPE, nor-Binaltorphimine (standard κ selective antagonist) and WIN 55,212-2 (novel, low potency CB2 receptor antagonist and CB1 receptor partial inverse agonist) were obtained from Tocris Bioscience (Ellisville, Missouri, U.S.A.).
Fungal Material
The fungus was collected in Tifton, GA in 1978, lyophilized, and stored at −20 °C. The fungus was determined to be Eurotium repens by sequence comparison of its β-tubulin partial gene sequence with the corresponding sequence from the NRRL 13 isolate, type of E. repens (100% homology). A voucher specimen (UM-031509) has been deposited in the culture collection of the Department of Medicinal Chemistry, University of Mississippi. The fungus was plated out on potato-dextrose agar (PDA) that was maintained at 24 °C until discrete fungal colonies appeared. 50 mL of potato-dextrose broth was inoculated with the fungus and incubated for two weeks in stationary phase at 24 °C; then the fungus was seeded onto a medium consisting of 100 g of shredded wheat, 100 g low-pH mycological broth, 40 g yeast extract, and 400 g sucrose in a 2.0 L Fernbach flask (10 flasks were used) followed by incubation for 22 days at 24 °C.
Extraction and Isolation
Following incubation, 300 mL of acetone was added to each flask and the culture was homogenized (Super Dispex, Tekmark Co., SD-45). The suspension was filtered and the filtrate concentrated under vacuum at 40 °C. The residue was mixed with H2O (200 mL) and extracted with EtOAc (500 mL × 3). The combined EtOAc extracts were dried using anhydrous Na2SO4 and concentrated under vacuum. The EtOAc extract (8.0 g) was chromatographed on silica gel 60, 70–230 mesh (400 g), and eluted stepwise with petroleum ether, diethyl ether, ethyl acetate, acetone and methanol, yielding five fractions. Bioassay showed that the ethyl acetate, diethyl ether and acetone fractions exhibited good opioid and cannabinoid receptors binding activity (> 40%). The ethyl acetate fraction was re-chromatographed over a silica gel 60 column eluted with CHCl3-EtOAc (0:100–100:0) to yield 10 fractions. Fractions 1–4, 9 and 10 exhibited good biological activities. Fractions 1 and 2 were combined and chromatographed by preparative TLC using n-hexane-CHCl3 (8:2) as the mobile phase, affording 3 (4 mg) and 4 (12 mg). Fractions 3 and 4 were combined and chromatographed by preparative TLC with n-hexane-CHCl3 (7:3), to yield 5 (4 mg) and 7 (3 mg). Fractions 9 and 10 were combined and purified on Sephadex LH-20 CC eluting with CHCl3-MeOH (2:1) to afford four subfractions. Subfraction 1 showed activity and was purified by C18-HPLC using gradient MeOH-H2O (50:50) to 100% MeOH to yield 1 (5.2 mg) and 9 (8.0 mg). Subfraction 2 was further chromatographed by preparative TLC with n-hexane-CHCl3 (5:5), affording 6 (6 mg) and 8 (10 mg). The diethyl ether fraction was purified by C18-HPLC using H2O-MeOH (4:6) as an eluent to give 2 (1.8 mg) and 5 (51 mg). The acetone fraction was re-chromatographed on Sephadex LH-20 CC eluting with CHCl3-MeOH (2:1) to afford 7 (24 mg).
Cell Culture
CHO-K1 cells (ATCC #CCL-61) were stably transfected via electroporation with full length human recombinant cDNA for cannabinoid receptor subtypes 1 and 2 (obtained from Origene). These cells were maintained in a Dulbecco’s Modified Eagles’s medium/F-12 (50/50) nutrient mixture supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and either 1–2% G418 sulfate (geneticin) or hygromycin B, dependent upon the cell line. Percentages are based on a total media volume of 500 mL. All opioid and cannabinoid cell lines were kept at 37 °C and 5% CO2. Membranes were prepared by scraping the cells in a 50 mM Tris-HCl buffer, homogenized via sonication and centrifuged for 40 minutes at 13650 rpm at 4 °C. These were kept at −80 °C until used for binding assays. Protein concentration was determined via Bio-Rad Protein Assay.8
Radio-ligand Binding for Cannabinoid and Opioid Receptor Subtypes
In the primary bioassay screen, compounds were tested at a final concentration of 10 μM for competitive binding to the respective receptor. For the cannabinoid receptor assays, test compounds were added into a 96-well plate followed by 0.6 nM [3H]CP-55,940 and 10 μg of cannabinoid membrane re-suspended in 50 mM Tris (pH 7.4), 154 mM NaCl, and 20 mM Di-Na-EDTA supplemented with 0.02% BSA. For the opioid receptor assays, saturation experiments were performed to determine optimal radioligand ([3H]enkephlin and [3H]DAMGO) and membrane concentrations.
The cannabinoid assay was allowed to incubate at 37 °C for 90 minutes while the opioid assay was incubated at 25 °C for 60 minutes. Both reactions were then terminated by rapid filtration using GF/C or GF/B filters (presoaked in 0.3% BSA) and washed with the buffer. Dried filters were then covered with scintillant and measured for the amount of radioligand retained using a Perkin Elmer Topcount (Perkin Elmer Life Sciences Inc., Boston, MA, USA). Non-specific binding, which was determined in the presence of 1μM CP-55,940 for cannabinoid receptors or 10μM DPDPE, nor-Binaltorphimine, or DAMGO for opioid receptors, was subtracted from the total binding to yield the specific-binding values. Compounds showing competitive inhibition of the labelled ligand to bind to the receptor at 40% or greater were tested in a dose response curve with concentrations of the test compound ranging from 300 μM to 1.7 nM.
(E)-2-(hept-1-enyl)-3-(hydroxymethyl)-5-(3-methylbut-2-enyl)benzene-1,4-diol (1): yellowish solid; [α]D25 +12 (c 0.5, CHCl3); UV (MeOH) λmax 265 nm; 1H and 13C NMR, see Table 1; Negative HRESIMS m/z 303.1956 [M-H]− (calcd for C19H27O3, 303.1960).
(E)-4-(hept-1-enyl)-7-(3-methylbut-2-enyl)-2,3-dihydrobenzofuran-2,5-diol (2): yellow oil; [α]D25 0 (c 0.4, CHCl3); UV (MeOH) λmax 265 nm; 1H and 13C NMR, see Table 2; Negative HRESIMS m/z 315.1968 [M-H]− (calcd for C20H27O3, 315.1960).
Supplementary Material
Acknowledgments
This project is supported by Grant Number 5P20RR021929 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. Furthermore, this investigation was conducted in a facility constructed with support from research facilities improvement program C06 RR-14503-01 from the NIH National Center for Research Resources. We are especially grateful to the lab of Dr. Bryan Roth, Department of Pharmacology, School of Medicine, University of North Carolina, Chapel Hill, for the generous donation of stably transfected opioid cell lines.
Footnotes
Supporting Information Available: The 1H, 13C, DEPT, HMQC, COSY, and HMBC spectra of compounds 1 and 2 are available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Kessler RC, Chiu WT, Demler O, Walters EE. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foley KM. N Engl J Med. 2003;348:1279–1281. doi: 10.1056/NEJMe030014. [DOI] [PubMed] [Google Scholar]
- 3.Eisenberg E, McNicol ED, Carr DB. JAMA. 2005;293:3043–3052. doi: 10.1001/jama.293.24.3043. [DOI] [PubMed] [Google Scholar]
- 4.Waldhoer M, Bartlett SE, Whistler JL. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 5.Onaivi ES. Int Rev Neurobiol. 2009;88:335–369. doi: 10.1016/S0074-7742(09)88012-4. [DOI] [PubMed] [Google Scholar]
- 6.Pan HL, Wu ZZ, Zhou HY, Chen SR, Zhang HM, Li DP. Pharmacol Ther. 2008;117:141–161. doi: 10.1016/j.pharmthera.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welch SP. Int Rev Psychiatry. 2009;21:143–151. doi: 10.1080/09540260902782794. [DOI] [PubMed] [Google Scholar]
- 8.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 9.Li DL, Li XM, Li TG, Dang HY, Proksch P, Wang BG. Chem Pharm Bull. 2008;56:1282–1285. doi: 10.1248/cpb.56.1282. [DOI] [PubMed] [Google Scholar]
- 10.Yoshihira K, Takahashi C, Sekita S, Natori S. Chem Pharm Bull. 1972;20:2727–2728. [Google Scholar]
- 11.Hamasaki T, Kimura Y, Hatsuda Y, Nagao M. Agric Biol Chem. 1981;45:313–314. [Google Scholar]
- 12.Allen CM., Jr J Am Chem Soc. 1973;95:2386–2387. [Google Scholar]
- 13.Habib E, León F, Bauer J, Hill RA, Carvalho P, Cutler HG, Cutler SJ. J Nat Prod. 2008;71:1915–1918. doi: 10.1021/np8003497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross RA, Gibson TM, Stevenson LA, Saha B, Crocker P, Razdan RK, Pertwee RG. Br J Pharmacol. 1999;128:735–743. doi: 10.1038/sj.bjp.0702836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA, Pertwee RG. Br J Pharmacol. 2005;146:917–926. doi: 10.1038/sj.bjp.0706414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.