

Abstract
Restoration of transcriptionally silenced genes by means of methyltransferases inhibitors plays a crucial role in the current therapy of myelodysplastic syndromes and certain types of leukemias. A comparative study of hypomethylating activities of a series of 5-azacytidine nucleosides: 5-azacytidine (AC), 2′-deoxy-5-azacytidine (DAC) and its α-anomer (α-DAC), 5,6-dihydro-5-azacytidine (DHAC), 2′-deoxy-5,6-dihydro-5-azacytidine (DHDAC, KP-1212) and its α-anomer (α-DHDAC), and of a 2-pyrimidone ribonucleoside (zebularine) was conducted. Methylation-specific PCR was employed to detect the efficiency of individual agents on cyclin-dependent kinase inhibitor 2B and thrombospondin-1 hypermethylated gene loci. Overall changes in DNA methylation level were quantified by direct estimation of 5-methyl-2′-deoxycytidine-5′-monophosphate by HPLC using digested genomic DNA. Flow cytometric analysis of cell cycle progression and apoptotic markers was used to determine cytotoxicity of the compounds. mRNA expression was measured using qRT-PCR. 2′-deoxy-5,6-dihydro-5-azacytidine was found to be less cytotoxic and more stable than 2′-deoxy-5-azacytidine at the doses that induce comparable DNA hypomethylation and gene reactivation. This makes it a valuable tool for epigenetic research and worth further investigations to elucidate its possible therapeutic potential.
Key words: epigenetic therapy; nucleoside analogs; DNA methylation; 2′-deoxy-5-azacytidine; 2′-deoxy-5,6-dihydro-5-azacytidine
Introduction
Epigenetics focuses on changes in gene expression caused by mechanisms that do not interfere with the DNA sequence. One of the most prominent epigenetic events is represented by DNA methylation,1,2 i.e., a covalent addition of a methyl group to carbon at position 5 of the cytosine within the CpG dinucleotides.3 This enzymatic modification is carried out by DNA methyltransferases (DNMTs). A certain level of DNA methylation is common in most cells and varies throughout the life of an individual. However, cancer cells often display aberrant hypermethylation and/or hypomethylation of gene promoters leading to their transcriptional inactivity or genomic instability.1–5 Methylated cytosines frequently undergo spontaneous deamination to thymines, which represents the first interference to the DNA sequence.1 Therefore, epigenetic mechanisms such as abnormal DNA methylation are considered being the first marker of tumorigenesis.4,5 Reversal of the unfavorable methylation status in malignant cells has been a subject for epigenetic chemotherapy of cancer using hypomethylating drugs. Importantly, hypomethylating agents do not target the cells for immediate death. The cells must be allowed to proliferate and reactivate genes that have been methylation-silenced for these drugs to take effect.1 This is why epigenetic therapy may be expected to be more specific, less toxic and more effective than standard chemotherapy.
Nucleoside analogs such 5-azacytidine, 2′-deoxy-5-azacytidine, 5,6-dihydro-5-azacytidine and zebularine are well established inhibitors of DNA methylation already tested in clinical or preclinical studies.1,6 It has been almost 50 years since the synthesis and antitumor activity of 5-azacytidine (AC) was described. 5-Azacytidine analogs are characterized by a presence of an extra nitrogen atom at position C5 of pyrimidine ring (Fig. 1). This modification leads to a blockade of cytosine methylation via a covalent trapping of DNA methyltransferases (DNMT). AC is believed to utilize a dual mechanism of action following its phosphorylation: (1) hypomethylation of DNA at low doses and (2) cytotoxicity due to the incorporation into RNA and apparent interaction with protein biosynthesis at high doses.7–10
Figure 1.
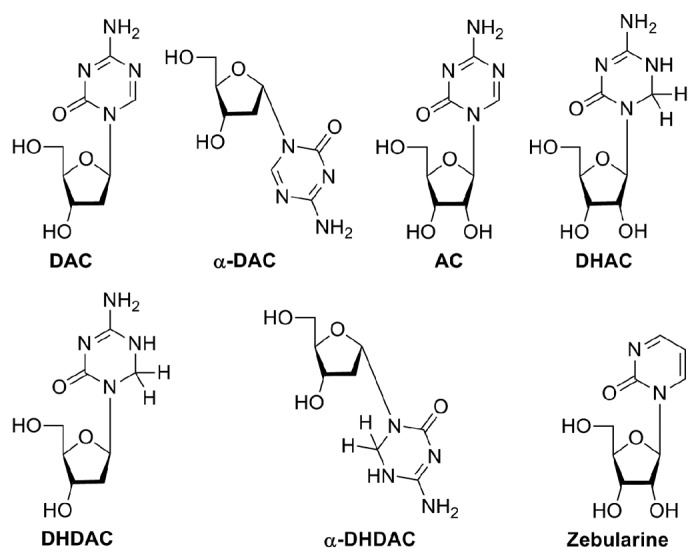
Structures of the compounds under investigation.
To overcome cytotoxicity issues, a deoxy analog of AC—2′-deoxy-5-azacytidine (DAC) was synthesized, which is incorporated only into the DNA following its phosphorylation.11 DAC significantly inhibits DNA methylation at lower concentrations and with less cytotoxicity10 in comparison with AC. Both, AC and DAC, are active against human leukemias but show very limited efficacy against solid tumors.12 Both were also demonstrated to deplete DNMT1 protein.10,13 Unfortunately both nucleosides possess high cytotoxicity at their maximal tolerated doses,12,14 and are unstable in aqueous solution. On the other hand, low doses of DAC induce reactivation of silenced genes in neoplastic cells with relatively minor side effects.12 AC and DAC have been launched to the market under the commercial names of Vidaza® and Dacogen®, respectively, for the treatment of myelodysplastic syndromes (approved by FDA in 2004 and 2006, respectively).
Another 5-azacytidine nucleoside, 5,6-dihydro-5-azacytidine (DHAC), surpasses the main disadvantages of both DAC and AC. It is hydrolytically stable, due to the saturation of 5,6-double bond preventing the nucleophilic attack of the position 6 by water,15 and less cytotoxic. Similarly to AC it can be incorporated into the RNA and inhibits RNA synthesis and DNA methylation in human lymphoid and leukemia cell lines.16,17 Unfortunately, clinical trials revealed significant side effects of the DHAC antitumor therapy including severe chest pain,16 low response rate and cardiotoxicity.17,18
2′-Deoxy-5,6-dihydro-5-azacytidine (DHDAC, KP-1212), a hydrolytically stable congener of DAC,19 and its prodrug KP-1461 are known for their anti-HIV activity via the mechanism of lethal mutagenesis of the viral genome. There is no evidence of significant genotoxicity and/or mitochondrial toxicity on mammalian cells.20,21 Clinical trials on HIV-1-infected patients demonstrated only minimal side effects of DHDAC's prodrug treatment.22 The first reference to a hypomethylating activity of DHDAC was reported by Sheikhnejad et al. The authors synthesized oligodeoxyribonucleotides containing 5,6-dihydro-5-azacytosine nucleobase and observed their ability to efficiently inhibit DNMT in vitro. Interestingly, hypomethylating potential of DHDAC as a free nucleoside analog at cellular level was not clarified to date.
There are two anomeric forms of nucleosides and only β-anomers are naturally found in nucleic acids. α-Anomers of nucleosides generally do not interfere with cellular metabolism and possess biological activity only in rare cases. Nevertheless, Fojtová et al. previously noted identical hypomethylating activities of DAC and α-DAC suggesting that α-anomers of nucleosides may also be of therapeutic interest.
Here we present a comparative study of hypomethylating activities of 5-azacytidine nucleosides: 5-azacytidine (AC), 2′-deoxy-5-azacytidine (DAC) and its α-anomer (α-DAC), 5,6-dihydro-5-azacytidine (DHAC), 2′-deoxy-5,6-dihydro-5-azacytidine (DHDAC, KP-1212) and its α-anomer (α-DHDAC), and of a 2-pyrimidone ribonucleoside zebularine, a cytidine analog lacking an amino group (Fig. 1), which ranks among the more recently described hypomethylating agents. However, its mechanism of action is believed to be similar to that of 5-azacytidine analogs.7,25–28 Contrary to DAC and AC it is stable in neutral and acidic solutions, making oral administration of the drug possible,29,30 with minimal toxicity.7,25,29–31 This allows a long-term treatment in low doses28,30,32 necessary to prevent resilencing of reactivated genes.28,32
All compounds of interest were investigated in parallel in two human leukemic cell lines (CCRF-CEM and HL-60) for their overall hypomethylating potential (genomic DNA) and their ability to reactivate two distinct epigenetically silenced genes. Using three independent methods to estimate the methylation level in the cells we arrived to the conclusion that DHDAC might represent a hypomethylating agent superior to the recently used DAC.
Results
DHDAC hypomethylates CDKN2B and THBS-1 gene loci similarly to DAC.
In a preliminary experiment we looked for a set of genes that would serve as a model of a hypermethylated promoter state. Out of 13 genes tested in both CCRF-CEM and HL-60 cell lines, cyclin-dependent kinase inhibitor 2B (CDKN2B, p15) and thrombospondin-1 (THBS-1) were finally identified as fully methylated in both cell lines and selected for further experimentation. Cells were cultivated with the nucleoside analogs for five days at a concentration corresponding to their respective IC50 determined by means of XTT cytotoxicity test. Where no cytotoxicity was detected, a 100 µM concentration was set as standard (CCRF-CEM cells: AC and DAC 1 µM, α-DAC 0.9 µM, DHAC 10 µM; zebularine, α- and β-DHDAC 100 µM; HL-60 cells: DAC 0.7 µM, AC 6.2 µM; zebularine, α-DAC, DHAC, α- and β-DHDAC 100 µM). As shown in Figure 2, all compounds under investigation were able to hypomethylate fully methylated gene locus of CDKN2B in CCRF-CEM cells. Using the methylation-specific PCR (MSP) and the primers specifically designed for the detection of the methylated and unmethylated form of CDKN2B gene, differences in hypomethylating capabilities of the compounds are evident (Fig. 2A). Zebularine and α-DHDAC seem to be the weakest hypomethylating agents in this assay. The same results were obtained using another set of primers for THBS-1 gene locus (data not shown). To be able to determine the relative efficiencies of the individual hypomethylating agents precisely, MSP was further modified for quantification.5 For this purpose we used primers recognizing the methylated form of the genes (see Methods).
Figure 2.
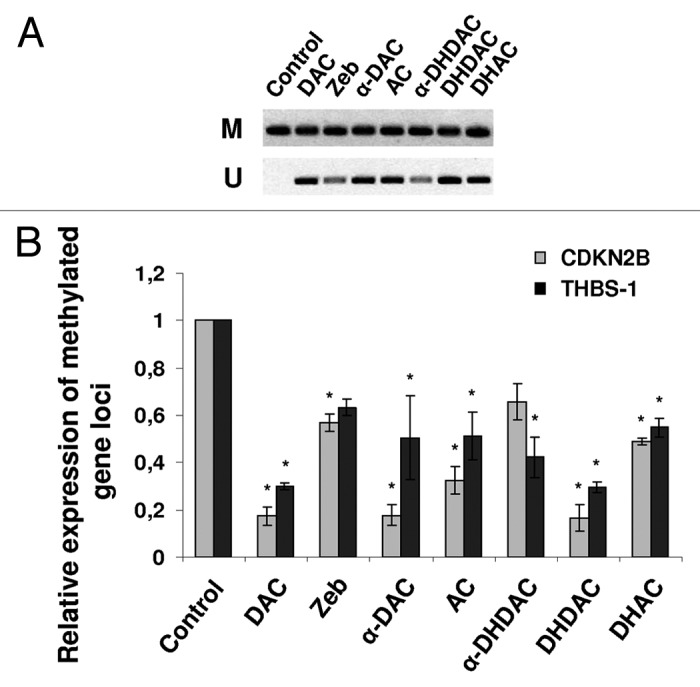
The hypomethylating effect of 5-azacytidine analogs and zebularine on CDKN2B and THBS-1 gene loci. CCRF-CEM cells were cultivated for 5 days with the compounds and the isolated DNA was bisulfite modifated. (A) Agarose gel electrophoresis of MSP products of CDKN2B gene. The PCR products in the lanes M and U indicate presence of methylated and unmethylated templates, respectively. The absence of PCR product in case of unmethylated set of primers in untreated control sample indicates a fully methylated gene locus. Both primer sets contain identical CpG islands. (B) Quantitative MSP analysis of CDKN2B and THBS-1 genes. Relative expression was quantified by means of ΔΔCt method. *p < 0.05 compared with untreated control cells (ANOVA).
Generally, CDKN2B gene locus appeared to be hypomethylated more easily than that of THBS-1 (Fig. 2B). DAC and DHDAC decreased the methylation level of THBS-1 and CDKN2B by 70 and 80%, respectively, compared to untreated control. Zebularine reduced the methylation level only by 40% and represented the weakest hypomethylating agents of all compounds tested. DHAC was similarly weak with the efficiency of about 50%. α-DHDAC decreased the methylation level in THBS-1 and CDKN2B genes to 60 and 70% of control, respectively. In case of α-DAC the decrease in methylation level was similar to the effect of DAC and DHDAC (80%) for CDKN2B gene while weaker effect, comparable to that of AC and DHAC (50%) was achieved in case of THBS-1.
Genomic DNA analysis confirms the hypomethylating capability of DHDAC.
Hypomethylating abilities of all tested nucleosides were verified at the level of genomic DNA to confirm that the effects of the compounds are not restricted to specific DNA sequences. The percentage of methylated 2′-deoxycytidine 5′-monophosphate in untreated control cells was set to 100%. Experiments were performed in both CCRF-CEM and HL-60 cells and the global hypomethylating activity was found to be independent on the cell type used (Fig. 3A). DAC, α-DAC, AC and DHDAC were similarly effective resulting in a 60-50% decrease in DNA methylation. Zebularine and DHAC induced only 20–25% hypomethylating effect while α-DHDAC was effective only for about 15% and it was identified as the weakest hypomethylating agent of the group. In addition, we looked at DHDAC hypomethylating efficiency depending on its concentrations used. CCRF-CEM cells were treated with 1, 10, 50 and 100 µM DHDAC for five days and HPLC analysis was then performed (Fig. 3B). Starting from 10 µM DHDAC, significant hypomethylation could be observed although the efficiency was only about 15%. A profound 40% decrease in DNA methylation was already caused by 50 µM DHDAC. Last, as we mentioned before, 1 µM DAC and 100 µM DHDAC were similarly effective resulting in a 60 and 50% decrease in DNA methylation, respectively.
Figure 3.

Changes in global DNA methylation level in leukemic cell lines treated with (A) a fixed concentration of 5-azacytidine analogs and zebularine or (B) various concentrations of DHDAC. Genomic DNA from treated and untreated cells was denaturated, hydrolyzed using nuclease P1 and subsequently injected onto HPLC column. The amount of methylated 2′-deoxycytidine 5′-monophosphate was calculated as follows: medCMP/(medCMP + dCMP) × 100 and its relative level in control cells was set to 100%. *p < 0.05 compared with untreated control cells (ANOVA).
The hypomethylating effects of DAC and DHDAC are comparable in methylation profiling array.
To further confirm the newly discovered hypomethylating activity of DHDAC we have used a DNA methylation profiling PCR array to compare the relative efficiencies of DAC and α- and β-anomers of DHDAC. This method allows investigation of the panel of 24 genes for their methylation status (see Methods). Again, both CCRF-CEM and HL-60 cell lines were used as models. The values of hyper- and intermediately methylated cytosine in untreated cells were set as 100% methylation. Hypomethylating effect of DAC was similar in both cell lines, resulting in about a 30% decrease in the methylation level (Fig. 4). DHDAC was less effective in CCRF-CEM cell line, for about 20%, whereas its activity in HL-60 cells was higher than that of DAC (40%). α-DHDAC did not induce any significant hypomethylating effect in this assay.
Figure 4.

Comparison of the hypomethylating activity of DAC and α- and β-DHDAC on 24 different genes (Methyl-Profiler PCR array). The DNA from treated and untreated CCRF-CEM and HL-60 cells was digested and quantified for methylation level using RT-PCR. Hyper- and intermediately methylated gene loci in untreated control cells were set as 100% methylation.
DHDAC is less cytotoxic than DAC.
CCRF-CEM cells were treated with the same concentrations of the compounds as used in methylation assays, i.e., 1 µM DAC and 100 µM DHDAC for three and five days to investigate the effect on the cell cycle distribution. After three days of DAC exposure, a regular distribution of cell cycle phases was still somewhat apparent (Fig. 5) but a marked increase of the proportion of the cells in the S phase and a dramatic rise of sub-G1 cell (apoptotic) population was observed. After five days of DAC exposure, G2/M phase was not detectable any more with further increase of sub-G1 phase. In contrast, in the cells treated with DHDAC we observed a cell cycle phase distribution similar to that of untreated control cells, with only a negligible amount of sub-G1 population.
Figure 5.

Effect of DAC and DHDAC on cell cycle distribution in CCRF-CEM cells. Untreated control cells and cells treated with DAC (1 µM) and DHDAC (100 µM) for 3 (A) and 5 (B) days were permeabilized and stained with PI. Cellular DNA content was analyzed by flow cytometry. Data are representative of two independent measurements.
To determine the ability of DAC and DHDAC to induce apoptosis in CCRF-CEM cells, phosphatidylserine externalization was monitored using flow cytometry. Already after three days of DAC treatment the majority of the cells (73%) were found to be in the apoptotic state and after a five-day exposure this proportion further increased to 93% (Fig. 6). On the other hand, DHDAC treatment resulted in only 28 and 34% apoptotic cells after 3 and 5 days of exposure, respectively.
Figure 6.

Apoptosis induced by DAC and DHDAC after 3 (A) and 5 (B) days exposure in CCRF-CEM. Annexin V-FITC-positive cells represents apoptotic population. Data are representative of two independent measurements.
Both DAC and DHDAC enhance THBS-1 mRNA expression.
Since hypomethylating agents such as DAC are considered to reactivate methylation-silenced genes we investigated the ability of DHDAC to increase mRNA expression of both hypermethylated genes of interest i.e., CDKN2B and THBS-1 by means of qRT-PCR. 1 µM DAC was used as a reference. Unfortunately, no CDKN2B mRNA expression was detected in neither control nor treated samples for up to 40 cycles of PCR indicating that the expression of this particular gene in CCRF-CEM cells is too low (below the detection limit of this method). On the other hand, THBS-1 mRNA was only undetectable in untreated control cells and cells treated with 1 and 10 µM DHDAC whereas 1 µM DAC, 50 and 100 µM DHDAC reproducibly decreased Ct values in a dose-dependent manner suggesting a reactivation of the THBS-1 gene (Table 1). Phospholipase A2 (PLA) and TATA-Box binding protein (TBP) were used as reference genes to normalize mRNA level between individual samples.
Table 1.
Analysis of THBS-1 mRNA expression
| Ct | |||
| THBS-1 | PLA | TBP | |
| Control | >40 | 24.8 ± 0.3 | 29.9 ± 0. 6 |
| 1 µM DAC | 35.8 ± 0.2 | 25.0 ± 0.2 | 29.8 ± 0.5 |
| 1 µM DHDAC | >40 | 24.4 ± 0.1 | 29.2 ± 0.4 |
| 10 µM DHDAC | >40 | 24.5 ± 0.1 | 29.6 ± 0.4 |
| 50 µM DHDAC | 38.8 ± 0.2 | 25.0 ± 0.3 | 29.9 ± 0.6 |
| 100 µM DHDAC | 36.9 ± 0.4 | 24.7 ± 0.1 | 29.9 ± 0.5 |
Cells were treated with 1 µM DAC and 1, 10, 50 or 100 µM DHDAC for 5 days prior to the RT-qPCR (40 cycles). PLA and TBP were used as reference genes for normalization purposes. Data represent mean Ct values ± SD out of three independent experiments.
Discussion
Aberrant DNA methylation and a consequent silencing of cancer-related genes are commonly found in human tumor cells. Inhibitors of DNA methyltransferases may represent a gentle therapeutic alternative to standard chemotherapy. They are incorporated into the DNA, reactivate methylated genes and protect them from re-methylation. Nevertheless, most of the currently used compounds suffer from significant disadvantages such as high toxicity and low stability. Therefore searching for new hypomethylating agents with an improved therapeutic window is a top-priority task.
In this study we present for the first time the hypomethylating capacity of DHDAC as a free nucleoside in a cellular model. So far, DHDAC was mostly recognized for its antiviral activity (KP-1212). It has been described to induce mutations in the HIV genome leading to G to A and A to G transitions.21,23 Importantly, its prodrug (KP-1461) exhibited only minimal toxicity in mammalian models in vitro and in vivo.21 The DHDAC prodrug was already introduced in a phase IIa clinical trial, HIV-1 infected patients were treated for 124 days and the prodrug was well tolerated.23
To disclose the hypomethylating potential of α- and β-anomer of DHDAC we used standard methylation-specific PCR method, which is the most analytically sensitive and specific assay for analyzing DNA methylation at single loci.5 We designed four primer sets for two cancer-related genes that are epigenetically silenced in CCRF-CEM cells—cyclin-dependent kinase inhibitor 2B (CDKN2B, p15) and thrombospondin-1 (THBS-1). Cyclin-dependent kinase inhibitors are involved in the regulation of cell cycle while thrombospondin-1 plays role in angiogenesis and tumorigenesis. All of the investigated 5-azacytidine nucleosides as well as zebularine were able to hypomethylate tested genes. Hypomethylating effects of DAC and DHDAC were comparable in both tested gene loci and seem to be the best hypomethylating agents of all tested. Zebularine and DHAC hypomethylated these genes only weakly, while α-DAC, AC and α-DHDAC exerted high variation between the two genes.
Due to the fact that the effect of hypomethylating agents is not specific for a set of genes, but results in a general decrease in the overall methylation level we employed an HPLC method to confirm our findings. This method provides several advantages in comparison to MSP: (1) enables to monitor changes in DNA methylation level using the whole genomic DNA and (2) allows to quantify the amount of methylated 2′-deoxycytidine 5′-monophosphate in digested DNA without the need for bisulfite modification. Both cell lines used, CCRF-CEM and HL-60, proved to be hypomethylated to the same extent by the tested compounds. Generally we can line up the compounds according to their declining hypomethylating potential as follows: DAC > α-DAC > DHDAC > AC > zebularine > DHAC > α-DHDAC.
Because 100 µM concentrations of drugs are usually not achievable in vivo, we tested the hypomethylation potential of the most promising compound of the study, DHDAC, also at lower concentrations. We found that after five days of cultivation, a concentration as low as 10 µM of DHDAC was able to hypomethylated DNA. Although this effect was only weak it proved to be reproducible and significant. Further increasing the concentration to 50 µM of DHDAC, we were able to reach a higher hypomethylating activity than 100 µM zebularine.
In agreement with Fojtová et al. we noted similar hypomethylating activity of α- and β-DAC. This can be explained by the observations of Hájek et al.33 who previously described spontaneous conversion of α-DAC to its β-anomer in the medium with the maximum reached after 6 hours of incubations. On the other hand α-DHDAC does not exert significant hypomethylating potency, which is in accordance with the fact that we did not detect any spontaneous conversion of α-DHDAC towards the corresponding β-anomer probably due to the absence of the 5,6-double bond (data not shown) and α-anomers are generally unlikely to be incorporated into the DNA.
DHDAC is not only a stable and efficient methylation inhibitor, but it has been suggested to possess a very low toxicity in human cells.21,23 To verify this, we compared the effect of DAC and DHDAC on cell cycle progression and apoptosis. Similarly to Schnekenburger et al. we observed a continuous increase of DAC toxicity in a time-dependent manner, while DHDAC, at 100 fold higher concentration did not affect the cell cycle progression. We found only a minor population of apoptotic cells, and the distribution of cell cycle phases was comparable to that of untreated control cells. Moreover, this effect did not progress in time indicating rather a reactivation of genes responsible for cellular repair mechanisms than the induction of programmed cell death as in the case of DAC. This suggestion is supported by Sheikhnejad et al.19 who presume a different mechanism of action of DHDAC and DAC in terms of inhibition of the DNMTs. Crystallographic analysis demonstrated the inability of DHDAC incorporated into the synthetic oligodeoxyribonucleotides to form covalent bond at the active site of the DNMT enzymes similarly to DAC due to the absence of the 5,6-double bond.
To confirm that the hypomethylating effect of DHDAC indeed have functional consequences in terms of gene reactivation, we performed qRT-PCR on hypermethylated genes in CCRF-CEM cells. Both genes tested, CDKN2B and THBS-1, were found to be transcriptionally silenced in untreated cells. Following their exposure to 50 or 100 µM DHDAC, THBS-1 was reactivated with similar efficiency as achieved with 1 µM DAC. Although the recorded Ct values were high in case of THBS-1 or even undetectable for CDKN2B after a five-day drug exposure, it is probable, that longer treatment would cause further increase in gene expression. High stability and low toxicity of DHDAC predestinate it to continuous treatment in order to reverse DNA hypermethylation, prevent gene remethylation and reactivate methylation-silenced genes with minimal side effects in comparison to DAC. From this point of view a relatively high dose of DHDAC required to exert its therapeutic effect may not be a critical obstacle.
In conclusion, we demonstrated that the antivirally active 5-azacytidine nucleoside DHDAC possesses a considerable hypomethylating activity while it is more stable and less toxic than DAC. This makes DHDAC a valuable tool for epigenetic research. Nevertheless, further in vitro and in vivo studies as well as clinical trials are clearly needed to verify whether DHDAC represents a feasible alternative to DAC.
Materials and Methods
Cell culture and drug treatments.
CCRF-CEM and HL-60 cells were cultivated in RPMI-1640 medium supplemented with 10% (v/v) heat-inactivated fetal calf serum, antibiotics (200 µgmL−1 of streptomycin and 200 UmL−1 of penicillin G) and 4 mM glutamine at 37°C under a humidified atmosphere containing 5% CO2. Nucleoside analogs were synthesized at IOCB according to previously published protocols: 5-azacytidine (AC),34 2′-deoxy-5-azacytidine (DAC) and its alpha anomer (α-DAC),35 5,6-dihydro-5-azacytidine (DHAC),15 2′-deoxy-5,6-dihydro-5-azacytidine (DHDAC) and its a anomer (α-DHDAC).20 Zebularine was commercially purchased (Sigma). 10 mM stock solutions in sterile H2O (5-azacytidine analogs) or DMSO (zebularine) were freshly prepared and stored at −80°C for not more than 3 days. Stock solutions were diluted in cell culture media at the concentration corresponding to their IC50 (50% reduction in cell proliferation after 3 days of growth in the drug-supplement medium). The cells were subcultured at day 3 and provided with fresh media and compounds. Cells were harvested after a five-day cultivation.
Cell proliferation and cell viability assay.
The growth inhibition IC50 concentration was determined using proliferation XTT test (Roche GmbH, Mannheim, Germany) after 72 h of cultivation in the inhibitor-supplemented media. After having received various experimental treatments 50 µL of cell suspension were mixed 1:1 with 0.4% Trypan blue solution, mixed well and counted using Countess™ Automated Cell Counter (Invitrogen). The number of viable (bright) cells that did not take up the dye was recorded for each sample.
DNA isolation.
Genomic DNA (gDNA) was isolated as previously described in reference 36, with an additional incubation with RNase T1 before DNA precipitation. The yield of extracted DNA was measured using NanoDrop® Spectrophotometer. One microgram of gDNA per sample were subjected to electrophoresis on a 0.7% agarose gel containing ethidium bromide and visualized under UV light.
Bisulfite modification, methylation-specific PCR (MSP) and quantitative MSP (qMSP) amplification.
1.5 µg of gDNA was bisulfite modified using an EpiTect® bisulfite kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol. The concentration of converted ssDNA was quantified using a NanoDrop® Spectrophotometer and 50 ng were used for PCR amplification. The primers were designed on exon-1 to distinguish between a methylated and unmethylated DNA and with respect to bisulfite modification as follows: CDKN2B methylated sense 5′-CGG TTG CGT TCG CGT TAG GCG T-3′; methylated antisense 5′-CAA CGA CCC GAA TAA TCC ACC G-3′; unmethylated sense 5′-GTT GTG TTT GAG TTG GTT TGT G-3′; unmethylated antisense 5′-TTA ATC CTC AAC AAC CAC CAA A-3′; THBS-1 methylated sense 5′-GTT GCG TTC GAG TTG GTT TGC G-3′; methylated antisense 5′-TTA ATC CTC GAC GAC CGC CGA A-3′; unmethylated sense 5′-GTT GTG TTT GAG TTG GTT TGT G-3′; unmethylated antisense 5′-TTA ATC CTC AAC AAC CAC CAA A-3′. CpG rich regions were detected using an EMBOSS CpGPlot/CpGReport/Isochore software available for free at www.ebi.ac.uk.
MSP was performed in 25 µl reactions containing 1x PCR buffer (Promega), 0.125 mM of each dNTP, 5.5 mM MgCl2, 200 nM of each primer and 1.3 U of hot start polymerase (Promega). In case of qMSP 1x SYBR® Green I was added. The thermocycling program included: an initial denaturation at 95°C for 2 min to ensure a complete reactivation of the hot start DNA polymerase, 40 cycles of denaturation at 95°C for 30 s, annealing at the indicated temperatures (THBS-1: M 69°C, U 62°C, CDKN2B: M 69°C, U 64°C) for 30 s and elongation at 72°C for 30 s.
The specificity of the amplified products was checked by agarose gel electrophoresis (standard MSP) and melting curve analysis (qMSP).
High-performance liquid chromatography analysis of methylated 2′-deoxycytidine 5′-monophosphate.
60 µg of DNA was resuspended in 50 µl redistilled H2O, heat-denaturated at 100°C for 10 min and cooled rapidly on ice for further 10 min. The denaturated DNA was mixed with nuclease P1 (Biomol, GmbH, Germany) and zinc chloride to a final concentration of 1.2 µg/µl and 1 mM, respectively and incubated at 42°C overnight. To remove any solid debris, the samples were extracted with 70% methanol at −20°C for 1 h. After centrifugation at 15,000x g at 5°C for 5 min supernatant was lyophilized and redissolved in 70 µl redistilled H2O. Aliquots of the hydrolyzed DNA were analyzed using an Aliance Waters HPLC system (996 PDA Detector, PDA Empower Pro Software, version 2) equipped with 15 cm × 4.6 mm SUPELCOSIL™ LC 18T, 3 µm reverse-phase column. A non-linear gradient of acetonitrile (0–30%) in 50 mM potassium phosphate, pH 5.1, 3 mM tetrabutylammoniumbisulfate at a flow rate 0.75 ml/min was used for the analysis. Peaks of the corresponding nucleotides dCMP, me-dCMP, dGMP, dTMP and dAMP were identified (UV-spectra library) and quantified with the aid of external standards.
DNA methylation profiling.
Screening for the methylation status of 24 gene promoters was performed using a Methyl-profiler™ DNA methylation PCR array system (SABiosciences, a Qiagen Company). One microgram of gDNA was digested with restriction enzyme according to manufacturer's protocol. The remaining DNA was quantified by real-time PCR in each individual enzyme reaction to detect the methylation status of the genes. Data were analyzed using a Hierarchical Clustering method (www.sabiosciences.com/dna_methylation_data_analysis.php, available for free) developed by the manufacturer.
Cell cycle analysis.
106 cells were washed twice with PBS and fixed with 70% ice-cold ethanol for 30 min. Fixed cells were washed in PBS, treated with RNase A (500 µg mL−1) at 37°C for 30 min and incubated with a staining solution containing 0.1% Triton X®-100 and propidium iodide (100 µg mL−1) in PBS for 1 h. Cellular DNA content was determined using a flow cytometer FACSAria (BD Biosciences, San Jose, CA).
Flow cytometric determination of apoptosis.
Apoptosis was quantified using an ApoAlert® Annexin V-FITC apoptosis kit (Clontech). Briefly, 106 cells were washed with PBS and resuspended in 0.2 mL of the Ca2+-containing binding buffer supplemented with 1 µgmL−1 of annexin V-FITC conjugate and incubated in the dark for 15 min. The cells were washed twice with the binding buffer, propidium iodide (5 gmL−1) was added and the cells were immediately subjected to flow cytometric analysis.
Quantitative RT-PCR.
Total RNA from 3 × 106 cells was extracted using RNeasy Mini isolation kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol. The purity of RNA samples was characterized by their 260/280 and 260/230 absorption ratios, which were at least 1.8. cDNA was synthesized from 0.1 µg RNA using SuperScript® VILO™ cDNA Synthesis kit (Invitrogen) according to the manufacturer's protocol. The primers sequences were as follows: THBS-1 sense 5′-TGT AGA GAT CCC TAA TCA-3′; antisense 5′-ATG CAA GCA CAG AAA AGA-3′; CDKN2B sense 5′-ATG ATG ATG GGC AGC GCC-3′; antisense 5′-AGC GTG TCC AGG AAG CCC-3′. Quantitative RT-PCR was performed in 25 µl reactions using RT2 qPCR Primer Assays (SABiosciences) according to the manufacturer's protocol employing DNA Engine Opticon® 2 thermocycler (Biorad). The thermocycling program included: an initial denaturation at 95°C for 10, 40 cycles of denaturation at 95°C for 30 s, annealing at the indicated temperatures (THBS-1 at 44°C, CDKN2B at 65°C) for 30 s and elongation at 72°C for 30 s.
Reference genes PLA and TBP to normalized mRNA level between individual samples were quantified.
Acknowledgments
This work was supported by the Project 1M0508 by the Ministry of Education, Youth and Sports of the Czech Republic. This work is a part of the research project of the Institute Z40550506. We would like to thank Mrs. K. Mullerova and D. Prášková for technical assistance.
Abbreviations
- AC
5-azacytidine
- DAC
2′-deoxy-5-azacytidine
- α-DAC
α-anomer of 2′-deoxy-5-azacytidine
- DHAC
5,6-dihydro-5-azacytidine
- DHDAC
KP-1212, 2′-deoxy-5,6-dihydro-5-azacytidine
- KP-1461
N4-heptyloxycarbonyl-5,6-dihydro-5-azacytidine, a prodrug of DHDAC
- α-DHDAC
α-anomer of 2′-deoxy-5,6-dihydro-5-azacytidine
- DNMT
DNA methyltransferase
- MSP
methylation-specific PCR
- qRT-PCR
quantitative reverse transcription PCR
- PLA
phospholipase A2
- TBP
TATA-Box binding protein
References
- 1.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaissière T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res: Re Mutat Res. 2008;659:40–48. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: The time is now. Crit Rev Oncol Hematol. 2008;68:1–11. doi: 10.1016/j.critrevonc.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Kristensen LS, Hansen LL. PCR-based method for detecting single-locus DNA methylation biomarkers in cancer diagnostics, prognostice and response to treatment. Clin Chem. 2009;55:1471–1483. doi: 10.1373/clinchem.2008.121962. [DOI] [PubMed] [Google Scholar]
- 6.Goffin J, Eisenhauer E. DNA methyltransferase inhibitors-state of the art. Ann Oncol. 2002;13:1699–1716. doi: 10.1093/annonc/mdf314. [DOI] [PubMed] [Google Scholar]
- 7.Mund C, Brueckner B, Lyko F. Reactivation of epigenetically silenced genes by DNA methyltransferase inhibitors. Epigenetics. 2006;1:7–13. doi: 10.4161/epi.1.1.2375. [DOI] [PubMed] [Google Scholar]
- 8.Šorm F, Pískala A, Cihák A, Veselý J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia. 1964;4:202–203. doi: 10.1007/BF02135399. [DOI] [PubMed] [Google Scholar]
- 9.Veselý J, Cihák A, Šorm F. Characteristics of mouse leukemic cells resistant to 5-azacytidine and 5-aza-2′-deoxycytidine. Cancer Res. 1968;28:1995–2000. [PubMed] [Google Scholar]
- 10.Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, et al. A comparison of azacytidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 2010;5:9001. doi: 10.1371/journal.pone.0009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veselý J, Cihák A. Incorporation of a potent antileukemic agent, 5-aza-2-deoxycytidine, into DNA of cells from leukemic mice. Cancer Res. 1977;37:3684–3689. [PubMed] [Google Scholar]
- 12.Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–1640. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 13.Schnekenburger M, Grandjenette C, Ghelfi J, Karius T, Foliguet B, Dicato M, et al. Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence and autophagy. Biochem Pharmacol. 2011;81:364–378. doi: 10.1016/j.bcp.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 15.Beisler JA, Abbasi MM, Kelley JA, Driscoll JS. Synthesis and antitumor activity of dihydro-5-azacytidine, a hydrolytically stable analogue of 5-azacytidine. J Med Chem. 1977;20:806–812. doi: 10.1021/jm00216a013. [DOI] [PubMed] [Google Scholar]
- 16.Carr BI, Rahbar S, Doroshow JH, Blayney D, Goldberg D, Leong L, et al. Fetal hemoglobin gene activation in a phase II study of 5,6-dihydro-5-azacytidine for Bronchogenic carcinoma. Cancer Res. 1987;47:4199–4201. [PubMed] [Google Scholar]
- 17.Vogelzang NJ, Herndon JE, Cirrincione C, Harmon DC, Antman KH, Corson JM, et al. Dihydro-5-azacytidine in malignant mesothelioma. Cancer. 1997;79:2237–2242. doi: 10.1002/(sici)1097-0142(19970601)79:11<2237::aid-cncr23>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 18.Samuels BL, Herndon JE, Harmon DC, Carey R, Aisner J, Corson JM, et al. Dihydro-5-azacytidine and cisplatin in the treatment of malignant mesothelioma. Cancer. 1998;82:1578–1584. [PubMed] [Google Scholar]
- 19.Pískala A, Cesnekovα B, Veselý J. Preparation and biological activity of 5,6-dihydro-5-azapyrimidine nucleosides. Nucleic Acids Res, Symposium Ser. 1987;18:57–60. [PubMed] [Google Scholar]
- 20.Harris KS, Brabant W, Styrchak S, Gall A, Daifuku R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 2005;67:1–9. doi: 10.1016/j.antiviral.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Murakami E, Basavapathruni A, Bradley WD, Anderson KS. Mechanism of action of a novel viral mutagenic covert nucleotide: molecular interaction with HIV-1 reverse transcriptase and host cell DNA polymerases. Antiviral Res. 2005;67:10–17. doi: 10.1016/j.antiviral.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Mullins JI, Heath L, Hughes JP, Kicha J, Styrchak S, Wong KG, et al. Mutation of HIV-1 genomes in a clinical population treated with the mutagenic nucleoside KP1461. PLoS One. 2011;6:15135. doi: 10.1371/journal.pone.0015135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheikhnejad G, Brank A, Christman JK, Goddard A, Alvarez E, Ford H, Jr, et al. Mechanism of inhibition of DNA (cytosine C5)-methyltransferases by oligodeoxyribonucleotides containing 5,6-dihydro-5-azacytosine. J Mol Biol. 1999;285:2021–2034. doi: 10.1006/jmbi.1998.2426. [DOI] [PubMed] [Google Scholar]
- 24.Fojtová M, Piskala A, Votruba I, Otmar M, Bartova E, Kovarik A. Efficacy of DNA hypomethylating capacities of 5-aza-2′-deoxycytidine and its alpha anomer. Pharmacol Res. 2007;55:16–22. doi: 10.1016/j.phrs.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Yoo CB, Cheng JC, Jones PA. Zebularine: a new drug for epigenetic therapy. Biochem Soc Trans. 2004;32:910–912. doi: 10.1042/BST0320910. [DOI] [PubMed] [Google Scholar]
- 26.Champion C, Guianvarc'h D, Sénamaud-Beaufort C, Jurkowska RZ, Jeltsch A, Ponger L, et al. Mechanistic insights on the inhibition of C5 DNA methyltransferases by zebularine. PLoS One. 2010;5:12388. doi: 10.1371/journal.pone.0012388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: A novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321:591–599. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, et al. Preferential response of cancer cells to zebularine. Cancer cell. 2004;6:151–158. doi: 10.1016/j.ccr.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 29.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst. 2003;95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- 30.Yoo CB, Chuang JC, Byun HM, Egger G, Yang AS, Dubeau L, et al. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumor in mice. Cancer Prev Res. 2008;1:233–240. doi: 10.1158/1940-6207.CAPR-07-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben-Kasus T, Ben-Zvi Z, Marquez VE, Kelley JA, Agbaria R. Metabolic activation of zebularine, a novel DNA methylation inhibitor, in human bladder carcinoma cells. Biochem Pharmacol. 2005;70:121–133. doi: 10.1016/j.bcp.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 32.Cheng JC, Weisenberger DJ, Gonzales FA, Liang G, Xu GL, Hu YG, et al. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol Cell Biol. 2004;24:1270–1278. doi: 10.1128/MCB.24.3.1270-1278.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hájek M, Votruba I, Holý A, Krecmerová M, Tlouštíová E. Alpha anomer of 5-aza-2′-deoxycytidine downregulates hTERT mRNA expression in human leukemia HL-60 cells. Biochem Pharmacol. 2008;75:965–972. doi: 10.1016/j.bcp.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 34.Pískala A, Šorm F. Nucleic acids components and their analogues. LI. Synthesis of 1-glycosyl derivatives of 5-azauracil and 5-azacytosine. Collect Czech Chem Commun. 1964;29:2060–2076. [Google Scholar]
- 35.Holý A, Otmar M, Pískala A. method of manufacturing 1-(2-deoxy-alpha-D-erythro-pentofuranosyl)-5-azacytosine. WO 2008101448 A2 20080828. [Google Scholar]
- 36.Mertlíková-Kaiserová H, Votruba I, Matoušová M, Holý A, Hájek M. Role of caspases and CD95/Fas in the apoptotic effects of a nucleoside analog PMEG in CCRF-CEM cells. Anticancer Res. 2010;30:2791–2798. [PubMed] [Google Scholar]