

Abstract
Backround
While the molecular entity responsible for the rewarding effects of virtually all drugs of abuse is known; that for ethanol remains uncertain. Some lines of evidence suggest that the rewarding effects of alcohol are mediated not by ethanol per se but by acetaldehyde generated by catalase in the brain. However, the lack of specific inhibitors of catalase has not allowed strong conclusions to be drawn about its role on the rewarding properties of ethanol. The present studies determined the effect on voluntary alcohol consumption of two gene vectors; one designed to inhibit catalase synthesis and one designed to synthesize alcohol dehydrogenase, to respectively inhibit or increase brain acetaldehyde synthesis.
Methods
The lentiviral vectors, which incorporate the genes they carry into the cell genome, were: (i) one encoding a shRNA anticatalase synthesis and (ii) one encoding alcohol dehydrogenase (rADH1). These were stereotaxically microinjected into the brain ventral tegmental area (VTA) of Wistar-derived rats bred for generations for their high alcohol preference (UChB), which were allowed access to an ethanol solution and water.
Results
Microinjection into the VTA of the lentiviral vector encoding the anticatalase shRNA virtually abolished (-94% p<0.001) the voluntary consumption of alcohol by the rats. Conversely, injection into the VTA of the lentiviral vector coding for alcohol dehydrogenase greatly stimulated (2-3 fold p<0.001) their voluntary ethanol consumption.
Conclusions
The study strongly suggests that to generate reward and reinforcement, ethanol must be metabolized into acetaldehyde in the brain. Data suggest novel targets for interventions aimed at reducing chronic alcohol intake.
Keywords: Alcoholism, Lentiviral vector, Brain catalase, Acetaldehyde, Reinforcement
It is widely accepted that some polymorphisms in genes that encode alcohol dehydrogenases (ADHs) and aldehyde dehydrogenases (ALDHs) protect individuals against alcoholism. These gene variants code for enzymes that allow the systemic accumulation of acetaldehyde when alcohol is consumed, which generates dysphoric reactions that deter their carriers from alcohol abuse (see Peng et al., 2009). It has, nevertheless, been proposed that acetaldehyde may have opposite effects in the brain; being not an aversive metabolite, but the actual molecule responsible for the rewarding and reinforcing effects of ethanol (Rodd-Henriks et al., 2002; Rodd et al., 2005; Hahn et al., 2006; Deng and Deitrich, 2008).
An important question in this context is whether blood acetaldehyde generated in the liver from ethanol enters the brain or whether acetaldehyde is generated in the brain itself. Brain is a protected organ, such that for blood acetaldehyde to enter the brain it must first enter the endothelial cells of brain capillaries, which degrade this metabolite by the action of aldehyde dehydrogenase. There is general agreement that under conditions of ethanol intoxication, little or no systemic acetaldehyde crosses into the brain (Tabakoff et al., 1976; Lindros and Hillbom, 1979; Petersen and Tabakoff, 1979; Ericksson 1977, Stowell et al., 1980). While alcohol dehydrogenase activity is not present in the brain, ethanol can be oxidized into acetaldehyde in this organ by the action of catalase (Tampier and Mardones 1979; Cohen et al., 1980; Aragon et al., 1992; Gill et al., 1992; Jamal et al., 2007) and cytochrome P4502E1 (CYP2E1) (Zimatkin et al., 2006). It is estimated that brain catalase accounts for 60-70% of acetaldehyde generated in this organ, while CYP2E1 accounts for 10-20% (Zimatkin et al., 2006; Zimatkin et al., 1998). Acetaldehyde generated in the brain is readily oxidized into acetate by aldehyde dehydrogenase, so that acetaldehyde is not detectable in extracellular microdialyzates (Jamal et al., 2007).
Rats have been shown to self-administer acetaldehyde into the cerebral ventricles, demonstrating that in the brain this metabolite has reinforcing effects (Amit et al., 1977). Further, animals bred to prefer alcohol self-administer acetaldehyde into the ventral tegmental area (VTA) at concentrations that are three-orders of magnitude lower than those needed to promote the self administration of ethanol (Rodd-Henriks et al., 2002), suggesting that in relation to reward and reinforcement ethanol might be a prodrug, of which the active form is its metabolite acetaldehyde.
When compared to other tissues, brain catalase levels are low (Haliwell, 2006), which may be surprising since the brain displays the greatest utilization of oxygen per unit tissue weight, which leads to superoxide and hydrogen peroxide formation. However, it has become clear that in the brain enzymes other than catalase are responsible for the elimination of hydrogen peroxide (Haliwell, 2006); namely most active glutathione peroxidases and peroxiredoxins (Turrens, 2003; Rhee et al., 2005).
Studies have addressed the role of catalase on a variety of central nervous system (CNS) effects of alcohol (Quertemont et al., 2004; Quertemont et al., 2005). The herbicide 3-amino-1,2,4-triazole (aminotriazol), an inhibitor of catalase, was shown to lower the voluntary intake of ethanol by rats (Aragon et al., 1992; Tampier et al., 1995). However, aminotriazole also reduced the voluntary intake of a highly preferred saccharin solution and of food (Tampier et al., 1995; Rotzinger et al., 1994), thus indicating that its effect on ethanol intake was nonspecific. Quertemont et al. (2005) reviewing the CNS effects of aminotriazol, concluded that the evidence to involve catalase in the actions of ethanol was not clear. Furthermore, Rodd et al., (2005) reported that aminotriazole did not inhibit ethanol self-administration into the VTA. Thus, to date a role for catalase in the rewarding properties of ethanol has not been established. Unraveling the contribution of catalase by gene manipulation would have major therapeutic implications, as it might be possible to specifically prevent the metabolism of ethanol into its reinforcing metabolite, without affecting other rewarding effects selected by evolution (e.g., food ingestion).
New methods of gene delivery to specifically increase or reduce gene expression have become valuable tools to elucidate major pharmacological and physiological mechanisms. Adeno-associated viral vectors can deliver genes that remain in human brain for up to one year (Feigin et al., 2007). Other viral vectors, such as the lentiviral vectors, can permanently incorporate the genes they carry into the genome of the cells they infect (Hu and Pathak, 2000). Both types have high efficiency and low immunogenicity. The newly developed self-inactivating lentiviral vectors are safe and most valuable for the delivery of genes to neurons (Kitagawa et al 2007).
The present studies used gene delivery technology to investigate the role in the brain of two enzymes able to oxidize ethanol into acetaldehyde. Two self-inactivating lentiviral vectors were generated: (i) one that encodes a shRNA against catalase synthesis and, (ii) one that encodes liver alcohol dehydrogenase (ADH1). These were microinjected into the VTA where dopamine cell bodies are located, projecting axons to the nucleus accumbens, thus releasing dopamine upon stimulation.
Materials and Methods
Generation of lentiviral vectors
A lentiviral vector expressing a rat catalase-targeting shRNA driven by the human U6 promoter (The RNAi Consortium, Broad Institute of MIT and Harvard TRCN0000120679) was packaged with the pPack Lentivector Packaging System (System Biosciences, Mountain View, CA) in HEK 293T cells, according to the manufacturer protocols. The lentiviral vector coding for rat ADH1 was constructed cloning the rat ADH1 cDNA sequence (Rivera-Meza et al., 2010) in pCDH1 (System Biosciences, Mountain View, CA) under the control of CMV promoter. The control viruses were generated from the same vectors but containing no shRNA or cDNA sequences. After lipofection of the vectors along with three other plasmids that code for the viral proteins GAG, Rev and VSV-G, the viruses were concentrated from the cell culture supernatant with 10% polyethylene glycol (PEG-8000, Sigma-Aldrich). After centrifugation to precipitate viral particles, these were resuspended in PBS plus 1% bovine serum albumin. The viruses were titrated by infection of HEK 293T cells, seeded in 35 mm dishes, with several viral dilutions. Forty eight hours post-infection, cellular genomic DNA was isolated with Trizol Reagent (Invitrogen, Carlsbad, CA) and quantitative PCR was performed to estimate the amount of viral genomes integrated into cellular DNA. The primers hybridize to the lentiviral sequences (forward primer: 5′-CAGCGCCCGACCGAAAGGAG-3′ for the shRNA anticatalase vector; forward primer: 5′-CTTCGCCTTCGCCCTCAGAC-3′ for pCDH1 and reverse primer: TGGTCTAACCAGAGAGACCCAGTA for both vectors). In parallel, qPCR reactions were also performed using lentiviral vector plasmid DNA of known concentration and the absolute amount of infective virus was calculated.
Inhibition of catalase activity in vitro by anticatalase-Lenti-shRNA
HEK 293 cells, which do not normally express the catalase gene, were seeded in 35 mm dishes at 50% of confluence and were lipofected with 0.1 μg of a plasmid that codes for rat catalase under the CMV promoter. Sixteen hours after transfection, medium was replaced and anticatalase-Lenti-shRNA, or control-Lenti was added (multiplicity of infection= 1 vector per cell). Three days post-infection, catalase activity was measured with a Catalase Assay Kit (Biovision, Mountain View, CA). The anticatalase-Lenti-shRNA vector reduced by 83% (n=3 p<0.001) the activity of catalase in the cells.
Alcohol dehydrogenase activity coded by Lenti-ADH vector
HEK 293 cells display a minimal alcohol dehydrogenase activity. Three days after transduction with the Lenti-ADH the activity of the cells increased over10-fold (two separate experiments), an activity that was fully inhibited by pyrazole, a known ADH inhibitor.
Intracerebral administration of lentiviral vectors
The studies were performed in Wistar-derived rats (UChB) bred for over 80 generations to ingest ethanol solutions in preference to water (Mardones and Segovia-Riquelme, 1983; Quintanilla et al., 2006). Naive UChB female rats (approximately 200g) were anesthetized with a mixture of air and isoflurane administered by a mask fitted over the nose of the animal, and placed in a Kopf stereotaxic frame with the skull oriented according to the atlas of Paxinos and Watson (1986). The skull was exposed and a 2μl Hamilton syringe with a conic tip (diameter at the insertion tip less than 0.2 mm), filled with lentiviral vectors expressing a rat anticatalase shRNA or rat ADH or the corresponding control vectors. The syringe was inserted into the left VTA (coordinates: B-5.2; L-0.8; V-7.2, from the dura mater). Two minutes after syringe implantation, 1 μl of the corresponding solution (anticatalase-Lenti-shRNA 8×104 virus/μl; or Lenti-ADH, 8×104, virus/μl, or the corresponding controls) was infused at the rate of 0.4μl/min. The syringe was kept in place for an additional 2min period, before removing it slowly. The skin was then sutured, and the rat left to recover in the surgery station before being transferred to individual cages at the animal station, with access to water and food ad libitum. Four days after the surgery, the rats were exposed 24 hours a day to a drinking paradigm, consisting of free availability to two bottles, one containing 10% or 5% (v/v) ethanol, as indicated, and the other containing distilled water only until the end of the experiments (30 to 50 days as indicated). Ethanol consumption was determined on a daily basis and expressed as g ethanol/kg body weight/ day. Following the drinking determinations rats were used for in vivo microdialysis and/or histochemistry.
In vivo Microdialysis
Approximately two months after the intracerebral administration of the anticatalase- Lenti- shRNA or control Lenti, animals were anaesthetized as above and stereotaxically implanted with a microdialysis probe (dialyzing length: 2 mm; diameter: 0.25 mm) (dialysis membrane, cat. 0318; Cuprophan, Idemsa, Spain) into the nucleus accumbens (shell) (coordinates: B1.7; L-0.7; V-8.2). The probe was fixed to the skull with dental acrylate anchored by two screws. The microdialysis experiment per se was carried out in awake animals two days after implantation in a microdialysis arena (34×40mm) equipped with a perfusion setup including a liquid swivel (CMA/Microdialysis AB, Stockholm, Sweden). A two hours perfusion period (artificial cerebrospinal fluid, aCSF, pH∼7, 2 μl/min) elapsed before starting sample collection (60 μl, using a microfraction collector CMA 140, CMA/Microdialysis AB, Stockholm, Sweden), assayed immediately for dopamine by HPLC-ED, according to Bustamante et al. (Bustamante et al., 2008). One hundred and twenty (120) min after the beginning of the microdialysis experiment, a bolus of 1g/kg i.p of ethanol (20%) was administered and further microdialysis samples were collected every 30 minutes. As previously reported (Imperato and di Chiara, 1986), systemic ethanol administration produced a significant increase in dopamine overflow in nucleus accumbens of animals treated with a control-Lenti probe. Three hours after ethanol administration, 100 μM of D-amphetamine diluted in the aCSF was perfused via the probe for 30 min (300-330 min period, after the beginning of the microdialysis experiment). Three subsequent aCSF alone samples were taken, and then 100 mM KCl was added to the perfusion medium to induce K+-depolarisation (390-420 min period). Changes of the perfusion medium were performed with a syringe selector (model CMA 111, CMA/Microdialysis AB, Stockholm, Sweden).
Immunohistochemistry
At the end of the experiments, rats were deeply anaesthetized with chloral hydrate (400 mg/kg i.p.) and perfused via the heart with 100 ml of 0.1M of PBS (pH 7.4), followed by 200 ml formalin solution (4% paraformaldehyde, PF; Sigma, in 0.1 M of PBS, pH 7.4). The brain was removed from the skull, post-fixed in a formalin solution overnight, and immersed in 30% sucrose in 0.1 M of PBS at 4°C for 2-3 days. Then, the tissue was embedded in cryomatrix (Thermo Electron Corp, Pittsburgh, PA) and stored at -70°C. Coronal sections (20 μm thick) were sliced and processed for immunocytochemistry (Morales et al., 2008). After rinsing cycles, endogenous peroxidase activity was blocked with 1% H2O2 for thirty min and rinsed again with PBS. The tissue was preincubated with 2% of bovine serum albumin (BSA) (Calbiochem, San Diego, CA), 0.3% triton X-100, in PBS, for 1 h at 37°C, and incubated for 72h with a mouse monoclonal antibody against tyrosine hydroxylase antibody (Sigma, St. Louis, MO, USA) (dilution 1:1000, 2% BSA, PBS/0.5% triton X-100). After rinsing, the slices were processed using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA), according to the instructions of the manufacturer, visualizing the reaction with Vector Nova Red (Vector Laboratories, Burlingame, CA). The sections were dehydrated through graded alcohols, cleared in xylene and coverslipped in Entellan mounting medium (Merck, Darmstadt, Germany) and examined by transmission microscopy (Morales et al., 2008). The tip of the cannula and the volume of injection (1 μl) cannula was on the left VTA, not differentiating between anterior or posterior VTA as reported by Rodd et al. (2005), who could differentiate between the two regions when administering pulses of 100 nl of ethanol. Nevertheless, when assaying the injection site in TH-labelled section, it was observed that it could be re-constructed in an area equivalent to that reported by Rodd et al (Bregma -5.0 to -5.8), but without any sign of TH-cell impairment as indicated by arrows heads comparing TH-positive cells on the right (control) and left (injected) site in a section from an animal injected with the anticatalase Lenti-shRNA (1 μl). However, the tract of the needle could be observed, always about the targeted tissue (arrows in Fig 3).
Figure 3. Immunocytochemistry against tyrosine hydroxylase (TH) of a coronal mesencephalic section.
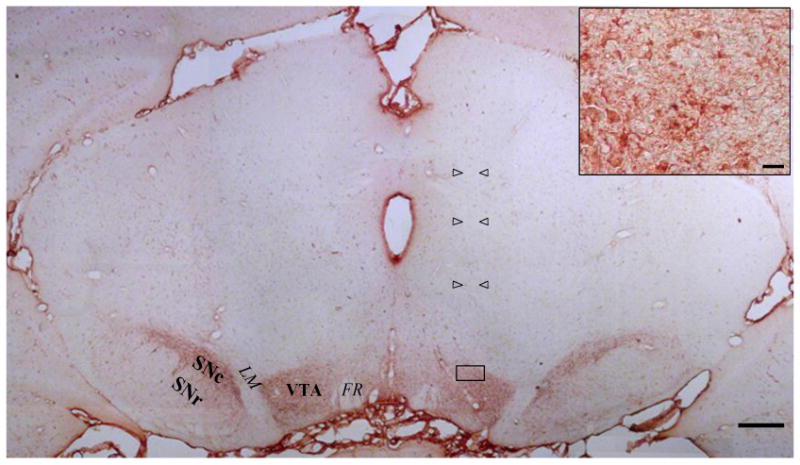
Rats were injected with a 1μl solution containing anticatalase-Lenti-shRNA (8×104 virus/μl) into the ventral tegmental area (coordinates: B-5.2; L-0.8; V-7.2), two months after the stereotaxic injection. Open arrow heads show the tract left by the injection cannula. VTA, ventral tegmental area; SNc, substantia nigra, pars compact; SNr, substantia nigra, pars reticulata; FR, fasciculus retroflexus; LM, medial lemniscus. Scale bar= 500μm. Inset: Magnification of the area within the rectangle, showing normal morphology of TH-positive cells in the vicinity of the tip of the injection cannula; scale bar= 20μm.
Results and Discussion
The in vitro effect of the lentiviral vectors developed to lower catalase activity or increase alcohol dehydrogenase activity is presented in Methods. One microliter of the gene-coding lentiviral vectors or their control vectors (8 ×104 viral particles) was microinjected into the VTA of naïve rats and 4 days later the animals were placed in separate cages and offered continuous access to fluid in two bottles, one containing an alcohol solution (either 5% or 10% as indicated) and one containing distilled water (shown as consumption Day 1 in Figures below).
UChB animals offered 10% ethanol and water to drink for 30-50 days on a 24 hour basis, consume and metabolize 7-8 g ethanol/kg/day (the equivalent of 500 g of pure ethanol/70 kg) (30). Figure 1A shows that the administration of the anticatalase-Lenti-shRNA into the VTA greatly reduced alcohol drinking. Fifty days after the administration of anticatalase-Lenti-shRNA, the inhibition of ethanol intake was 94% (p<0.001). Along the complete 50-day study the inhibition of alcohol intake was of the order of 85% (p<0.0001). This small difference may relate to an initial gene expression in the episomal state of the DNA generated by reverse transcription (see Haas et al 2000) and the expression that follows its permanent transduction into the genome, with a brief period of reduced expression between the two processes. Body weights and the relative growth of animals were virtually identical along treatment for both the control and the shRNA treated animals, (Figure 1B). Also, there were no differences in water intake, indicating that the effect was specific for ethanol consumption, and after discontinuing the ad-libitum availability of ethanol, several behavioral and neurological parameters scored (e.g., grooming, rearing, leg tapping, sidedness) were unaltered in the animals in the anticatalase-Lenti-shRNA treated animals versus control-Lenti animals (data not shown).
Figure 1. Anticatalase-Lentiviral vector administration into the ventral tegmental area reduces voluntary alcohol intake in rats bred as alcohol drinkers.

(A) Rats significantly (p<0.001) reduced their alcohol intake when injected with a lentiviral vector coding for a shRNA against catalase (Anticatalase-Lenti) into the VTA (n=10) compared to that after the injection of an empty lentiviral vector (control-Lenti) (n=10). Animals were allowed free availability of 10% (v/v) ethanol and water four days after the administration of the lentiviral vectors (B). No differences in body weight were observed along the experiment when Anticatalase-treated rats were compared to controls, indicating a good general health of the animals. Abscissa: days of ethanol availability. Deviations shown are S.E.M.
The marked inhibition of voluntary ethanol intake shown in animals administered the anticatalase-Lenti –shRNA vector into the VTA prompted us to determine if such a reduction in ethanol intake had a parallel in reducing the release of dopamine in the nucleus accumbens induced by the acute administration of ethanol (1g/kg i.p). It has been previously shown (Diana et al., 2008) that acetaldehyde administration into the VTA leads to the release of dopamine in the nucleus accumbens. Thus, it was postulated that animals treated with control-Lenti would respond to ethanol releasing dopamine in nucleus accumbens, while such an effect would be obliterated or greatly reduced in animals treated with anticatalase-Lenti-shRNA. Indeed, while the acute administration of ethanol to animals treated with control-Lenti resulted (Figure 2A) in significantly increases in dopamine outflow in nucleus accumbens versus its basal level (152 ± 10 %; p<0.05), such an alcohol-induced increase virtually disappeared in anticatalase-Lenti-shRNA treated animals (107 ± 2 %; N.S.). To determine the physiological intactness of the dopamine release system we perfused the nucleus accumbens with D-amphetamine (10-4 M). Dopamine levels in microdialyzate were increased by D-amphetamine both in control-Lenti treated animals (301 ± 62 %) and also in anticatalase-Lenti-shRNA treated animals (546 ± 84%), indicating that the mechanism of dopamine release from a de novo synthesis pool was intact (Figure 2B). The slightly greater, although not significant, release of dopamine observed in anticatalase-Lenti-shRNA treated animals during D-amphetamine perfusion may be due to the lower basal release of dopamine seen when ethanol was administered prior to the amphetamine perfusion (see Methods). To assess the release of dopamine via a calcium-dependent vesicular release, KCl (0.1M) was infused in the microdialysis fluid. Dopamine levels in the dialyzate were increased by KCl both in the control-Lenti (207± 50%) and the anticatalase-Lenti-shRNA treated animals (277± 55%; N.S. between lentiviral treatment), also indicating that dopaminergic innervation was unaffected by the anticatalase vector (Figure 2C). Further, the administration of the anticatalase vector did not induce histological changes in dopaminergic VTA neurons, as shown by immunohistochemical examination based on an anti-tyrosine hydroxylase monoclonal antibody (Figure 3).
Figure 2. Anticatalase-Lentiviral vector administration into the ventral tegmental area.

(A) Inhibition by anticatalase-lentiviral vector of dopamine efflux into the microdialysis fluid of nucleus accumbens (shell) induced by the systemic administration of ethanol (1g/kg i.p.). (B) Anticatalase-lentiviral vector does not affect dopamine efflux into the microdialysis fluid of nucleus accumbens (shell) induced by D-amphetamine (0.1mM) addition to the microdialysis fluid. (C) Anticatalase-lentiviral vector does not affect dopamine efflux into the microdialysis fluid of nucleus accumbens (shell) induced by KCl (100 mM) addition to the microdialysis fluid (n=5 to 7). Respective control values (100%) ranged 0.57 to 0.82 nM dopamine and were not influenced by the viral vectors.
In order to determine the effect of the administration into the VTA of (ectopic) liver alcohol dehydrogenase we lowered the spontaneous preference for ethanol by reducing the concentration of ethanol offered to 5%, rather than the 10% offered in the earlier study. As seen in Figure 4A, the consumption of ethanol in controls offered ethanol at 5% was about one half of that seen at 10% (see also Fig 1A). However, in as little as 10 days, animals treated with Lenti-ADH increased their alcohol intake by 2 to 3 fold (p<0.0001) versus their respective control. Their intakes tended to stabilize at 15 days, being still 25-30% higher at the end of the 30-day study. It has previously been reported that CYP2E1, which is also able to metabolize ethanol, is elevated in the VTA following chronic ethanol intake (Sanchez-Catalán et al., 2008), which may generate additional acetaldehyde and promote the near equalization of ethanol intakes after prolonged periods in animals treated with Lenti-ADH versus Control-Lenti. As also seen in the previous anticatalase-Lenti-shRNA study, lentiviral-ADH administration did not alter animal body weights or relative growth (Fig 4B). It is noteworthy that the systemic transduction of the alcohol dehydrogenase gene (ADH1) by a vector that targets the liver, but does not penetrate the brain, results in a clear reduction of alcohol intake by the animals (Rivera-Meza et al 2010), consistent with the view that in the periphery, unlike in brain, an increased metabolism of ethanol is aversive rather than rewarding.
Figure 4. Induction of alcohol dehydrogenase synthesis increases voluntary alcohol intake in rats bred as alcohol drinkers.

(A) Rats significantly (p<0.001) increase their alcohol intake when injected with a lentiviral vector encoding alcohol dehydrogenase (ADH) (Lenti-ADH) into the ventral tegmental area (n=7), compared to that observed after treatment with an empty lentiviral vector (control-Lenti) (n=7) (A). Four days after the injection of the lentiviral vectors, animals were allowed free availability of 5% (v/v) ethanol and water. (B) No differences were observed in body weight along the experiment when Lenti-ADH-treated rats were compared versus control-empty-Lenti virus-treated rats. Abscissa: days of ethanol availability. Deviations shown are S.E.M.
It is also noted that both in the 5% and 10% ethanol access studies, animals receiving the control vectors increased their alcohol intake as they consumed ethanol, akin to tolerance, reaching a plateau only at the 5% concentration. It is suggested that such an effect may relate to the learning process in which the rewarding effects of acetaldehyde lead to a reinforced behavior, which increases ethanol intake.
Overall, the studies presented strongly suggest that in relation to reward and reinforcement, ethanol acts as a prodrug, which gives rise to acetaldehyde, the active drug. This concept should allow new therapeutic avenues to reduce chronic alcohol use, whether by inhibiting the generation of brain acetaldehyde or by reducing its levels by sequestration or by increasing its degradation.
Acknowledgments
We thank Fondecyt 1095021; 1080447, the Millennium Scientific Initiative, P05 001-F and NIAAA R01AA0142 for financial support, Dr. Harold Kalant for editorial advice and Mr. Juan Santibáñez and Mrs. Carmen Almeyda to their technical help.
References
- Amit Z, Brown ZW, Rockman GE. Possible involvement of acetaldehyde, norepinephrine and their tetrahydroisoquinoline derivatives in the regulation of ethanol self-administration. Drug Alcohol Depend. 1977;2:495–500. doi: 10.1016/0376-8716(77)90049-7. [DOI] [PubMed] [Google Scholar]
- Aragon CM, Amit Z. The effect of 3-amino-1,2,4-triazole on voluntary ethanol consumption: evidence for brain catalase involvement in the mechanism of action. Neuropharmacol. 1992;31:709–712. doi: 10.1016/0028-3908(92)90150-n. [DOI] [PubMed] [Google Scholar]
- Aragon CM, Rogan F, Amit Z. Ethanol metabolism in rat brain homogenates by a catalase-H2O2 system. Biochem Pharmacol. 1992;44:93–98. doi: 10.1016/0006-2952(92)90042-h. [DOI] [PubMed] [Google Scholar]
- Bustamante D, Quintanilla ME, Tampier L, Gonzalez-Lira V, Israel Y, Herrera-Marschitz M. Ethanol induces stronger dopamine release in nucleus accumbens (shell) of alcohol-preferring (bibulous) than in alcohol-avoiding (abstainer) rats. Eur J Pharmacol. 2008;591:153–158. doi: 10.1016/j.ejphar.2008.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen G, Sinet PM, Heikkila R. Ethanol oxidation by rat brain in vivo. Alcohol Clin Exp Res. 1980;4:366–370. doi: 10.1111/j.1530-0277.1980.tb04833.x. [DOI] [PubMed] [Google Scholar]
- Deng XS, Deitrich RA. Putative role of brain acetaldehyde in ethanol addiction. Curr Drug Abuse Rev. 2008;1:3–8. doi: 10.2174/1874473710801010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana M, Peana AT, Sirca D, Lintas A, Melis M, Enrico P. Crucial role of acetaldehyde in alcohol activation of the mesolimbic dopamine system. Ann NY Acad Sci. 2008;1139:307–317. doi: 10.1196/annals.1432.009. [DOI] [PubMed] [Google Scholar]
- Eriksson CJ. Acetaldehyde metabolism in vivo during ethanol oxidation. Adv Exp Med Biol. 1977;85A:319–341. doi: 10.1007/978-1-4899-5181-6_21. [DOI] [PubMed] [Google Scholar]
- Feigin A, Kaplitt MG, Tang C, Lin T, Mattis P, Dhawan V, During MJ, Eidelberg D. Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson's disease. Proc Natl Acad Sci USA. 2007;104:19559–9564. doi: 10.1073/pnas.0706006104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill K, Menez JF, Lucas D, Deitrich RA. Enzymatic production of acetaldehyde from ethanol in rat brain tissue. Alcohol Clin Exp Res. 1992;16:910–915. doi: 10.1111/j.1530-0277.1992.tb01892.x. [DOI] [PubMed] [Google Scholar]
- Hahn CY, Huang SY, Ko HC, Hsieh CH, Lee IH, Yeh TL, Yang YK, Lee JF, Lin WW, Lu RB. Acetaldehyde involvement in positive and negative alcohol expectancies in Han Chinese persons with alcoholism. Arch Gen Psychiatry. 2006;63:817–823. doi: 10.1001/archpsyc.63.7.817. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Haas DL, Case SS, Crooks GM, Kohn DB. Critical factors influencing stable transduction of CD34+ cells with HIV-1 derived lentiviral vectors. Mol Therapy. 2000;2:78–80. doi: 10.1006/mthe.2000.0094. [DOI] [PubMed] [Google Scholar]
- Hu WS, Pathak VK. Design of retroviral vectors and helper cells for gene therapy. Pharmacol Rev. 2000;52:493–511. [PubMed] [Google Scholar]
- Imperato A, Di Chiara G. Preferential stimulation of dopamine release in the nucleus accumbens of freely moving rats by ethanol. J Pharmacol Exp Ther. 1986;239:219–228. [PubMed] [Google Scholar]
- Jamal M, Ameno K, Uekita I, Kumihashi M, Wang W, Ijiri I. Catalase mediates acetaldehyde formation in the striatum of free-moving rats. Neurotoxicology. 2007;28:1245–1248. doi: 10.1016/j.neuro.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Kitagawa R, Miyachi S, Hanawa H, Takada M, Shimada T. Differential characteristics of HIV-based versus SIV-based lentiviral vector systems: Gene delivery to neurons and axonal transport of the expressed gene. Neurosci Res. 2007;57:550–558. doi: 10.1016/j.neures.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Lindros KO, Hillbom ME. Acetaldehyde in cerebrospinal fluid: its near-absence in ethanol-intoxicated alcoholics. Med Biol. 1979;57:246–247. [PubMed] [Google Scholar]
- Mardones J, Segovia-Riquelme N. Thirty two years of selection of rats by ethanol preference: UChA and UChB strains. Neurobehav Toxicol Teratol. 1983;5:171–178. [PubMed] [Google Scholar]
- Morales P, Fiedler JL, Andrés S, Berrios C, Huaiquín P, Bustamante D, Cardenas S, Parra E, Herrera-Marschitz M. Plasticity of hippocampus following perinatal asphyxia: effects on postnatal apoptosis and neurogenesis. J Neurosci Res. 2008;86:2650–2662. doi: 10.1002/jnr.21715. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 1986. [DOI] [PubMed] [Google Scholar]
- Peng GS, Yin SJ. Effect of allelic variants of aldehyde dehydrogenase ALDH2*2 and alcohol dehydrogenase ADH1B*2 on blood acetaldehyde concentrations. Hum Genomics. 2009;3:121–127. doi: 10.1186/1479-7364-3-2-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen DR, Tabakoff B. Characterization of brain acetaldehyde oxidizing systems in the mouse. Drug Alcohol Depend. 1979;4:137–144. doi: 10.1016/0376-8716(79)90054-1. [DOI] [PubMed] [Google Scholar]
- Quertemont E, Tambour S. Is ethanol a pro-drug? The role of acetaldehyde in the central effects of ethanol. Trends Pharmacol Sci. 2004;25:130–134. doi: 10.1016/j.tips.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Quertemont E, Tambour S, Tirelli E. The role of acetaldehyde in the neurobehavioral effects of ethanol: a comprehensive review of animal studies. Prog Neurobiol. 2005;75:247–274. doi: 10.1016/j.pneurobio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Quintanilla ME, Israel Y, Sapag A, Tampier L. The UChA and UChB rat lines: metabolic and genetic differences influencing ethanol intake. Addict Biol. 2006;11:310–323. doi: 10.1111/j.1369-1600.2006.00030.x. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Rivera-Meza M, Quintanilla ME, Tampier L, Mura CV, Sapag A, Israel Y. Mechanism of Protection against alcoholism by an alcohol dehydrogenase polymorphism: development of an animal model. FASEB J. 2010;24:266–274. doi: 10.1096/fj.09-132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodd ZA, Bell RL, Zhang Y, Murphy JM, Goldstein A, Zaffaroni A, Li TK, McBride WJ. Regional heterogeneity for the intracranial self-administration of ethanol and acetaldehyde within the ventral tegmental area of alcohol-preferring (P) rats: involvement of dopamine and serotonin. Neuropsychopharmacology. 2005;30:330–338. doi: 10.1038/sj.npp.1300561. [DOI] [PubMed] [Google Scholar]
- Rodd-Henriks ZA, Melendez RI, Zaffaroni A, Goldstein A, McBride WJ, Li TK. The reinforcing effects of acetaldehyde in the posterior ventral tegmental area of alcohol preferring rats. Pharmacol Biochem Behav. 2002;72:55–64. doi: 10.1016/s0091-3057(01)00733-x. [DOI] [PubMed] [Google Scholar]
- Rotzinger S, Smith BR, Amit Z. Catalase inhibition attenuates the acquisition of ethanol and saccharin-quinine consumption in laboratory rats. Behav Pharmacol. 1994;5:203–209. doi: 10.1097/00008877-199404000-00012. [DOI] [PubMed] [Google Scholar]
- Sánchez-Catalán MJ, Hipólito L, Guerri C, Granero L, Polache A. Distribution and Differential Induction of CYP2E1 by Ethanol and Acetone in the Mesocorticolimbic System of Rat. Alcohol Alcohol. 2008;43:401–407. doi: 10.1093/alcalc/agn012. [DOI] [PubMed] [Google Scholar]
- Stowell A, Hillbom M, Salaspuro M, Lindros KO. Low acetaldehyde levels in blood, breath and cerebrospinal fluid of intoxicated humans as assayed by improved methods. Adv Exp Med Biol. 1980;132:635–645. doi: 10.1007/978-1-4757-1419-7_66. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Anderson RA, Ritzmann RF. Brain acetaldehyde after ethanol administration. Biochem Pharmacol. 1976;25:1305–1309. doi: 10.1016/0006-2952(76)90094-0. [DOI] [PubMed] [Google Scholar]
- Tampier L, Mardones J. Catalase mediated oxidation of ethanol by rat brain homogenates. IRCS Medical Science. 1979;7:389. [Google Scholar]
- Tampier L, Quintanilla ME, Mardones J. Effects of aminotriazole on ethanol, water, and food intake and on brain catalase in UChA and UChB rats. Alcohol. 1995;12:341–344. doi: 10.1016/0741-8329(95)00014-i. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(part 2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimatkin SM, Liopo AV, Deitrich RA. Distribution and kinetics of ethanol metabolism in rat brain. Alcohol Clin Exp Res. 1998;22:1623–1627. [PubMed] [Google Scholar]
- Zimatkin SM, Pronko SP, Vasiliou V, Gonzalez FJ, Deitrich RA. Enzymatic mechanisms of ethanol oxidation in the brain. Alcohol Clin Exp Res. 2006;30:1500–1505. doi: 10.1111/j.1530-0277.2006.00181.x. [DOI] [PubMed] [Google Scholar]