

Abstract
Cholinergic neurons are widespread, and pharmacological modulation of acetylcholine receptors affects numerous brain processes, but such modulation entails side effects due to limitations in specificity for receptor type and target cell. As a result, causal roles of cholinergic neurons in circuits have been unclear. We integrated optogenetics, freely moving mammalian behavior, in vivo electrophysiology, and slice physiology to probe the cholinergic interneurons of the nucleus accumbens by direct excitation or inhibition. Despite representing less than 1% of local neurons, these cholinergic cells have dominant control roles, exerting powerful modulation of circuit activity. Furthermore, these neurons could be activated by cocaine, and silencing this drug-induced activity during cocaine exposure (despite the fact that the manipulation of the cholinergic interneurons was not aversive by itself) blocked cocaine conditioning in freely moving mammals.
Acetylcholine is an important and widely studied neurotransmitter, which acts on a variety of receptors and target cells (1–5). Pharmacological and genetic studies have elucidated the complex and often opposing influences of the individual subtypes of muscarinic and nicotinic acetylcholine receptors on numerous biological processes, but no study has yet resolved the question of the causal role of cholinergic neurons themselves within a central nervous system tissue (6–11). Addressing such a question would require a novel paradigm for selective and temporally precise control (activation and inhibition) of cholinergic neurons within living mammalian tissues, because previous investigations have resulted in contradictory findings linked to challenges with specificity and temporal resolution. For example, elegant in vivo pharmacological approaches have shown (12–14) that cholinergic transmission in the nucleus accumbens (NAc) [a structure involved in natural reward-related behaviors and responses to drugs such as cocaine (15–19)] is required for reward learning, but novel studies of molecular ablation of cholinergic interneurons within the NAc instead have reported enhanced reward learning (20). Cholinergic interneurons within the NAc are particularly intriguing because they constitute less than 1% of the local neural population (21), yet they project throughout the NAc and provide its only known cholinergic input (22). Relevant cholinergic receptors are expressed locally, and nicotinic and muscarinic pharmacological agonists can exert complex influences on medium spiny neurons (MSNs, which represent >95% of the local neuronal population and constitute the output of the NAc) (23–25). However, the net effect (if any) of the cholinergic interneurons on any aspect of NAc physiology or behavior is unknown.
We undertook an optogenetic approach to resolve this question by selectively driving or blocking action potential firing in these cells. To express microbial opsin genes specifically in cholinergic interneurons, we employed a transgenic mouse line expressing Cre-recombinase under the choline acetyltransferase (ChAT) promoter (26). We stereotaxically injected into the NAc (Fig. 1A) a Cre-inducible adeno-associated virus (AAV) vector carrying the opsin gene fused in-frame with coding sequence for enhanced yellow fluorescent protein (eYFP) (27, 28); the opsin gene encoded either the blue-light gated cation channel channelrhodopsin-2 (ChR2) (29) or the yellow-light gated third-generation chloride pump halorhodopsin (eNpHR3.0) (30). eYFP expression was specific to neurons that expressed ChAT; moreover, the majority of neurons that expressed ChAT also expressed eYFP (Fig. 1, B and C). Both opsins were expressed on the surface membranes of ChAT neurons (Fig. 1D), and the targeted neurons responded to current injection in a manner corresponding to previously established responses of cholinergic interneurons in the dorsal striatum (Fig. 1E) (31). Both the resting membrane potential (Vm) and input resistance (Rinput) were higher for ChAT neurons (YFP+ neurons) than for MSNs [identified as YFP− neurons; table S1; P < 10−4 for VM and P = 0.004 for Rinput, two-tailed t test]. Finally, both opsins were functional in ChATcells, as eNpHR3.0 drove large hyperpolarizations (Fig. 1F; mean ± SEM: −83.8 ± 11.9 mV, n = 4) and ChR2 reliably drove spiking up to 20 to 30 Hz (Fig. 1, G and H).
Fig. 1.

Specificity, membrane targeting, and functionality of ChR2 and eNpHR3.0 in ChAT inter-neurons of the NAc. (A) Cre-dependentAAV[expressing either eNpHR3.0-eYFP or ChR2(H134R)-eYFP] was injected into the medial portion of the NAc.(B) Confocal image of an injected slice demonstrates colocalization of eYFP expression with the ChAT antibody, costained with 4′,6′-diamidino-2-phenylindole (DAPI). (C) 91.3 ± 1.3% of neurons that expressed YFP also stained for the ChAT antibody (n = 418); 93.5 ± 2.8% of neurons that stained for the ChAT antibody also expressed YFP (n = 413). Error bars indicate SEM. (D)High-magnification view reveals membrane localization of eNphR3.0-eYFP (left) and ChR2-eYFP (right), costained with ChAT antibody. (E) Membrane potential changes induced by current injection in a ChR2-eYFP-expressing ChAT neuron. VM = −48 mV. Current steps: −60, −20, +20 pA. (F) Membrane potential changes induced by 1 s of 580-nm light in an eNpHR3.0-eYFP-expressing ChAT neuron (peak hyperpolarization: −103 mV). VM = −49 mV. (Inset) Population-averaged peak hyperpolarization (mean ± SEM: −83.8 ± 11.9 mV; n =4). (G) Consecutive action potentials in a ChR2-eYFP-expressing ChAT neuron evoked by a 470-nm pulse train (5 ms pulse width;10Hz).(H) Average success probability for generating action potentials in ChR2-eYFP-expressing ChAT neurons at different stimulation frequencies (n = 4; mean ± SEM; 470-nm pulse train, 5-ms pulse width).
ChAT interneurons are thought to be tonically active in vivo (3 to 10 Hz) (32, 33), but it has remained mysterious how (or even if) this slow activity in the sparse ChAT cells could be causally involved in affecting local circuit activity or behavior. We used optogenetics to address this question with a combination of slice electrophysiology, in vivo electrophysiology, and freely moving behavior. First, we monitored postsynaptic currents in MSNs (ChR2-eYFP non-expressing cells) in acute NAc slices during optogenetic photostimulation of ChAT cells expressing ChR2-eYFP (Fig. 2A), targeted as in Fig. 1. Stimulating ChAT neurons in this setting increased the frequency of γ-aminobutyric acid type A (GABAA) receptor–mediated inhibitory postsynaptic currents (IPSCs) recorded in MSNs (Fig. 2, B and C). Evoked inhibitory currents were generally synchronized to the light pulse, with a modal latency of 6 ms (Fig. 2D), coupled with a smaller enhancement of asynchronous IPSCs (fig. S1, A to C). Across all recorded cells, the mean frequency of IPSCs observed in the MSNs increased by 525.8 ± 154.3% during light stimulation of the ChAT neurons (n = 7; mean ± SEM, P = 0.01, paired t test), whereas the mean IPSC amplitude was unaffected (P > 0.05, paired t test; n = 7, Fig. 2E). This effect was attenuated by the nicotinic antagonist mecamylamine (fig. S3, n = 5, P < 0.05, paired t test).
Fig. 2.

Optogenetic photoactivation of ChAT interneurons increases frequency of inhibitory currents and suppresses MSN spiking. (A)ChAT neurons transduced with ChR2-eYFP were activated with blue light (470 nm) in brain slices, and nearby MSNs (eYFP− cells) were whole-cell patch-clamped. (B) (Left) Spontaneous synaptic currents were observed in an MSN in a slice expressing ChR2-eYFP in ChAT neurons. (Middle) Synaptic currents increased in frequency in response to 470-nm light pulses (5-ms pulse width; 10 Hz). (Right) These currents were blocked by GABAA receptor antagonist SR-95531 (5 mM) and are thus considered IPSCs. 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline-2,3-dione (NBQX) (5 mM) and (RS)-3-(2-Carboxypiperazin-4-yl)-propyl-1-phosphonic acid (RS-CPP) (5 μM) were present in all experiments. (C)Time course of IPSC frequencies for this MSN, showing the effect of light pulses (blue dashed bars) and SR-95531 (black bar). (D)Average percentage increase in IPSC frequency during the light-on periods (normalized to that of light-off periods) as a function of time relative to light pulses (n = 6). The blue dashed line indicates the onset of light pulses; error bars denote SEM. (E) Light pulses increased the frequency of IPSCs by 525.8 ± 154.3% (n =6, P = 0.01, paired two-tailed t test), whereas the average amplitudes of spontaneous IPSCs were changed by 21.3 ± 28.9% (P >0.05). (F) An optrode (optical fiber attached to a tungsten electrode) was stereotaxically positioned in vivo into a NAc that expressed ChR2-eYFP in ChAT cells. (G) (Top) Voltage trace of an isolated unit that is inhibited by blue light stimulation. (Middle) Raster plot displaying the response of the same unit to five repetitions of the light stimulation, with each action potential represented by a dot. (Bottom) Average and SEM of the firing rate over time for the same unit. (H) Fraction of sites that were inhibited versus excited by light stimulation. (I) Population summary of the time course of response to light stimulation for sites that were inhibited (left; n = 13 of 16) or excited (right; n =3 of 16)by light. Solid lines represent average firing rate across sites as a function of time; each dot represents the average firing rate of an individual site. All firing rates are normalized to the mean rate before light stimulation. (F to I) Duration of photostimulation, 10 s; pulse duration, 5 ms; wavelength, 470 nm; frequency, 10 Hz. Epochs of light stimulation are represented by blue dashed lines.
We next asked if and how these changes in inhibitory current frequency would translate into changes in MSN spiking in vivo. We recorded neural activity extracellularly with an optrode in the NAc during optogenetic activation of the ChAT interneurons in vivo (Fig. 2F). At sites where single units were not isolated, we observed neural population firing that tracked the light stimulation at 10 Hz but not 100 Hz (fig. S1D), probably representing population spiking across the sparse but synchronously activated ChAT cells in the neighborhood of the electrode. In contrast to these population spikes, the isolated units in the NAc displayed a markedly different response to the optogenetic photostimulation. In agreement with the observed increase in IPSC frequency in MSNs in slices, we observed inhibition of background firing during stimulation of the ChATcells in vivo (representative cell, Fig. 2G). Across the population, most significantly modulated sites showed a suppression of background firing, although a few responded with an increase in firing (Fig. 2, H and I), consistent with known recurrent inhibition among MSNs and corresponding release from inhibition, during ChAT neuron drive, that had been previously exerted by the broader MSN population.
We next explored the consequences of specifically inhibiting ChAT interneurons, employing Cre-dependent eNpHR3.0 expression in vivo. In contrast to what was observed with ChAT neuron excitation, firing of NAc neurons was typically increased in likely MSNs by optogenetic inhibition of the ChAT cells (a representative cell is shown in Fig. 3A). Power spectral analysis revealed a frequency peak in the firing pattern at ~4 Hz in these in vivo recordings (Fig. 3B). Summary data are presented in Fig. 3C; across the population of significantly modulated sites, most neurons were excited by the optogenetic inhibition of ChAT neurons (n = 17). We were able to obtain a single-unit recording from a rare putative ChAT interneuron, which was completely shut down by eNpHR3.0 (Fig. 3D) and displayed the long action-potential duration characteristic of ChAT interneurons (22) (2.0 ms for this cell, whereas spike durations for MSNs in our recordings ranged from 1.1 to 1.7 ms). Summary data (Fig. 3E) show the dynamics of excitation and inhibition for all recorded sites, illustrating the dominant pattern of excitation (firing increased by 130.5 ± 17.5% in sites that were excited by light).
Fig. 3.
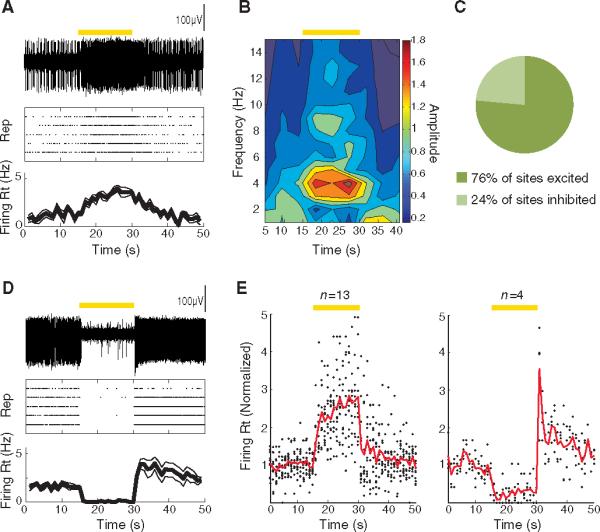
Optogenetic photoinhibition of ChAT interneurons enhances MSN spiking in vivo. (A) (Top) Voltage trace of an isolated unit (recorded from the NAc in vivo) that was excited by optogenetic photoinhibition of the ChAT interneurons with eNpHR3.0. (Middle) Raster plot displaying the response of the same unit to five repetitions of the light stimulation, with each action potential represented by a dot. (Bottom) Average and SEM of the firing rate over time for the same unit. (B) Wavelet analysis reveals power of spiking as a function of frequency and time (average across five repetitions) for the same unit as in (A). (C) Fraction of sites that were inhibited versus excited by light stimulation. (D) Sameas (A), for a unit that was inhibited by light stimulation. (E) Population summary of the time course of response to light stimulation for sites that were inhibited (left; n = 13 of 17) or excited (right; n = 4 of 17) by light. Solid lines represent the average firing rate across sites as a function of time; each dot represents the average firing rate of an individual site. All firing rates are normalized to the mean value before light stimulation. (A to E) Duration of photostimulation, 15 s (constant illumination); wavelength, 560 nm. Epochs of light stimulation are represented by yellow bars.
Finally, we sought to test if this potent NAc control mechanism was relevant to accumbens-dependent reward behavior in freely moving mice. We first tested the effect of acutely administered cocaine on activity of these identified ChAT neurons in acute NAc slices. In ventro-medial NAc ChAT cells, cocaine tended to increase spontaneous firing (representative ChAT neuron shown in Fig. 4, A and B). Summary data revealed that cocaine increased firing rates from 0.60 ± 0.41 Hz to 1.74 ± 0.56 Hz at 10 min in ChAT neurons (n = 7; P < 0.005, paired t test), whereas in the control group of cells receiving only vehicle, firing rates decreased from 0.69 ± 0.24 Hz to 0.09 ± 0.09 Hz over the same time period (n = 6; P < 0.05 comparing the two groups, two-tailed t test) (Fig. 4C).
Fig. 4.

ChAT interneurons can be activated by cocaine in slice and required for cocaine conditioning in vivo. (A) The frequency of spontaneous action potentials in a ChAT neuron increased 10 min after bath application of cocaine (5 μM). ACSF, artificial cerebrospinal fluid. (B) Firing rate over time for this ChAT neuron. Horizontal gray bar, application of cocaine; vertical dotted line, 10 min after cocaine application, the time point illustrated in detail in (A) and (C). (C) Population data illustrating the cocaine-induced increase in firing in ChAT neurons, comparing the baseline firing rate (averaged over the 2.5 min before cocaine application) with the rate after cocaine infusion (averaged between 10 and 12.5 min after onset of cocaine application; gray bars, cells receiving cocaine; white bars, control cells receiving only ACSF; P < 0.005, paired two-tailed t test for cocaine-treated group before versus after cocaine; P < 0.05 unpaired two-tailed t test comparing cocaine versus control cells after cocaine or vehicle). (D) Schematic illustration of a bilateral cannula system with double fibers inserted to illuminate the medial portion of the NAc. (Left inset) Endpoint of cannula track for all mice used in (H). (Right inset) eYFP expression in NAc of a ChAT∷Cre+ mouse injected with Cre-dependent eNpHR3.0-eYFP. (E) Conditioning paradigm for cocaine CPP (H). Mice were conditioned with ip cocaine (20 mg/kg), along with ChAT cell inhibition with eNpHR3.0 (wavelength: 590 nm). (F) Tracking data from representative ChAT∷Cre+ and ChAT∷Cre− mice on the testing day after cocaine conditioning (day 3). On the previous day (day 2), the mice had received cocaine and light in one left chamber, whereas in the other they received saline. The ChAT∷Cre− mouse (but not the ChAT∷Cre+ mouse) exhibited a preference for the conditioned chamber. (G) (Left) Fold change in time in conditioned chamber during day 3 versus day 1 of cocaine CPP (conditioning with cocaine and light). Comparison of ChAT∷Cre+ and ChAT∷Cre− littermates; in both cases injected with Cre-dependent eNpHR3.0 (n = 10 ChAT∷Cre+, n = 12 ChAT∷Cre−; P < 0.01 for two-tailed t test; three cohorts). (Right) Fold change in time in conditioned chamber during day 3 versus day 1 for conditioning with light alone (no cocaine; n = 9 ChAT∷Cre+, n = 7 ChAT∷Cre; P > 0.05 for two-tailed t test; three cohorts). Error bars indicate SEM. n.s., not significant. (H) Velocity of virus-injected (Cre-dependent eNpHR3.0) and photostimulated ChAT∷Cre+ and ChAT∷Cre− mice in the open field (n = 10 ChAT∷Cre+, n = 10 ChAT∷Cre−; P > 0.05 for two-tailed t test; three cohorts). (I) Same as (H) for track length in open field (n = 10 ChAT∷Cre+, n = 10 ChAT∷Cre−; P > 0.05 for two-tailed t test; three cohorts). (J) Same as (H) for time in center of open field (n = 10 ChAT∷Cre+, n = 10 ChAT∷Cre; P > 0.05 for two-tailed t test; three cohorts). (A to J) *P < 0.05; **P < 0.01; ***P < 0.005.
We next used eNpHR3.0 to test for causal roles in either this cocaine-induced activity or baseline activity of ChAT cells in the reward-related behavior of cocaine conditioned place preference (CPP), in which animals learn to associate an environment with cocaine. After injecting virus and implanting cannulae bilaterally (Fig. 4D) to silence ChAT neurons during cocaine exposure (Fig. 4E), mice that expressed eNpHR3.0 in the ChAT cells exhibited significantly less cocaine-induced CPP than did their control (Cre recombinase-negative) littermates that had received the same virus, surgery, and light-delivery protocol [20 mg/kg intraperitoneally(ip), Fig.4, F and G; n = 10 ChAT∷Cre+, n = 12 ChAT∷Cre− (left panel); P < 0.01 for two-tailed t test; three cohorts; see also fig. S2A]. We observed no behavioral effect of inhibiting the ChAT cells in the absence of cocaine, and ChAT neuron inhibition by itself was not aversive, as conditioning with eNpHR3.0 alone did not affect place preference (Fig. 4G, right panel; n = 9 ChAT∷Cre+, n = 7 ChAT∷Cre−; P > 0.05 for two-tailed t test; three cohorts; fig. S2B; see also fig. S4A for cocaine dose-response curve). Activation of the cells with ChR2 at 10 Hz was not sufficient to drive place preference by itself or enhance cocaine place preference (10 and 20 mg/kg ip, fig. S4, B to D), with our data from ChAT cell inhibition instead demonstrating necessity of these cells. Finally, in control experiments, we found that ChAT neuron inhibition by itself had no effect on mobility or anxiety in the open field (Fig. 4, H and J), and contextual- and auditory-cued fear conditioning were not disrupted by inhibition of the ChAT cells (fig. S5).
Together, these data demonstrate that selectively inhibiting ChAT interneurons in the NAc with high temporal precision has the overall effect of increasing MSN activity and blocking cocaine conditioning in freely moving mammals. These behavioral results do not support conclusions arising from chronic ablation of the cholinergic interneurons (20); instead they are more consistent with interpretations arising from faster but less cellularly targeted pharmacological modulation in the NAc(12–14). Ablation of the cholinergic interneurons might lead to indirect effects, such as a compensatory increase in dopamine in the NAc, which, in turn, could enhance cocaine reward. In fact, a fundamental difference between acute and chronic manipulations could explain clinically relevant apparent contradictions in our understanding of the acetylcholine/dopamine balance in the brain. For example, an acute increase in nicotine (presumably acting on cholinergic receptors) causes a corresponding acute increase in dopamine (34), whereas chronic changes in dopamine or acetylcholine levels can cause opposing changes in the levels of the other neuromodulators (35), as seen in the dopamine depletion of Parkinson'sdisease(36).
Because cocaine increases dopamine levels in the NAc, the multiple classes of dopamine receptors expressed on the various cell types within the NAc will give rise to substantial complexity. Although the neural encoding of both cocaine and natural stimuli in the NAc is heterogeneous (37), the predominant effect of appetitive stimuli may be to decrease activity in the MSNs (inhibitory projection neurons), thereby gating directed behavior through disinhibition of target brain regions. Consistent with this picture, a pause in NAc activity (which we have found that ChAT neurons are well-suited to implement) may be required for reward-related conditioning (38, 39); in contrast, the predominant effect of aversive stimuli may be to increase MSN activity (40, 41). The fact that acute silencing of ChAT interneurons disrupts drug-related learning without affecting conditioning in the absence of drug suggests that control over this microcircuit could be used to selectively disrupt effects of drugs of abuse without affecting appetitive or aversive responses in general, a possibility that would be of substantial clinical benefit. Together, our results point to a powerful role for these sparsely distributed neurons in controlling local circuit activity and implementing behavioral conditioning in freely moving mammals.
Supplementary Material
Acknowledgments
We thank the entire Deisseroth lab for their support. I.B.W. is supported by the Helen Hay Whitney Foundation; S.-C.L. is supported by the National Institute of Neurological Disorders and Stroke; I.D. is supported by DAAD and the Human Frontier Science Program; P.A. is supported by the Stanford Dean's fellowship; V.G. is supported by Bio-X SIGF; K.D. is supported by the Keck, Snyder, Woo, Yu, and McKnight Foundations, as well as by CIRM, the National Institute of Mental Health, and the National Institute on Drug Abuse.
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/330/6011/1677/DC1 Materials and Methods
References and Notes
- 1.Changeux JP. C. R. Biol. 2009;332:421. doi: 10.1016/j.crvi.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Kilgard MP, Merzenich MM. Science. 1998;279:1714. doi: 10.1126/science.279.5357.1714. [DOI] [PubMed] [Google Scholar]
- 3.Bakin JS, Weinberger NM. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11219. doi: 10.1073/pnas.93.20.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maskos U. Br. J. Pharmacol. 2008;153(suppl. 1):S438. doi: 10.1038/bjp.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picciotto MR, et al. Nature. 1998;391:173. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 6.Aosaki T, et al. J. Neurosci. 1994;14:3969. doi: 10.1523/JNEUROSCI.14-06-03969.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furey ML, Pietrini P, Haxby JV, Drevets WC. Neuropsychopharmacology. 2008;33:913. doi: 10.1038/sj.npp.1301461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anagnostaras SG, et al. Nat. Neurosci. 2003;6:51. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- 9.Ikemoto S, Glazier BS, Murphy JM, McBride WJ. Physiol. Behav. 1998;63:811. doi: 10.1016/s0031-9384(98)00007-9. [DOI] [PubMed] [Google Scholar]
- 10.Williams MJ, Adinoff B. Neuropsychopharmacology. 2008;33:1779. doi: 10.1038/sj.npp.1301585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Everitt BJ, Robbins TW. Annu. Rev. Psychol. 1997;48:649. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- 12.Pratt WE, Spencer RC, Kelley AE. Behav. Neurosci. 2007;121:1215. doi: 10.1037/0735-7044.121.6.1215. [DOI] [PubMed] [Google Scholar]
- 13.Crespo JA, Sturm K, Saria A, Zernig G. J. Neurosci. 2006;26:6004. doi: 10.1523/JNEUROSCI.4494-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pratt WE, Kelley AE. Behav. Neurosci. 2004;118:730. doi: 10.1037/0735-7044.118.4.730. [DOI] [PubMed] [Google Scholar]
- 15.Pontieri FE, Tanda G, Orzi F, Di Chiara G. Nature. 1996;382:255. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- 16.Chen BT, Hopf FW, Bonci A. Ann. N.Y. Acad. Sci. 2010;1187:129. doi: 10.1111/j.1749-6632.2009.05154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wise RA. NIDA Res. Monogr. 1984;50:15. [PubMed] [Google Scholar]
- 18.Robbins TW, Ersche KD, Everitt BJ. Ann. N.Y. Acad. Sci. 2008;1141:1. doi: 10.1196/annals.1441.020. [DOI] [PubMed] [Google Scholar]
- 19.Nestler EJ, Aghajanian GK. Science. 1997;278:58. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 20.Hikida T, et al. Proc. Natl. Acad. Sci. U.S.A. 2001;98:13351. doi: 10.1073/pnas.231488998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rymar VV, Sasseville R, Luk KC, Sadikot AF. J. Comp. Neurol. 2004;469:325. doi: 10.1002/cne.11008. [DOI] [PubMed] [Google Scholar]
- 22.Zhou FM, Wilson CJ, Dani JA. J. Neurobiol. 2002;53:590. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]
- 23.Zhou FM, Wilson C, Dani JA. Neuroscientist. 2003;9:23. doi: 10.1177/1073858402239588. [DOI] [PubMed] [Google Scholar]
- 24.Koós T, Tepper JM. J. Neurosci. 2002;22:529. doi: 10.1523/JNEUROSCI.22-02-00529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Rover M, Lodder JC, Kits KS, Schoffelmeer ANM, Brussaard AB. Eur. J. Neurosci. 2002;16:2279. doi: 10.1046/j.1460-9568.2002.02289.x. [DOI] [PubMed] [Google Scholar]
- 26.Materials and methods are available as supporting material on Science Online.
- 27.Tsai H-C, et al. Science. 2009;324:1080. doi: 10.1126/science.1168878. 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atasoy D, Aponte Y, Su HH, Sternson SM. J. Neurosci. 2008;28:7025. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Nat. Neurosci. 2005;8:1263. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 30.Gradinaru V, et al. Cell. 2010;141:154. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawaguchi Y. J. Neurosci. 1993;13:4908. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson CJ, Chang HT, Kitai ST. J. Neurosci. 1990;10:508. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inokawa H, Yamada H, Matsumoto N, Muranishi M, Kimura M. Neuroscience. 2010;168:395. doi: 10.1016/j.neuroscience.2010.03.062. [DOI] [PubMed] [Google Scholar]
- 34.Nisell M, Marcus M, Nomikos GG, Svensson TH. J. Neural Transm. 1997;104:1. doi: 10.1007/BF01271290. [DOI] [PubMed] [Google Scholar]
- 35.Hrabovska A, et al. Chem. Biol. Interact. 2010;183:194. doi: 10.1016/j.cbi.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 36.Hoebel BG, Avena NM, Rada P. Curr. Opin. Pharmacol. 2007;7:617. doi: 10.1016/j.coph.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carelli RM, Deadwyler SA. J. Neurosci. 1994;14:7735. doi: 10.1523/JNEUROSCI.14-12-07735.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taha SA, Fields HL. J. Neurosci. 2006;26:217. doi: 10.1523/JNEUROSCI.3227-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krause M, German PW, Taha SA, Fields HL. J. Neurosci. 2010;30:4746. doi: 10.1523/JNEUROSCI.0197-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlezon WA, Jr., Thomas MJ. Neuropharmacology. 2009;56(suppl. 1):122. doi: 10.1016/j.neuropharm.2008.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peoples LL, West MO. J. Neurosci. 1996;16:3459. doi: 10.1523/JNEUROSCI.16-10-03459.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.