

Abstract
Background
We reported recently that endotoxemia promotes microvascular thrombosis in cremaster venules of wild type mice, but not in mice deficient in toll-like receptor-4 (TLR4) or von Willebrand factor (VWF).
Objective
To determine whether the clinically relevant model of polymicrobial sepsis induced by cecal ligation/perforation (CLP) induces similar responses via the same mechanisms as endotoxemia.
Methods
We used a light/dye injury model of thrombosis in the cremaster microcirculation of wild type mice and mice deficient in toll-like receptor-4 (C57BL/10ScNJ), toll-like receptor-2 (TLR-2), or VWF. Mice underwent CLP or sham surgery, or an intraperitoneal injection of endotoxin (LPS) or saline. In the CLP model, we assessed the influence of fluid replacement on thrombotic responses.
Results
Both CLP and LPS enhanced thrombotic occlusion in wild type mice. In contrast to LPS, CLP enhanced thrombosis in TLR4- and VWF-deficient strains. While TLR-2-deficient mice did not demonstrate enhanced thrombosis following CLP, LPS enhanced thrombosis in these mice. LPS, but not CLP, increased plasma VWF antigen relative to controls. Septic mice, particularly those undergoing CLP, developed significant hemoconcentration. Intravenous fluid replacement with isotonic saline prevented the hemoconcentration and prothrombotic responses to CLP, though fluids did not prevent the prothrombotic response to LPS.
Conclusions
Polymicrobial sepsis induced by CLP and endotoxemia promote microvascular thrombosis via distinct mechanisms; enhanced thrombosis induced by CLP requires TLR-2 but not TLR4 or VWF. The salutary effects of intravenous fluid replacement on microvascular thrombosis in polymicrobial sepsis remain to be characterized.
Keywords: Endotoxin, sepsis, platelets, venules, intravital microscopy
INTRODUCTION
Sepsis is a significant contributor to mortality in intensive care units[1] associated with endothelial dysfunction and a prothrombotic state[2]. Microvascular thrombosis has been documented in human sepsis and in animal models[3–7], and is suggested to contribute to sepsis-associated organ dysfunction[8]. One commonly used model of experimental sepsis involves systemic administration of endotoxin (lipopolysaccharide, LPS), a cell wall component of gram-negative bacteria. Endotoxemia results in platelet adhesion to microvessels in vivo, and promotes microvascular thrombosis in a photochemical injury model[9–11]. We reported previously that the enhanced microvascular thrombosis induced by endotoxemia required the endotoxin signaling receptor toll-like receptor 4 (TLR4)[11], and von Willebrand factor (VWF)[12]. Whether these mechanisms promote microvascular thrombosis in other experimental sepsis models is unclear.
While studies in endotoxemic animals have generated significant knowledge, the clinical relevance of this model has been questioned since a bolus of endotoxin does not reflect the complex pathophysiology of most cases of human sepsis[13]. Alternative sepsis models have been proposed[14], including the well-characterized model of polymicrobial sepsis induced by cecal ligation and perforation (CLP), which involves different signaling mechanisms and cytokine profiles than endotoxemia[15–17]. The main objective of this study was to determine whether polymicrobial sepsis induced by CLP promotes microvascular thrombosis, via similar mechanisms as those reported previously in endotoxemic mice. Specifically, we assessed the role of TLR4 and VWF on microvascular thrombosis in CLP, and compared hemodynamic, hematologic and cytokine profiles between the models. Since TLR-2 mediates certain inflammatory responses to CLP in some models[18, 19], we assessed the role of TLR-2 on microvascular thrombosis in CLP and LPS. The data demonstrate that both polymicrobial sepsis and endotoxemia promote microvascular thrombosis, with distinct contributions of TLR4, VWF, and TLR-2, and with a differential response to intravenous fluid replacement.
MATERIAL AND METHODS
Male mice, ~25–30 g of weight, were studied; all protocols were approved by the Animal Care and Use Committee of Baylor College of Medicine. We studied C57BL/6 mice(wild type), VWF-deficient mice[20] and their littermate controls, TLR4-deficient mice (C57BL/10ScNJ)[21] and their controls, C57Bl/10J, and TLR-2 deficient mice. VWF-deficient mice and their littermate controls (backcrossed onto C57BL/6 for >8 generations) were generated by heterozygous breeding; TLR-2 deficient mice (backcrossed onto C57BL/6 for >9 generations) were generated by homozygous breeding. All strains originated from the Jackson Laboratories; VWF strains were genotyped by PCR analysis of tail clippings, and the phenotype of TLR-4 deficient mice was demonstrated by evaluating their response to endotoxin.
Animal preparation
Mice were anesthetized with an intraperitoneal (IP) injection of pentobarbital (50 mg/kg), with additional doses (12.5–25 mg/kg) as needed. They were placed on a custom Plexiglas tray and maintained at 37 °C with a homeothermic blanket, monitored with a rectal temperature probe (F.H.C., MA). A tracheotomy was performed to facilitate breathing, a jugular vein was cannulated for intravenous drug administration, and a carotid artery was cannulated for heart rate and blood pressure measurement. The cremaster muscle was exteriorized and prepared for intravital microscopy as described[11, 22].
Intravital microscopy
The preparation was placed under an upright microscope (BX-50, Olympus, NY), observed with a 40X water-immersion objective (N.A. 0.8) and allowed to equilibrate for 30 minutes. After equilibration, FITC-dextran (150 kD, 10 ml/kg of a 5% solution) was injected via the venous catheter and allowed to circulate for ~10 minutes. Thereafter, venular diameter was measured (image 1.6, NIH, public domain software) as well as mean blood cell velocity (Vdoppler, using an optical Doppler velocimeter, Cardiovascular Research Institute, Texas A&M University). Venular wall shear rate (γ) was calculated as 8(Vdoppler/1.34)/diameter[23].
Following those measurements, light/dye-induced injury was begun by exposing ~100 µm of vessel length to epi-illumination, with a 175W xenon lamp (Lambda LS, Sutter, CA) and a fluorescein filter cube (HQ-FITC, Chroma, VT). Excitation light was monitored daily (IL 1700 Radiometer, SED-033 detector, International Light, MA) and maintained at 0.6 W/cm2 as described[11]. Epi-illumination was applied continuously and the following times were recorded: 1) Time of onset of platelet aggregates and 2) Time of flow cessation, for at least 60 seconds.
Cecal ligation/perforation protocol
Cecal ligation and perforation (CLP) was performed using techniques comparable to those described by others[15, 16]. Briefly, animals were anesthetized as described above, a midline laparotomy was performed to expose the cecum, which was then ligated at its base with 2-0 silk suture and perforated three times with a 20-gauge needle. A small amount of feces were gently squeezed and spread on the cecum with a cotton swab. The incision was closed and the animals received subcutaneous buprenorphine (1.0 mg/kg) for post-operative analgesia and isotonic saline (1 ml) for fluid replacement. Sham-operated CLP mice underwent laparotomy and cecal exposure but no ligation or perforation; they received buprenorphine and isotonic saline as above. Mice were allowed to recover from anesthesia under a heating lamp with continuous monitoring. They were re-anesthetized ~4.5 hours later to prepare them for thrombosis experiments as described above. An additional group of mice underwent CLP or sham surgery for blood collection at 24 hours. These mice received additional doses of buprenorphine (1.0 mg/kg subcutaneously every 6 hours) and isotonic saline (1.0 ml subcutaneously 12 hours after surgery).
Experimental groups
To determine the influence of CLP on thrombosis, photochemical-induced thrombosis was assessed in CLP and sham-operated mice 6 hours following surgery. For endotoxemia experiments, mice were injected intraperitoneally with either endotoxin (lipopolysaccharide, LPS) from Escherichia coli serotype 0111:B4 (Sigma, endotoxin content 1 × 106 EU/mg) at 5 mg/kg in 0.5 ml of sterile, pyrogen-free isotonic saline or 0.5 ml saline, 4 hours prior to photochemical injury. This dose and preparation of LPS is shown to be sublethal in C57BL/6 mice; the reported LD50 is more than one order of magnitude higher[24].
Within each group, the investigator performing intravital microscopy was blinded to genotype and intervention (saline vs. LPS, or CLP vs. sham). Blinding was not feasible between the sepsis models since an abdominal incision was evident on CLP/sham mice but not LPS/saline mice.
In some experiments, mice received intravenous (I.V.) fluid replacement (isotonic saline, 16 ml/kg, via slow intravenous injection over a period of 10–15 minutes). I.V. fluids were administered prior to exteriorization of the cremaster muscle (~ 3 hours following LPS or saline injection, and ~5 hours following CLP or sham surgery).
Hematologic and cytokine measures
In separate wild-type mice in each sepsis model, blood was collected via a vascular catheter into EDTA-coated tubes for platelet counts and hematocrit, performed with a Coulter counter in a reference laboratory (Center for Comparative Medicine, Baylor College of Medicine, TX). These mice did not undergo intravital microscopy or photochemical injury. Platelet-poor plasma from such mice was collected by centrifugation, stored at −80°C, and later analyzed for tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), VWF antigen, and thrombin-antithrombin complexes (TAT). We used commercially available ELISA kits for TNF-α and IL-6 (BD Biosciences, CA), VWF (Ramco Laboratory, TX), and TAT (Enzygnost, Siemens), according to the manufacturer’s instructions. The TAT assay uses anti-human antibodies but has cross-reactivity with murine specimens. The investigator performing ELISA assays was blinded to the experimental conditions.
Statistics
All data are expressed as mean±SE; comparisons by genotype within test groups (i.e. LPS vs. saline, or CLP vs. sham) were done with one-way analysis of variance with Fisher’s post-hoc test, using Statview 5.01 statistical software (SAS Institute, NC). A p<0.05 was considered statistically significant. Comparisons between more than 2 groups used a Bonferroni correction, and the p value was adjusted accordingly.
RESULTS
Intravital microscopy was performed on 211 mice; average weight was 28.6±0.2 g, average venule diameter was 45±0.2 µm. There were no statistical differences in weight, microvessel diameters, and shear rates within each comparison group (data not shown).
Polymicrobial sepsis and endotoxemia promote microvascular thrombosis
Figure 1 depicts the influence of CLP and endotoxemia on kinetics of light/dye-induced thrombosis in venules of wild-type (WT) mice. Both CLP and endotoxin enhanced microvascular thrombosis, manifest as a significant reduction in the time to flow cessation relative to their respective controls. The time of onset of thrombosis was significantly reduced in LPS-treated mice, but not in mice undergoing CLP. The magnitude of the prothrombotic response to LPS was comparable to that reported previously by us[11, 12].
FIGURE 1.
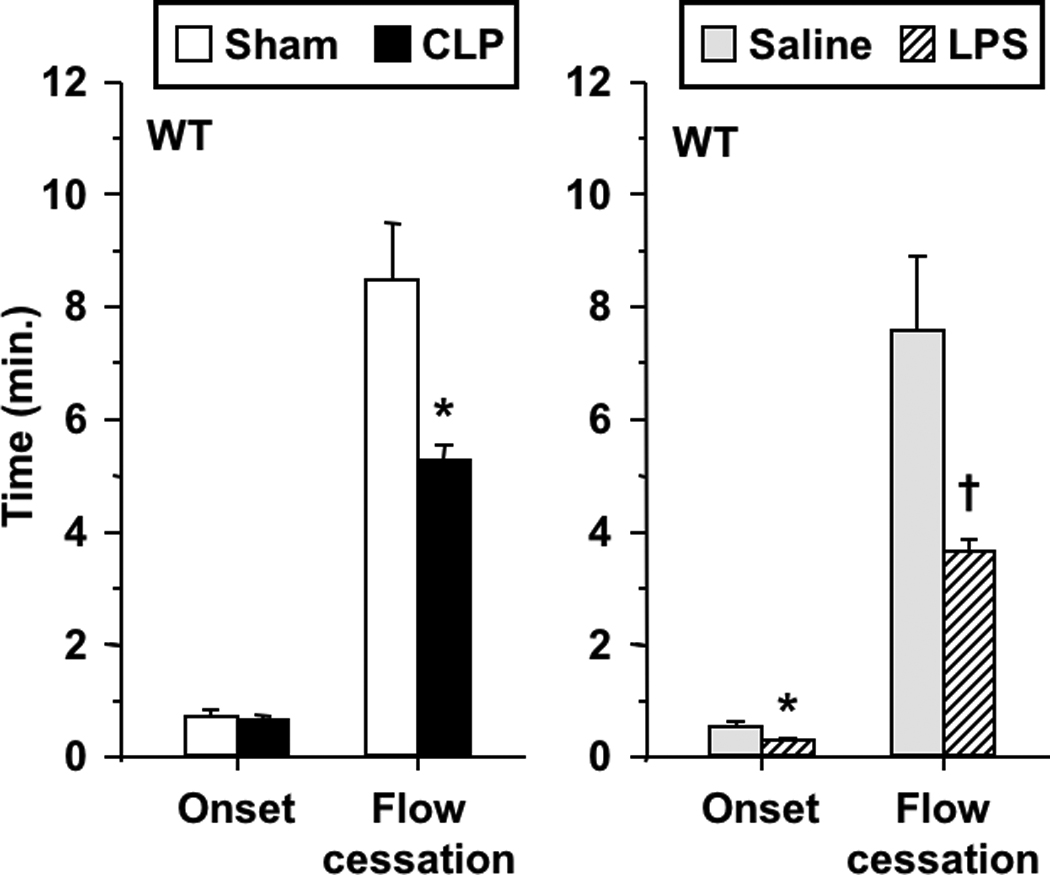
Thrombotic responses in wild type mice in both sepsis models: cecal ligation/perforation (CLP) and sham-operated controls (left), and saline- and LPS-treated mice (right). Data shown as mean±SE, n=8–12 per group. *: p<0.05, †: p<0.005, for comparison with the respective control group.
Role of TLR4 and VWF in the prothrombotic responses to polymicrobial sepsis
Figure 2 depicts the influence of CLP and endotoxemia on kinetics of light/dye-induced thrombosis in venules of TLR4-deficient mice. As in the case of WT mice, CLP enhanced thrombosis in TLR4-deficient mice, manifest by a reduction in the time to thrombotic occlusion, with no change in time to onset of thrombosis. Similarly, CLP resulted in 25% reduction in time to flow cessation in the control strain for TLR4-deficient mice (C57BL/10J, n=8–9 per group, p<0.05). In contrast, endotoxin had no effect on the kinetics of thrombosis in TLR4-deficient mice, as anticipated by their lack of sensitivity to LPS[21], demonstrated previously in this model[11]. Based on our recent report that VWF was required for LPS-enhanced thrombosis[12], we determined whether VWF was required for enhanced thrombosis induced by CLP. As shown in Figure 3, CLP promoted thrombosis in both VWF-deficient mice (VWF−/−) and their littermate controls (VWF+/+) with a similar pattern: reduced time to thrombotic occlusion without effect on onset of thrombosis. As shown previously in this model[12], VWF−/− mice had prolonged times to thrombotic occlusion under control conditions (sham) as compared to littermates.
FIGURE 2.
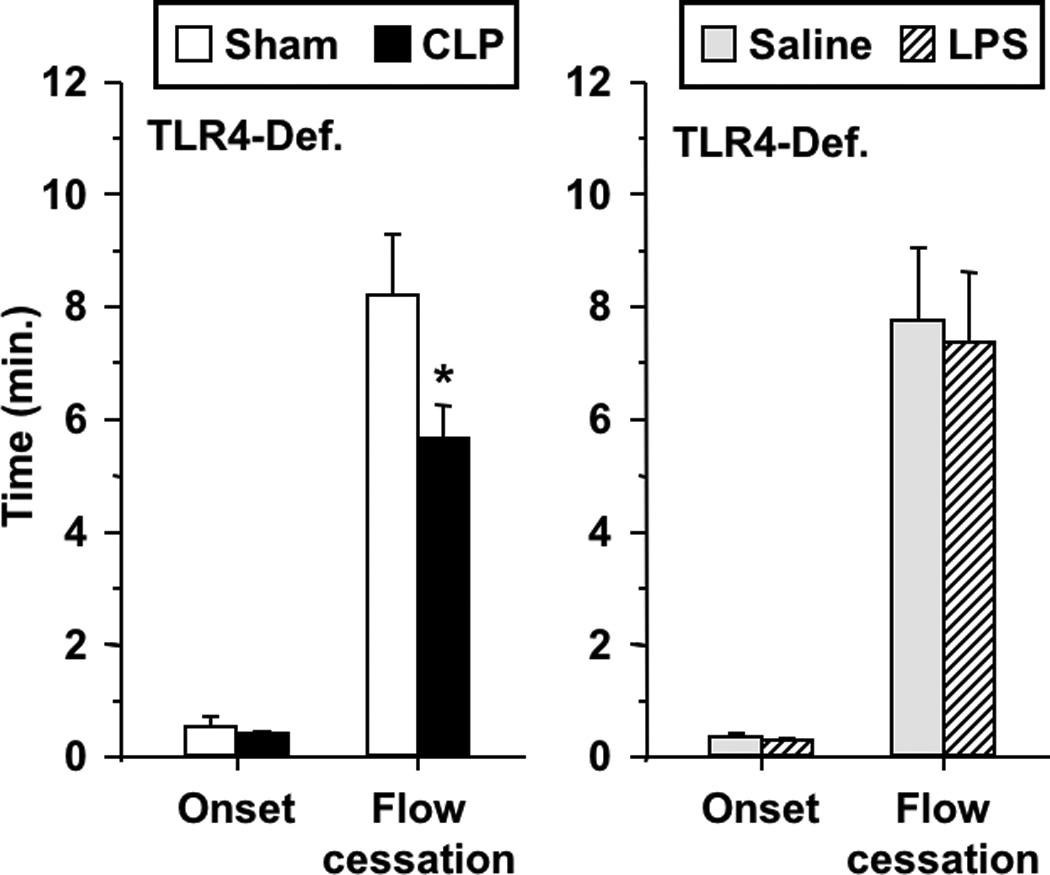
Thrombotic responses in TLR-4 deficient mice in both sepsis models: CLP and sham-operated (left), and saline- and LPS-treated mice (right). Data shown as mean±SE, n=9–11 per group. *: p<0.05, for comparison between sham-operated and CLP groups.
FIGURE 3.
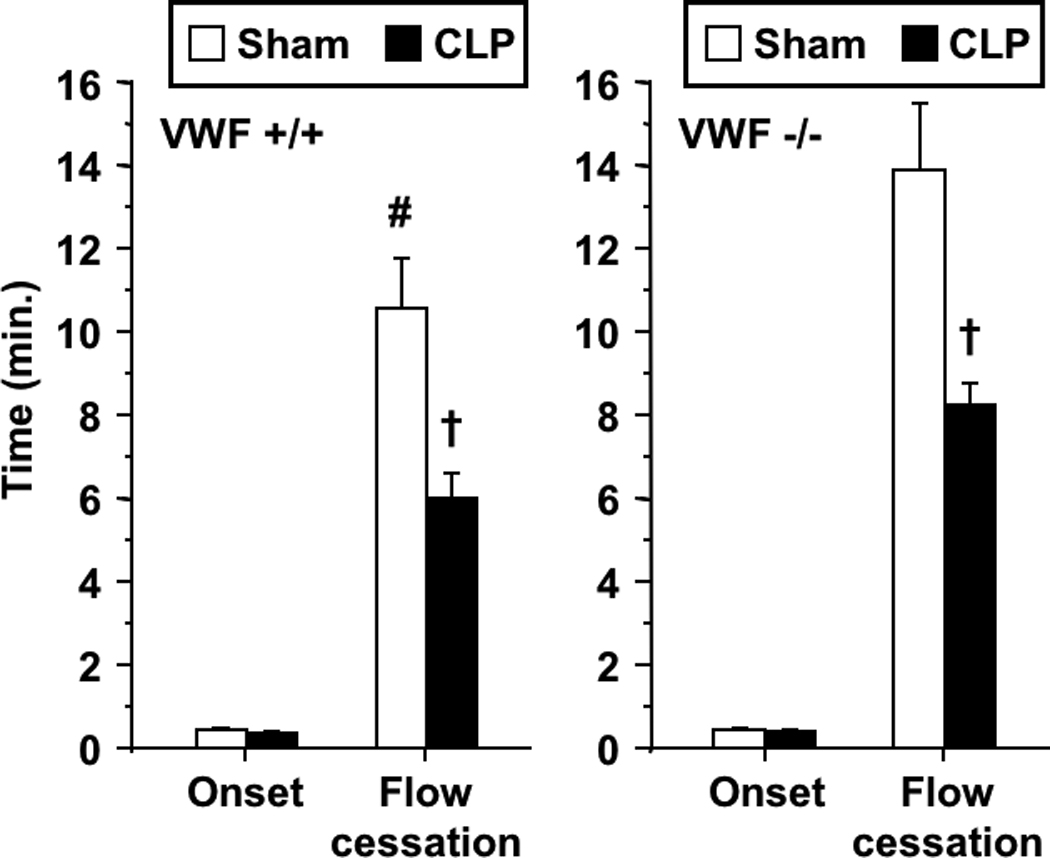
Thrombotic responses in CLP and sham-operated VWF-deficient mice (VWF−/−) and their littermate controls (VWF+/+). Data shown as mean±SE, n=8–12 per group. †: p<0.005 for comparison between sham-operated and CLP groups, #: p<0.05 for comparison between sham-operated VWF+/+ and VWF−/− mice; †: p<0.005, for comparison between sham-operated and CLP groups. Note different ordinate scale as compared to all other figures.
Role of TLR-2 in Microvascular Thrombosis in Experimental Sepsis
Figure 4 illustrates the influence of CLP and endotoxemia on light/dye-induced thrombosis in venules of TLR-2 deficient mice. In these mice, CLP did not alter either onset of thrombosis or thrombotic occlusion relative to sham-operated controls. In contrast, LPS enhanced thrombosis in TLR-2 deficient mice, reducing both the times to onset and to flow cessation.
FIGURE 4.
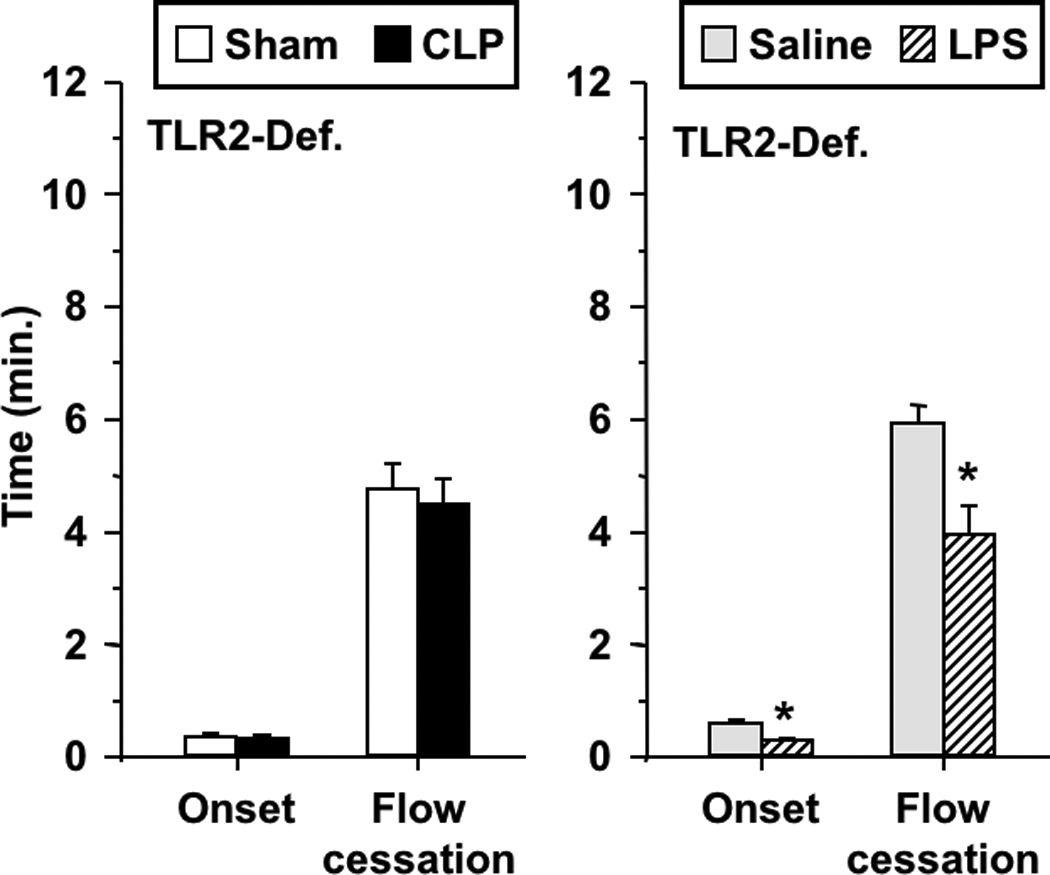
Thrombotic responses in TLR-2 deficient mice in both sepsis models: CLP and sham-operated (left), and saline- and LPS-treated mice (right). Data shown as mean±SE, n=9–12 per group. *:p<0.05, for comparison between LPS- and saline-treated mice.
VWF antigen and TAT levels in experimental sepsis
To determine the influence of experimental sepsis on plasma VWF, we measured plasma VWF antigen levels in sham- and CLP-operated mice, as well as in mice treated with saline or LPS. As shown in Table 1, plasma VWF antigen levels did not differ between sham- and CLP-operated mice. However, LPS enhanced VWF antigen, comparable to our prior findings[12]. Similarly, the LPS and CLP models also differed in their activation of systemic coagulation, as determined by plasma TAT complexes; LPS, but not CLP, resulted in a statistically significant increase in TAT (Table 1). To determine whether our CLP model increases plasma VWF at later time points, as reported by others[25], we measured VWF antigen levels 24 hours after sham or CLP surgery. However, VWF did not differ between sham and CLP mice at 24 hours (10.1±1.1% vs. 10.1±0.6%, respectively, n=14–16 per group, N.S.).
Table 1.
Hematologic and cytokine parameters in C57BL/6 mice
| Platelets (x 106/µl) |
Hematocrit (%) |
VWF Antigen (%) |
TAT (µg/L) |
TNF-α (pg/ml) |
IL-6 (pg/ml) |
|
|---|---|---|---|---|---|---|
| Sham | 1.02±0.03 | 44.7±0.7 | 9.5±1.1 | 10.7 ±2.5 | 26.3±0.5 | 103.6±45.7 |
| CLP | 1.01±0.08 | 53.8±1.5 (†) | 10±0.5 | 14.6 ±1.8 | 43.2±4.6 | 1180.5 ±357.3 (†) |
| CLP + I.V. fluids | 0.99±0.03 | 45.9±1.3 | N/A | N/A | N/A | N/A |
| Saline | 1.17±0.06 | 44.9±1.4 | 8.85±0.4 | 21.2±7.1 | 24.8±0.9 | 20.4±5.4 |
| LPS | 0.87±0.06 (†) | 48.8±0.7 (*) | 13.5±0.6(†) | 64.2±7.9 (†) | 1166.1±136.0 (†) | 1079.8±47.4 (†) |
Values represent mean ± standard error.
p<0.05,
p<0.005
Hematologic and cytokine responses to endotoxemia and polymicrobial sepsis
To compare the systemic responses in each of the two sepsis models, we assessed various hematologic and plasma cytokine parameters in WT mice undergoing CLP or LPS injection. As illustrated in Table 1, both models induced significant increases in plasma interleukin-6 and hematocrit. The hemoconcentration was particularly pronounced in the CLP model. In contrast, endotoxemia but not polymicrobial sepsis, was associated with significant thrombocytopenia and increased plasma levels of tumor necrosis factor-α (TNF-α).
Effects of intravenous fluid replacement on thrombotic responses
Although CLP mice received 1.0 ml of saline subcutaneously at the time of surgery, used commonly in this model, Table 1 demonstrates that these mice developed significant hemoconcentration, consistent with intravascular volume depletion. Thus, we assessed the influence of intravenous fluid replacement on the thrombotic responses. First, we demonstrated that isotonic saline, at a dose used by others in rodent models of experimental sepsis (16 ml/kg)[26] prevented hemoconcentration in these mice (Table 1). Subsequently, we assessed the effects of CLP and LPS on microvascular thrombosis in mice receiving intravenous saline at that dose. As shown in Figure 5, I.V. fluid replacement prevented the enhanced microvascular thrombosis induced by CLP, but did not prevent the prothrombotic manifestations in LPS-treated mice (reduced time to onset of thrombosis and flow cessation). The effects of I.V. fluids on microvascular thrombosis in the CLP model were not accompanied by changes in systemic hemodynamics (mean arterial pressure, heart rate) or wall shear rate of the venules we studied (Table 2).
FIGURE 5.
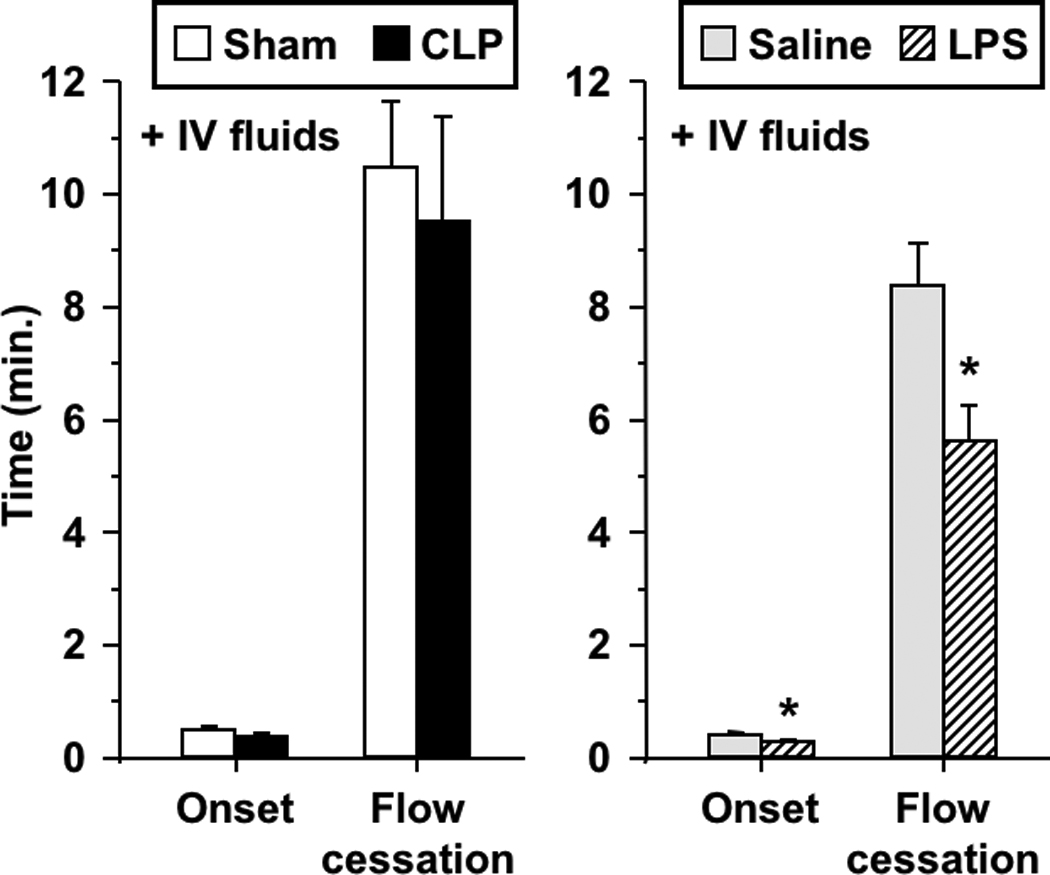
Thrombotic responses in wild type mice following intravenous (I.V.) fluid replacement with isotonic saline (16 ml/kg) in both sepsis models: CLP and sham-operated (left), and saline- and LPS-treated (right). Data shown as mean±SE, n=6 per group. *: p<0.05, for comparison between LPS- and saline-treated mice.
Table 2.
Hemodynamic parameters in C57BL/6 mice.
| Wall shear rate (s−1) |
MAP (mmHg) |
Heart Rate (beats/min) |
||
|---|---|---|---|---|
| CONTROL | ||||
| Sham | 507.8±29.6 | 70.1±3.8 | 496.2±25.7 | |
| CLP | 479.7±30.7 | 61.6±5.6 | 536.0±22.6 | |
| Saline | 483.6±26.5 | 80.3±2.7 | 434.0±7.9 | |
| LPS | 397.8±30.2 | 83.9±3.4 | 599.8±11.8 (†) | |
| + I.V. FLUIDS | ||||
| Sham | 520.8±25.3 | 70.7±4.4 | 431.2±25.8 | |
| CLP | 477.8±41.1 | 71.2±4.8 | 492.2±26.1 | |
| Saline | 497.7±37.2 | 82.2±3.4 | 441.7±9.4 | |
| LPS | 470.8±25.5 | 105.4±8.7 (*) | 651.0±20.9 (†) | |
Values represent mean ± standard error.
p<0.05,
p<0.005
DISCUSSION
The primary finding of this study is that polymicrobial sepsis and endotoxemia promote light/dye-induced microvascular thrombosis in mouse cremaster venules by distinct mechanisms. Our data suggest a mechanistic role for TLR-2 in the enhanced thrombosis in polymicrobial sepsis, though TLR4 and VWF were not required in our model. In contrast, TLR4 and VWF (but not TLR2), mediate enhanced thrombosis in endotoxemia, as shown in this manuscript and our recent reports [11, 12]. Further, the enhanced thrombosis induced by CLP, but not by LPS, was prevented by intravenous fluid replacement.
Polymicrobial sepsis and endotoxemia have been reported to have differing temporal cytokine responses, innate defense mechanisms, and signaling molecules involved[15–17]. Intraperitoneal administration of endotoxin to mice induces peak cytokine responses of TNF-α, interleukin-1β and interleukin-6 as early as 1.5–2 hours after injection, decreasing by 4 hours[27], in contrast to a delayed profile in CLP, continuing to increase 8 hours following surgery[17]. The plasma cytokine data shown in Table 1 exemplify these differences. Although we studied CLP mice at a later time point than LPS (6 vs. 4 hours), both models represent early stages of the inflammatory process and demonstrate that prothrombotic manifestations occur early in experimental sepsis.
The finding that intravenous fluid replacement prevented the prothrombotic responses induced by CLP but not by endotoxemia is intriguing. As shown in Table 2, I.V. fluid replacement did not change mean arterial pressure, heart rate or venular wall shear rate. However, since fluid resuscitation of mice undergoing CLP improves cardiac output[28], we cannot exclude changes in microvascular hemodynamics in other regions of the microcirculation. In addition, hemodilution induced by fluid replacement, and its associated rheologic effects, may improve tissue perfusion[29]. Conceivably, enhanced microvascular perfusion may result in shear-dependent release of agonists such as nitric oxide or prostacyclin, known to impact platelet-microvessel interactions[30]. Further, reversal of hemoconcentration may account for the beneficial effects of I.V. fluids on microvascular thrombosis, since increased hematocrit promotes hemostasis ex vivo[31, 32], and hemoconcentration associated with water deprivation enhances photochemical injury-induced thrombosis in mice[33]. Intravenous fluids have also been shown to improve microvascular dysfunction and survival in rodent sepsis[26, 28], and they represent the mainstay of initial resuscitation in patients with severe sepsis and septic shock[34]. The mechanisms by which fluid replacement prevented the prothrombotic manifestations of polymicrobial sepsis in our study remain to be clarified.
Our data support a differential role of toll-like receptors in microvascular thrombosis in CLP and LPS models. Toll-like receptors are components of the innate immune response responsible for recognizing pathogen-associated molecular patterns and initiating intracellular signaling, ultimately enhancing expression of a variety of inflammatory genes[35, 36]. Enhanced TLR-2 expression has been shown in monocytes of septic humans[37, 38] and has been implicated in the pathogenesis of various mouse models of inflammation, including CLP-induced sepsis[39, 40], procoagulant responses to antiphospholipid antibodies[35], and atherosclerosis[41, 42]. To our knowledge, the role of TLR-2 in thrombosis in experimental sepsis has not been described previously. Our findings in TLR-2-deficient mice shown in Figure 4 support the notion that TLR-2 mediates enhanced thrombosis in polymicrobial sepsis, but not endotoxemia. Although Figures 1 and 4 seem to suggest that thrombosis kinetics may differ between TLR-2-deficient and wild-type mice under control conditions (e.g., lower values for saline and sham groups in TLR-2 deficient mice), future studies using littermate controls will be required to address this question.
As shown in Figure 2, TLR4-deficiency did not influence the prothrombotic manifestations induced by CLP. While the pathogenic role of TLR4 in endotoxemia is well established, its role in polymicrobial sepsis is less clear, as conflicting data exist. For example, mice with defective TLR4 were reported to have either similar[16] or reduced[43] mortality following CLP as compared to controls. In addition, while TLR4 was not required for CLP-induced microvascular recruitment of inflammatory cells in some studies[15, 18], others reported TLR4-dependent neutrophil and renal inflammatory responses following CLP[43, 44]. Thus, we investigated the role of TLR4 in microvascular thrombosis induced by polymicrobial sepsis, demonstrating that TLR4 was not required for the CLP-enhanced thrombosis.
We also found that VWF was not required for the enhanced microvascular thrombosis induced by CLP, in contrast with our recent report that endotoxemia failed to enhance microvascular thrombosis in the same strain of VWF-deficient mice[12]. In the present manuscript (Figure 3) and in our prior report, VWF-deficient mice demonstrated delayed microvascular thrombosis under control conditions, consistent with data obtained in a ferric chloride model of thrombosis with this strain[20], and those obtained with mice deficient in the VWF receptor GPIbα[45]. We anticipated a role for VWF in microvascular thrombosis in polymicrobial sepsis, based not only on our prior findings in endotoxemia, but also on clinical data. Administration of endotoxin to healthy humans enhances plasma VWF and its more adhesive/pro-thrombotic ultra-large (UL) multimeric form (ULVWF)[46]. Similarly, septic patients with organ failure demonstrate enhanced levels of ULVWF[47, 48], suggesting a possible role for this molecule in clinical sepsis. Our data demonstrate that enhanced microvascular thrombosis induced by CLP at an early time point is independent of VWF. Interestingly, we did not detect elevation of VWF at a later time point (24 hours after surgery); these findings contrast with those reported by Lerolle et al., that VWF antigen increased significantly in mice 16 hours following CLP[25]. The reasons for the different results are not entirely clear, though several methodologic differences between the studies (CLP technique, postoperative analgesia, fluid replacement, VWF assays, etc.) tend to limit direct comparisons of these findings. Although we could not detect an increase in VWF antigen at 6 or 24 hours following CLP, we cannot exclude a possible role of this molecule in microvascular thrombosis at later time points in polymicrobial sepsis.
In summary, we report that polymicrobial sepsis and endotoxemia promote microvascular thrombosis in mouse cremaster venules, assessed with a light/dye-injury model, via distinct mechanisms. In the case of CLP, enhanced thrombosis required TLR-2, but not VWF or TLR4; these two molecules mediate endotoxin-enhanced microvascular thrombosis. The salutary effects of intravenous fluid replacement on microvascular thrombosis in polymicrobial sepsis remain to be characterized.
Acknowledgments
FUNDING
This work was supported by National Institutes of Health grant HL-079368 (to R.E.R.), a Merit Review grant from The Department of Veterans Affairs (to R.E.R.), and the Curtis Hankamer Basic Research Fund (to S.H.S).
REFERENCES
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 3.Dalldorf FG, Jennette JC. Fatal meningococcal septicemia. Arch Pathol Lab Med. 1977;101:6–9. [PubMed] [Google Scholar]
- 4.Goodman ML, Way BA, Irwin JW. The inflammatory response to endotoxin. J Pathol. 1979;128:7–14. doi: 10.1002/path.1711280103. [DOI] [PubMed] [Google Scholar]
- 5.Jaimes EA, del Castillo D, Rutherford MS, Raij L. Countervailing influence of tumor necrosis factor-alpha and nitric oxide in endotoxemia. J Am Soc Nephrol. 2001;12:1204–1210. doi: 10.1681/ASN.V1261204. [DOI] [PubMed] [Google Scholar]
- 6.Schaub RG, Ochoa R, Simmons CA, Lincoln KL. Renal microthrombosis following endotoxin infusion may be mediated by lipoxygenase products. Circ Shock. 1987;21:261–270. [PubMed] [Google Scholar]
- 7.Taylor FB., Jr Staging of the pathophysiologic responses of the primate microvasculature to Escherichia coli and endotoxin: examination of the elements of the compensated response and their links to the corresponding uncompensated lethal variants. Crit Care Med. 2001;29:S78–S89. doi: 10.1097/00003246-200107001-00026. [DOI] [PubMed] [Google Scholar]
- 8.Dixon B. The role of microvascular thrombosis in sepsis. Anaesth Intensive Care. 2004;32:619–629. doi: 10.1177/0310057X0403200502. [DOI] [PubMed] [Google Scholar]
- 9.Anthoni C, Russell J, Wood KC, Stokes KY, Vowinkel T, Kirchhofer D, Granger DN. Tissue factor: a mediator of inflammatory cell recruitment, tissue injury, and thrombus formation in experimental colitis. J Exp Med. 2007;204:1595–1601. doi: 10.1084/jem.20062354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerwinka WH, Cooper D, Krieglstein CF, Feelisch M, Granger DN. Nitric oxide modulates endotoxin-induced platelet-endothelial cell adhesion in intestinal venules. Am J Physiol Heart Circ Physiol. 2002;282:H1111–H1117. doi: 10.1152/ajpheart.00391.2001. [DOI] [PubMed] [Google Scholar]
- 11.Rumbaut RE, Bellera RV, Randhawa JK, Shrimpton CN, Dasgupta SK, Dong JF, Burns AR. Endotoxin enhances microvascular thrombosis in mouse cremaster venules via a TLR4-dependent, neutrophil-independent mechanism. Am J Physiol Heart Circ Physiol. 2006;290:H1671–H1679. doi: 10.1152/ajpheart.00305.2005. [DOI] [PubMed] [Google Scholar]
- 12.Patel KN, Soubra SH, Bellera RV, Dong JF, McMullen CA, Burns AR, Rumbaut RE. Differential role of von Willebrand factor and P-selectin on microvascular thrombosis in endotoxemia. Arterioscler Thromb Vasc Biol. 2008;28:2225–2230. doi: 10.1161/ATVBAHA.108.175679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;24 Suppl 1:7–11. doi: 10.1097/01.shk.0000191384.34066.85. [DOI] [PubMed] [Google Scholar]
- 14.Schultz MJ, van der Poll T. Animal and human models for sepsis. Ann Med. 2002;34:573–581. doi: 10.1080/078538902321117797. [DOI] [PubMed] [Google Scholar]
- 15.Bauer P, Lush CW, Kvietys PR, Russell JM, Granger DN. Role of endotoxin in the expression of endothelial selectins after cecal ligation and perforation. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1140–R1147. doi: 10.1152/ajpregu.2000.278.5.R1140. [DOI] [PubMed] [Google Scholar]
- 16.Echtenacher B, Freudenberg MA, Jack RS, Mannel DN. Differences in innate defense mechanisms in endotoxemia and polymicrobial septic peritonitis. Infect Immun. 2001;69:7271–7276. doi: 10.1128/IAI.69.12.7271-7276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock. 2000;13:110–116. doi: 10.1097/00024382-200013020-00004. [DOI] [PubMed] [Google Scholar]
- 18.Singer G, Houghton J, Rivera CA, Anthoni C, Granger DN. Role of LPS in the hepatic microvascular dysfunction elicited by cecal ligation and puncture in mice. J Hepatol. 2007;47:799–806. doi: 10.1016/j.jhep.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ha T, Lu C, Liu L, Hua F, Hu Y, Kelley J, Singh K, Kao RL, Kalbfleisch J, Williams DL, Gao X, Li C. TLR2 ligands attenuate cardiac dysfunction in polymicrobial sepsis via a phosphoinositide 3-kinase-dependent mechanism. Am J Physiol Heart Circ Physiol. 298:H984–H991. doi: 10.1152/ajpheart.01109.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Cullere M, Hynes RO, Wagner DD. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95:9524–9529. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 22.Rumbaut RE, Randhawa JK, Smith CW, Burns AR. Mouse cremaster venules are predisposed to light/dye-induced thrombosis independent of wall shear rate, CD18, ICAM-1, or P-selectin. Microcirculation. 2004;11:239–247. doi: 10.1080/10739680490425949. [DOI] [PubMed] [Google Scholar]
- 23.Davis MJ. Determination of volumetric flow in capillary tubes using an optical Doppler velocimeter. Microvasc Res. 1987;34:223–230. doi: 10.1016/0026-2862(87)90055-0. [DOI] [PubMed] [Google Scholar]
- 24.Purswani MU, Eckert SJ, Arora HK, Noel GJ. Effect of ciprofloxacin on lethal and sublethal challenge with endotoxin and on early cytokine responses in a murine in vivo model. J Antimicrob Chemother. 2002;50:51–58. doi: 10.1093/jac/dkf091. [DOI] [PubMed] [Google Scholar]
- 25.Lerolle N, Dunois-Larde C, Badirou I, Motto DG, Hill G, Bruneval P, Diehl JL, Denis CV, Baruch D. von Willebrand factor is a major determinant of ADAMTS-13 decrease during mouse sepsis induced by cecum ligation and puncture. J Thromb Haemost. 2009;7:843–850. doi: 10.1111/j.1538-7836.2009.03313.x. [DOI] [PubMed] [Google Scholar]
- 26.Anning PB, Finney SJ, Singh S, Winlove CP, Evans TW. Fluids reverse the early lipopolysaccharide-induced albumin leakage in rodent mesenteric venules. Intensive Care Med. 2004;30:1944–1949. doi: 10.1007/s00134-004-2385-3. [DOI] [PubMed] [Google Scholar]
- 27.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D. Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol. 2005;12:60–67. doi: 10.1128/CDLI.12.1.60-67.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zanotti-Cavazzoni SL, Guglielmi M, Parrillo JE, Walker T, Dellinger RP, Hollenberg SM. Fluid resuscitation influences cardiovascular performance and mortality in a murine model of sepsis. Intensive Care Med. 2009;35:748–754. doi: 10.1007/s00134-008-1360-9. [DOI] [PubMed] [Google Scholar]
- 29.Pries AR, Secomb TW, Sperandio M, Gaehtgens P. Blood flow resistance during hemodilution: effect of plasma composition. Cardiovasc Res. 1998;37:225–235. doi: 10.1016/s0008-6363(97)00226-5. [DOI] [PubMed] [Google Scholar]
- 30.Egbrink MG, Van Gestel MA, Broeders MA, Tangelder GJ, Heemskerk JM, Reneman RS, Slaaf DW. Regulation of microvascular thromboembolism in vivo. Microcirculation. 2005;12:287–300. doi: 10.1080/10739680590925628. [DOI] [PubMed] [Google Scholar]
- 31.Eugster M, Reinhart WH. The influence of the haematocrit on primary haemostasis in vitro. Thromb Haemost. 2005;94:1213–1218. doi: 10.1160/TH05-06-0424. [DOI] [PubMed] [Google Scholar]
- 32.Peerschke EI, Silver RT, Weksler B, Grigg SE, Savion N, Varon D. Ex vivo evaluation of erythrocytosis-enhanced platelet thrombus formation using the cone and plate(let) analyzer: effect of platelet antagonists. Br J Haematol. 2004;127:195–203. doi: 10.1111/j.1365-2141.2004.05190.x. [DOI] [PubMed] [Google Scholar]
- 33.Tanira MO, el-Sabban FF, Fahim MA, Wasfi IA. Acetyl salicylic acid alleviates increased susceptibility to thrombosis in pial microvessels of dehydrated mice. J Vet Med Sci. 1994;56:245–248. doi: 10.1292/jvms.56.245. [DOI] [PubMed] [Google Scholar]
- 34.Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, Calandra T, Dhainaut JF, Gerlach H, Harvey M, Marini JJ, Marshall J, Ranieri M, Ramsay G, Sevransky J, Thompson BT, Townsend S, Vender JS, Zimmerman JL, Vincent JL. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008;34:17–60. doi: 10.1007/s00134-007-0934-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satta N, Dunoyer-Geindre S, Reber G, Fish RJ, Boehlen F, Kruithof EK, de Moerloose P. The role of TLR2 in the inflammatory activation of mouse fibroblasts by human antiphospholipid antibodies. Blood. 2007;109:1507–1514. doi: 10.1182/blood-2005-03-024463. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 37.Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. Role of Toll-like receptors in the development of sepsis. Shock. 2008;29:315–321. doi: 10.1097/SHK.0b013e318157ee55. [DOI] [PubMed] [Google Scholar]
- 38.Viemann D, Dubbel G, Schleifenbaum S, Harms E, Sorg C, Roth J. Expression of toll-like receptors in neonatal sepsis. Pediatr Res. 2005;58:654–659. doi: 10.1203/01.PDR.0000180544.02537.FD. [DOI] [PubMed] [Google Scholar]
- 39.Williams DL, Ha T, Li C, Kalbfleisch JH, Schweitzer J, Vogt W, Browder IW. Modulation of tissue Toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med. 2003;31:1808–1818. doi: 10.1097/01.CCM.0000069343.27691.F3. [DOI] [PubMed] [Google Scholar]
- 40.Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A. 2009;106:4018–4023. doi: 10.1073/pnas.0900196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinohara M, Hirata K, Yamashita T, Takaya T, Sasaki N, Shiraki R, Ueyama T, Emoto N, Inoue N, Yokoyama M, Kawashima S. Local overexpression of toll-like receptors at the vessel wall induces atherosclerotic lesion formation: synergism of TLR2 and TLR4. Arterioscler Thromb Vasc Biol. 2007;27:2384–2391. doi: 10.1161/ATVBAHA.106.139253. [DOI] [PubMed] [Google Scholar]
- 42.Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, Thuillez C, Richard V. Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27:1064–1071. doi: 10.1161/ATVBAHA.107.140723. [DOI] [PubMed] [Google Scholar]
- 43.Alves-Filho JC, de Freitas A, Russo M, Cunha FQ. Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis. Crit Care Med. 2006;34:461–470. doi: 10.1097/01.ccm.0000198527.71819.e1. [DOI] [PubMed] [Google Scholar]
- 44.El-Achkar TM, Plotkin Z, Marcic B, Dagher PC. Sepsis induces an increase in thick ascending limb Cox-2 that is TLR4 dependent. Am J Physiol Renal Physiol. 2007;293:F1187–F1196. doi: 10.1152/ajprenal.00217.2007. [DOI] [PubMed] [Google Scholar]
- 45.Konstantinides S, Ware J, Marchese P, Almus-Jacobs F, Loskutoff DJ, Ruggeri ZM. Distinct antithrombotic consequences of platelet glycoprotein Ibalpha and VI deficiency in a mouse model of arterial thrombosis. J Thromb Haemost. 2006;4:2014–2021. doi: 10.1111/j.1538-7836.2006.02086.x. [DOI] [PubMed] [Google Scholar]
- 46.Reiter RA, Varadi K, Turecek PL, Jilma B, Knobl P. Changes in ADAMTS13 (von-Willebrand-factor-cleaving protease) activity after induced release of von Willebrand factor during acute systemic inflammation. Thromb Haemost. 2005;93:554–558. doi: 10.1160/TH04-08-0467. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen TC, Liu A, Liu L, Ball C, Choi H, May WS, Aboulfatova K, Bergeron AL, Dong JF. Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica. 2007;92:121–124. doi: 10.3324/haematol.10262. [DOI] [PubMed] [Google Scholar]
- 48.Ono T, Mimuro J, Madoiwa S, Soejima K, Kashiwakura Y, Ishiwata A, Takano K, Ohmori T, Sakata Y. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006;107:528–534. doi: 10.1182/blood-2005-03-1087. [DOI] [PubMed] [Google Scholar]