

Abstract
We systematically generated large-scale data sets to improve genome annotation for the nematode Caenorhabditis elegans, a key model organism. These data sets include transcriptome profiling across a developmental time course, genome-wide identification of transcription factor–binding sites, and maps of chromatin organization. From this, we created more complete and accurate gene models, including alternative splice forms and candidate noncoding RNAs. We constructed hierarchical networks of transcription factor–binding and microRNA interactions and discovered chromosomal locations bound by an unusually large number of transcription factors. Different patterns of chromatin composition and histone modification were revealed between chromosome arms and centers, with similarly prominent differences between autosomes and the X chromosome. Integrating data types, we built statistical models relating chromatin, transcription factor binding, and gene expression. Overall, our analyses ascribed putative functions to most of the conserved genome.
Complete genome sequences provide a view of the full instruction set of an organism. However, understanding the functional content of a genome requires more than DNA sequence. To address this need, in 2003 the U.S. National Human Genome Research Institute (NHGRI) initiated the Encyclopedia of DNA Elements (ENCODE) project in order to study the human genome in greater depth (1). Recognizing the importance of well-annotated model genomes, in 2007 the NHGRI initiated the model organism ENCODE (modENCODE) project on Caenorhabditis elegans and Drosophila melanogaster so as to systematically annotate the functional genomic elements in these organisms (2).
Given its intermediate complexity between single-celled eukaryotes and mammals, C. elegans offers an outstanding system for studies of genome organization and function. C. elegans was the first multicellular organism with a fully defined cell lineage, a nervous system reconstructed through serial-section electron microscopy, and a sequenced genome (3–5). Its 100.3-Mb genome is only about eight times larger than that of S. cerevisiae, and yet it contains almost as many genes as a human and all of the information necessary to specify the major tissues and cell types of metazoans.
From the project start in 2007 (2), the C. elegans modENCODE groups had by February 2010 collected 237 genome-wide data sets (table S1) bearing on gene structure, RNA expression profiling, chromatin structure and regulation, and evolutionary conservation. To ensure the completeness and standardization of modENCODE data, all data sets were submitted to the modENCODE Data Coordinating Center; hand curated with extensive, structured metadata; validated for completeness; and checked for consistency before release at www.modencode.org.
Analyses of these data reveal (i) directly supported protein-coding genes containing 5′ and 3′ ends and alternative splice junctions; (ii) sets of noncoding RNAs, including RNAs belonging to known classes and previously unknown types; (iii) gene expression and transcription factor (TF)–binding profiles across developmental stages; (iv) genomic locations bound by many of the TFs analyzed, designated as HOT (high-occupancy target) regions; (v) a hierarchy of candidate regulatory interactions among TFs and its relationship to the network of microRNAs (miRNAs) and their targets; (vi) differences in histone modifications and nuclear-envelope interactions between the centers and arms of autosomes and between autosomes and the X chromosome; (vii) evidence for chromatin-mediated epigenetic transmission of the memory of gene expression from adult germ cells to embryos; and (viii) predictive models that relate chromatin state to TF-binding sites and to expression levels of protein- and miRNA-encoding genes.
The summation of features annotated through these functional data sets provides a potential explanation for most of the conserved sequences in the C. elegans genome and lays the foundation for further study of how the genome of a multicellular organism accurately directs development and maintains homeostasis.
The Transcriptome
Accurate and comprehensive annotation of all RNA transcripts (the transcriptome) provides a framework for interpreting other genomic features, such as TF-binding sites and chromatin marks. At the project's inception [WS170; WormBase versions used for specific analyses can be found in (6)], the C. elegans genome lacked direct experimental support for about one third of predicted splice junctions, and some of these predictions were erroneous (7, 8). Many genes lacked transcript start sites and polyadenylate [poly(A)] addition sites; exons and even whole genes were missing. To address these deficiencies, cDNA-based evidence was obtained through high-throughput sequencing (RNA-seq), reverse transcription polymerase chain reaction (RT-PCR)/rapid amplification of cDNA ends (RACE), and tiling arrays from a variety of stages, conditions, and tissues (tables S1, S3, and S4). Analysis of the data yielded previously unrecognized protein-coding genes, refined the structure of known protein-coding genes, revealed the dynamics of expression and alternative splicing, provided evidence of pseudogene transcription, and suggested previously unknown noncoding RNAs (ncRNAs). Through mass spectrometry, we verified predicted proteins and distinguished short single-exon protein-coding transcripts from ncRNAs.
Protein-coding genes
We used RNA-seq to generate more than 1 billion uniquely aligned short sequence reads from 19 different nematode populations, including all major developmental stages (embryonic, larval, dauer, and adult), embryonic and late L4 males, animals exposed to pathogens, and selected mutants (fig. S3) (9, 10). Data sets targeting the 3′ ends of poly(A)-plus transcripts were also collected, and additional sequence tags representing polyadenylated 3′ ends that were acquired by using 3P-Seq [poly(A)-position profiling by sequencing] were made available to the consortium (11, 12).
RNA-seq reads were mapped exhaustively and, together with the 3P-Seq data, allowed us to detect with nucleotide resolution features of protein-coding genes independently of previous WormBase models (fig. S7). The number of confirmed splice junctions increased from 70,028 at project start to 111,786, with 8174 of these not previously represented in WormBase (Fig. 1A and fig. S8). The number of genes with a trans-spliced leader (either Spliced Leader 1 or 2) at the 5′ end increased from 6012 to 12,413, covering 20,515 different trans-spliced transcript start sites (TSSs), and the number of poly(A) sites associated with genes increased from 1330 to 28,199 (table S2A) (13). RT-PCR/RACE and mass spectrometry provided direct support for 40,114 splice junctions (6). About 95% of these overlapped with those detected with RNA-seq, providing independent support for 37,830 of these features (fig. S9). In addition, mass spectrometry proved that of 359 tested, 73 single-exon genes produced protein.
Fig. 1.
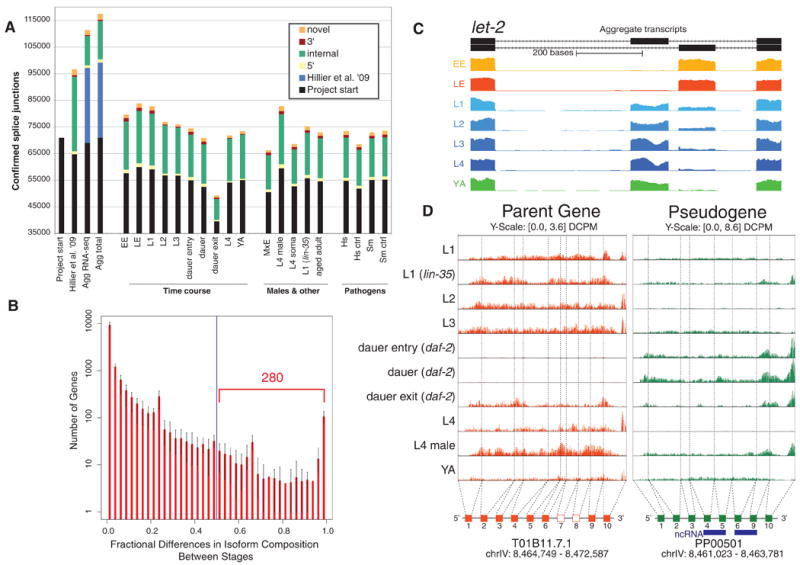
Transcriptome features and alternative splicing. (A) Bar graphs indicate the number of confirmed splice junctions categorized by type. The leftmost bars show the progression from project start (6) to the aggregate integrated transcript set. The three other groups provide data for the various developmental stages, males, mutants, and populations exposed to pathogens. Specific sample names are described in table S3. (B) Histogram of fractional differences in isoform composition for 12,875 genes with multiple isoforms in 21 pair-wise comparisons across seven developmental stages. A fractional difference close to 1 indicates large differences in the relative composition. (C) Representative example (F01G12.5; let-2), illustrating alternative exon usage across stages. (D) Example of a differentially transcribed pseudogene creating a ncRNA. Rows are normalized signal tracks for the various developmental stages, showing the expression pattern of the parent gene (T01B11.7.1; orange) and an associated duplicated pseudogene (PP00501, green).
We used several avenues to estimate how many features of protein-coding genes remain to be supported in C. elegans. Of predicted WormBase transcripts, only 1108 (5%) do not have support through RNA-seq (table S2B). Of these, 369 are members of rapidly evolving gene families implicated in environmental response and may be nonfunctional or only expressed under specific conditions. The yield of new features discovered with additional RNA-seq samples is clearly diminishing, and features such as newly discovered exons are approaching saturation (fig. S10). Intersection of the data sets produced here with previous evidence from WormBase suggests as few as 2000 to 3000 exons (2 to 3%) remain undetected (fig. S10). However, we continue to detect rare splice-junction and spliced-leader events, particularly those associated with more abundantly expressed genes. These could be biologically important but might also result from RNA-processing errors.
Gene models
We built probable gene models from the results of transcript sequencing, allowing for multiple transcripts (isoforms) from a given region (10). These models, called genelets because they could be fragments of full genes, were initiated with the most highly represented splice junction in a region and extended in each direction so as to incorporate regions covered by above-threshold sequence reads and splice junctions (6). The model was terminated when either a transcript start or stop signal was encountered or when coverage was interrupted (fig. S5). By iterating the process, we generated alternative isoforms. We used the longest open reading frame to annotate protein-coding sequences (CDSs) and 5′ and 3′ untranslated regions (UTRs).
For each of the 19 stages and conditions, we built transcript sets purely on the basis of RNA-seq data from a given stage (stage-specific RNA-seq–only genelets), along with three aggregate sets: (i) aggregate RNA-seq–only genelets; (ii) aggregate integrated genelets, which combined RNA-seq data with available ESTs (expressed sequence tags), cDNAs, and OSTs (open reading-frame sequence tags) (7, 8, 11) as well as the RT-PCR/RACE and mass spectrometry data produced in the project; and (iii) aggregate integrated transcripts, which incorporates all evidence from “(ii)” above and allows WormBase predictions to fill small coverage gaps within exons. The last set incorporates all of the splice junctions and spliced-leader sites, as well as multiple poly(A) addition sites, and thus often contains multiple isoforms. Altogether, we generated 64,824 transcripts from 21,733 genes, as compared with 23,710 transcripts from 20,082 genes in WormBase at the project start. Our gene models, which come from direct experimental evidence, exactly match the internal splice junction pattern for 10,123 WormBase transcripts, but we provide revised 5′- or 3′UTRs for many of these. For 6418 models, the internal gene structure was unchanged from WormBase, but new 5′ or 3′ exons and associated splice junctions were added. The remaining fall into three categories: Our models overlap WormBase transcripts but differ in splice junctions (3292); they fail to cover all of the splice junctions (2235); or they are not represented in WormBase at all (1952).
Expression dynamics
To determine the dynamics of gene expression during development and in specific cell types, we analyzed tiling array data from 42 biological samples, comprising 17 different growth stages and conditions from whole animals, and 25 samples from different isolated cell and tissue types (table S3) (6). For almost all whole-animal samples, RNA-seq data were also obtained from the same or similarly prepared samples. Calibration and processing were done to facilitate the integration of sequencing and arrays for both RNA-seq and for chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-seq), allowing them to be used for a merged data set (figs. S1, S2, and S4) (6, 14). Overall, we found that only a small number of genes (∼100 per stage) showed strong stage-specific expression in the whole-animal samples, but fewer than half of the genes were detectably expressed in all stages by means of RNA-seq, and tiling arrays suggest that >75% of genes show a greater than twofold range of expression across all the tissues (figs. S11 and S12) (15).
To investigate the relationship between gene expression and developmental stages in greater detail, we correlated the RNA-seq expression profiles at a given stage with all other stages. For simplicity, we focused on a set of 8428 genes with non-overlapping transcripts and found that profiles over the time course cluster into distinct embryo and larval phases (Fig. 2A) (6). This division was consistent with a principal-components analysis on the tiling-array data from matched tissues from embryo and L2 (Fig. 2C) (6). The RNA for the embryos and larvae was isolated through different procedures, but on the basis of a number of controls and comparisons these differences are unlikely to confound the analysis (6).
Fig. 2.

Expression and binding dynamics. (A) Spearman correlations of gene expression and RNA Pol II binding across seven stages. Expression-level correlations are shown above the diagonal; RNA Pol II–binding correlations appear below. For both expression and binding, there is a notable transition between embryonic and larval stages. (B) Correlation of RNA Pol II–binding levels with gene expression. Although RNA Pol II–binding in embryonic stages shows low correlation with gene expression in larval and young adult stages, expression in the embryo correlates moderately well with RNA Pol II–binding later. (C) Principal components analysis (PCA) of six matched tissue samples from mixed embryo (MxE) and L2 (7). GABA, γ-aminobutyric acid.
Alternative splicing
Alternative mRNA processing, including selection of alternative splice junctions, promoters, or poly(A) addition signals, provides another mechanism for differential transcript generation. To discover prominent stage-specific alternative isoforms among the aggregate integrated transcript models, we identified genes with two or more isoforms whose abundance changed more than fivefold during development; differential splice junction usage ranged from simple alternative exons to more complicated patterns, such as splicing or retention of an entire series of introns in different stages (Fig. 1C and fig. S6).
We also developed algorithms that infer quantitative transcript-level expression by distributing sequence reads among alternative isoforms in a probabilistic manner (6). Pairwise comparisons of staged samples showed that overall, isoform usage does not change dramatically between stages: Of 12,875 genes with multiple isoforms, 280 on average switch isoform usage between any two stages, totaling 1324 genes with switching (Fig. 1B and fig. S14) (6). Using a different approach, we grouped transcript-level expression profiles across many stages into 48 distinct clusters (figs. S15 and S16). We identified 1320 genes for which one isoform fell into a separate cluster from all the others and then classified these according to the type of processing events that distinguish them (figs. S17 and S18) (6). These analyses illustrate the range of alternative mRNA processing that takes place during development.
Pseudogenes
Several gene models derived from RNA-seq fell in regions previously annotated as pseudogenes. Pseudogenes are DNA sequences similar to protein-coding genes that are generally thought not to produce functioning proteins (16). However, some pseudogenes are transcribed and may potentially act as endo-siRNA (endogenous small-interfering RNA) regulators of their parent genes (17). Using computational methods, we identified 1293 probable pseudogenes in the C. elegans genome, adding 173 to and removing 213 from the previous annotation set (WS170), and established the probable source (parent) gene for 1198 of them (fig. S19) (6). Using RNA-seq data, we found evidence of transcription for 323 pseudogenes (6). For 191 of the 323, we determined that the transcription was clearly independent of the parent gene, ruling out potential mismapping artifacts. Of these 191, 104 had a discordant expression pattern across stages relative to the parent (Fig. 1D), and 87 were greater than two times more expressed than the parent (6). Intriguingly, 17 of the transcribed pseudogenes have a unique peptide match through mass spectrometry, suggesting that they are translated and may create novel short peptides.
ncRNAs
The genome produces a variety of transcripts that do not code for proteins but instead function directly as noncoding RNA (ncRNA). At the start of the project, there were 1061 known ncRNAs in C. elegans (table S5). These include small nucleolar RNAs (snoRNAs), RNAs involved in mRNA translation and splicing [such as ribosomal RNAs (rRNAs) and tRNAs], miRNAs, piwi-associated RNAs (piRNAs, called 21U-RNAs in C. elegans), and multiple classes of endo-siRNAs (18).
To provide a more comprehensive annotation of small ncRNAs, we profiled small-RNA gene expression using RNA-seq on size-fractionated total RNA. In particular, using 81 million aligned reads from 11 different stages enabled us to identify 154 out of 174 previously annotated miRNA genes (19, 20). Most of these are products of the canonical Drosha-Dicer cleavage pathway. However, four are mirtrons—miRNAs for which the precursor hairpins are generated directly by intron splicing (21). Our computational and experimental analysis validated 13 previously unidentified mirtrons (6, 22). Small-RNA data also defined 102 additional candidate canonical miRNAs and thousands of 21U-RNAs, although these latter were from previously identified loci (6, 19, 23).
To identify other candidate ncRNAs, particularly ones longer than those discussed above, we combined all the transcriptome data sets to integrate both tiling-array and RNA-seq data. We found that in comparison to other genomic “elements” (such as well-curated CDSs, UTRs, or intergenic regions), the known ncRNAs tend to have a higher small RNA-seq signal and very little poly(A)-plus RNA-seq signal. However, no single transcriptome feature was able to reliably distinguish them (fig. S21A) (24). Therefore, we developed a multivariate machine-learning model combining all the transcriptome data sets and found support for 21,521 previously unknown ncRNAs (4.3 Mb in total), which we call the 21k-set of ncRNAs (tables S6 to S8 and fig. S20) (6).
Because identifying ncRNAs by using tiling arrays can be problematic (14), we added conservation and RNA secondary structure to our model. However, doing so restricted the predictions of this second model to only the ∼15% of the C. elegans genome that was readily alignable to C. briggsae. Overall, the second model predicted 7237 previously unidentified ncRNA candidates (the 7k-set, comprising 1.0 Mb), with an estimated positive-predictive value of 91% (from testing against an independent validation set of known ncRNAs) (24). Of these, 1678 ncRNA candidates (181 kb) fell in intergenic regions, with the remainder in introns, pseudogenes, or regions antisense to exons (fig. S21B). We tested a number of these intergenic candidates to validate expression: RT-PCR detected RNA products for 14 of 15, and Northern blots detected expression for three of five (24).
The 7k-set contains many RNA structural motifs, including some not found in known RNA secondary structure families (24). Additionally, these ncRNA candidates tend to be differentially expressed across development (24), with many preferentially expressed in the embryo. Comparing the expression profiles of the 7k-set with those of well-characterized genes allowed us to identify putative functions for some candidate ncRNAs (table S9) (6). Lastly, in comparing the 7k and 21k sets of ncRNAs the overlap was small, with just 1259 overlaps. Thus, when conservation and structure were considered we detected candidate ncRNAs not found from the expression data alone; conversely, many previously uncharacterized transcripts in C. elegans may occur in nonconserved parts of the genome. Thus, the 7k and 21k sets provide complementary types of ncRNA candidates for further study.
In summary, the improved annotation of transcribed portions of the genome from these data sets provides the community with new substrates for further experimentation. However, gaps remain in some transcript models, some protein-coding genes remain to be discovered, and direct evidence is needed to support the candidate ncRNAs.
Regulatory Sites and Interactions
Accurate annotation of sites bound by TFs is central to understanding the regulatory networks underlying development and homeostasis. However, at the start of the project very few TF-binding sites had been annotated in the nematode genome, in part because of a lack of suitable methods with which to assay binding sites in whole animals (25). We developed these methods and have applied them to map the binding sites for 23 green fluorescent protein (GFP)–tagged fusion proteins and RNA polymerase II (RNA Pol II) using ChIP-seq (table S10) (6, 26). Most factors were assayed at their stage of highest expression, but both PHA-4 (a well-studied factor required for pharyngeal development) and RNA Pol II were analyzed at six developmental stages. Some of the factors were expressed in as few as 10% of the cells in the whole animal.
TF-binding sites, motifs, and targets
Binding sites were identified by first finding relatively broad regions of enrichment and then, for some analyses, refining these to narrow [≤200 base pairs (bp)] peak summits (figs. S24 and S46). Most TF-binding sites defined by means of ChIP-seq peaks for protein-coding genes lie within 500 bp upstream of transcript start sites. Binding sites assigned to known ncRNAs are even closer to the 5′ end of the transcript (fig. S22C). On the basis of their proximity to the TSS, we were able to assign most sites to specific protein-coding or known ncRNA genes, creating a set of candidate targets for each TF (6); however, some sites were ambiguously located and remain unassigned. Although most factors target both protein-coding and known ncRNA genes, GEI-11 preferentially targets ncRNAs (Fig. 3D and fig. S22, A and B). Analysis of TF-binding sites adjacent to ncRNA candidates from the 7k-set showed that 59% are potential targets of the 22 TFs examined, which is significantly more than would be expected by chance (P < 0.001, derived from a z score assuming a normal distribution of random sequences) (6, 24). Pairwise correlation of target genes revealed that factors with related functions often show substantial overlap in their protein-coding gene targets (fig. S23A). Three homeobox (HOX) genes involved in establishing the body plan provide particularly striking examples (mab-5, lin-39, and egl-5) (26). In contrast, pairwise correlation of targeted miRNAs shows that the factors bound to them tend to cluster together more by stage than by factor type (fig. S23B), which is consistent with observations that expression of miRNAs tends to show strong stage-specific enrichment (19).
Fig. 3.

Integrated miRNA-TF regulatory network. (A) TFs are organized hierarchically, and those miRNAs either regulating or being regulated by the TFs are shown. (TF names are in fig S36.) All larval TF-TF interactions in HOT regions were removed. Tissue specificity and number of protein-protein interactions are shown for each of the hierarchical levels (6). (B) TF network after filtering out edges that do not show a significant correlation in their expression patterns. Also shown is a schematic representation of the target genes of the 18 larval TFs. (C) One of the three significantly enriched network motifs (other two are in fig. S37). (D) Enrichment of binding targets and signal of TFs in noncoding versus coding genes. Max signal equals the ratio of maximum binding signal of a TF at noncoding versus coding genes. Target fraction represents the ratio of target percentage in noncoding genes to that in coding genes (fig. S22A).
To further characterize TF-binding sites, we searched for 8- to 12-bp cis-regulatory motifs within the ChIP-seq peaks (6) and found strong motifs for eight TFs (BLMP-1, CEH-14, CEH-30, EGL-5, HLH-1, LIN-39, NHR-6, and PHA-4) (fig. S35). Two of these are similar to previously described motifs (PHA-4 and HLH-1).
The binding sites (defined from narrow peaks) cover a total of 5,165,949 bp (5.2% of the genome) and target 8859 protein-coding genes, as well as 652 known ncRNAs, indicating that each gene may have sites for many factors.
Clustered binding in HOT regions
We identified 304 short binding regions (average length of ∼400 bp) that were significantly enriched (q value < 1e-5) in most TF ChIP-seq experiments despite the fact that the 22 analyzed factors have diverse functions and expression patterns. These regions, which we term HOT regions, were bound by 15 or more factors (Fig. 4, A and B, and fig. S25A) (6). Control experiments revealed that these regions are not enriched in input DNA, nor do they appear in control ChIPs from strains lacking GFP-tagged TFs (fig. S26) (6). The number of factors bound to HOT regions was relatively insensitive to the width of the peaks used to identify them because peak summits occur within 100 bp for over 80% of HOT regions (fig. S25B) (6).
Fig. 4.
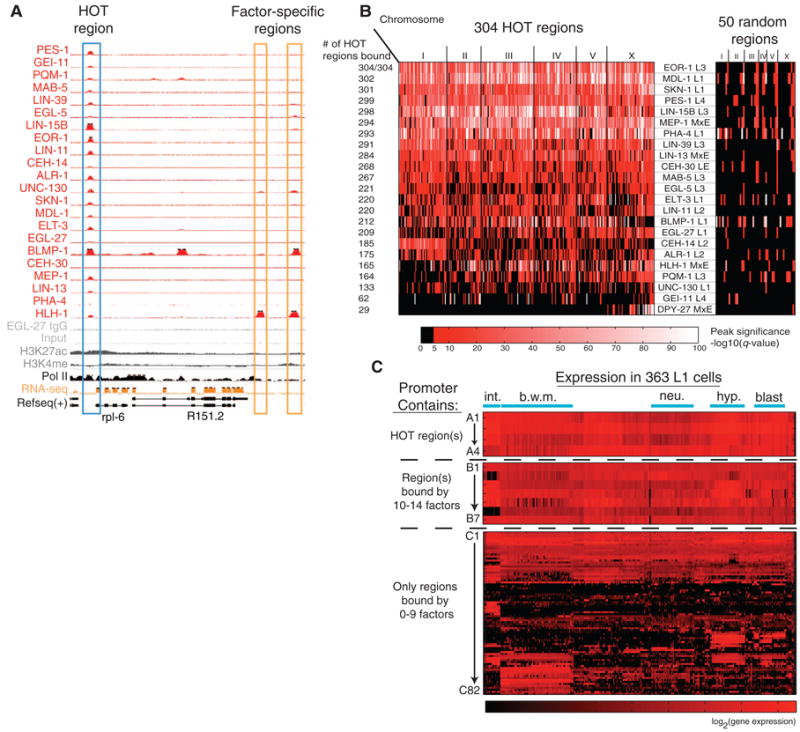
HOT regions. (A) TF-binding peaks at a HOT region and two “factor-specific regions” on chromosome III: 7,206,000 to 7,220,000. Top tracks show read density (scaled based on the total mapped reads) from 22 ChIP-seq experiments. Bottom tracks show ChIP-seq controls, RNA-seq expression levels, and ChIP-chip signals for two histone modifications. (B) 304 HOT regions bound by 15 or more factors and 50 randomly chosen TF-bound regions. Each row represents a TF, and each region is colored by enrichment q value (6). (C) Genes associated with HOT regions are broadly expressed. Single-cell gene expression measurements of 93 mCherry reporters (30) are shown separated by whether the promoter contains a HOT region, contains a region bound by 10 to 14 factors, or contains only regions bound by 0 to 9 factors (gene names are in fig. S29). The x axis represents 363 specific cells present in L1-stage animals.
In addition to the HOT regions, most TFs also cross-link to “factor-specific” DNA regions (bound by one to four total factors) (Fig. 4A). Using HLH-1, a typical factor with both known tissue specificity and a known binding motif, we compared these two different classes of sites (HOT and factor-specific) for functional differences. HLH-1 drives muscle development in C. elegans (27) and is associated with 598 factor-specific and 165 HOT regions. Relative to HOT regions, factor-specific HLH-1 ChIP-seq regions were over twofold enriched for the HLH-1–binding motif (Fisher's exact test, P < 0.0001) (28), and genes associated with these regions were more than ninefold enriched for muscle-specific expression (Fisher's exact test, P < 0.01) (fig. S27, A and C) (29). Similar enrichment for motifs and tissue-specific expression of targets was also observed for other TFs when factor-specific sites were compared with HOT regions (fig. S27B) (6), suggesting that factor-specific and HOT regions are functionally distinct.
Genes associated with HOT regions are distinguished by several other measures. HOT-region genes assayed for expression at the individual-cell level in L1 larvae are expressed in most or all cell types, whereas other genes mostly showed tissue-specific expression (Fig. 4C and fig. S29) (30). Genes associated with HOT regions were also expressed at higher levels in whole-animal and tissue-enriched measurements and were less likely to be stage-specific (fig. S28) (6). Compared with 3% of genes associated with factor-specific regions, 21% of the HOT region–associated genes are essential (P < 1e-40; χ2 test) (fig. S27C) (6, 31). Gene Ontology (GO) (32) analysis revealed a variety of biological processes highly represented in HOT-associated genes, including growth, reproduction, and larval and embryonic development (each P < 1e-15), as well as 19 ribosomal protein genes (>12× enrichment, P < 1e-12) (table S11). In comparison, GO analysis of the remaining (non-HOT) targeted genes identified functional terms that are consistent with the known tissue specificity and function of the individual TFs (26).
Extensive overlap in binding sites between TFs with disparate functions has previously been observed in both limited (33) as well as whole-genome ChIP-chip experiments (34, 35). Using ChIP-seq data, we have shown that hundreds of regions in C. elegans are bound by the majority of TFs within a 100-bp window. Our results suggest that many TFs that are cross-linked to HOT regions are not directly associated with DNA via specific binding, which is consistent with findings for highly occupied regions in Drosophila (34). Rather, they suggest that association with HOT regions may be driven by protein-protein interactions to a currently unknown set of HOT region–associated DNA-binding factors. We searched for sequence motifs that might be broadly associated with HOT regions and found a few that were significantly enriched (fig. S35), but the protein factors that bind directly to these motifs are currently unknown.
Building a TF hierarchy
Following the assignment of binding sites to target genes, we investigated the resulting “binding network,” as had previously been done in yeast and Escherichia coli (36). The network for 18 factors assayed in larval stages (Fig. 3, A and B, and fig. S36) is relatively dense, with each TF bound to an average of 828 genes, including TFs and other gene targets. We pruned the network to the strongest interactions, using the fact that the expression profile of a TF tends to be more strongly correlated over the time course with that of its targets than nontargets, being positive for activators and negative for repressors (table S12) (6). The pruned network shows a high level of autoregulation among the factors.
Within the network, we organized TFs hierarchically according to the degree to which they target other TFs (top of the hierarchy) or are themselves targets for other TFs (bottom) (37). We observed clear differences between the TFs at each level (Fig. 3, A and B). TFs at the lower levels tended to be more uniformly expressed across multiple tissues (P = 0.07, Student's t test) (6). Consistent with this, TFs at the bottom level were essential more often than those at the top. In contrast, members of the Hox family were more often at the top of the hierarchy—among the six Hox TFs examined, four were at the top layer of nine TFs—perhaps reflecting their role in modulating specific developmental processes across multiple tissues. Lastly, TFs showed connectivity in the existing C. elegans protein-protein interaction network so that those at the hierarchy top tended to have significantly fewer protein-protein interactions than those below (P = 0.002, Student's t test) (38). This suggests that TFs in the middle and bottom layers act as “mediators” or “effectors,” more likely to exchange information with other proteins. Although the predicted larval-stage TF network here is small and one cannot make strong statistical statements, these conclusions follow a pattern that is consistent with regulatory hierarchies in yeast and E. coli, in which essential and highly connected “workhorse” regulators tend to occupy lower levels whereas overall modulators are on the top (37).
An integrated miRNA-TF network and its motifs
Next, we added miRNAs to our TF hierarchy in order to enable us to explore the interplay between transcriptional and posttranscriptional regulation. In particular, we identified the targets of miRNAs on the basis of annotated 3′UTRs and sequence conservation (table S13) (6). We then constructed an integrated network between miRNAs expressed during larval stages and the above 18 TFs (all assayed in the same stages). For simplicity in this network, we describe connections between two entities as “A regulates B”—though more properly, we should describe them as “A is predicted to bind near B and regulate it.” In the integrated network, the level of a miRNA was assigned according to the highest-level TF it regulates or, if it does not regulate a TF, the lowest-level TF that regulates it. The miRNAs fall into distinct levels, paralleling the arrangement of TFs (Fig. 3A). Moreover, the network reveals two different classes of miRNAs: those that are more strongly regulated by TFs versus those that predominantly regulate TFs (Fig. 3A, bottom right versus top left, respectively).
We can further analyze our integrated network in terms of motifs, which is a common approach used to decompose a complex network into simple building blocks (36). Many different types of network motifs exist; as a simple example, we observed miRNA-TF loops in our integrated network, in which a miRNA regulates a TF and the same TF regulates the miRNA (39). Of particular interest are patterns that are overrepresented as compared with randomized, rewired null models (6). We observed three overrepresented motifs in the integrated miRNA-TF network (fig. S37) (6). One example is a miRNA-mediated feed-forward loop, in which a TF regulates a miRNA and, together with the miRNA, regulates a target coding gene (Fig. 3C). This particular motif structure is potentially responsible for buffering noise and maintaining target protein homeostasis (40).
RNA Pol II binding and expression
We profiled RNA Pol II and the specific factor PHA-4 in each of the main stages of C. elegans development and compared their binding profiles with the corresponding RNA-seq data. Similar to the above approach for gene-expression dynamics, for RNA Pol II we focused on a set of 8428 genes with non-overlapping transcripts and used the binding profiles at promoters to generate correlation matrices between each stage. We found a similar differential clustering of the embryonic and larval stages (Fig. 2A). This embryonic-larval division was also observed for PHA-4 binding across stages (fig. S30) and presumably reflects the different transcriptional programs between embryos and larvae.
Next, we correlated the RNA Pol II–binding profiles with expression profiles across all the stages. As expected, the same-stage correlation was fairly high (0.64 to 0.70) (Fig. 2B) but was notably lower for embryonic stages than for larval ones, perhaps reflecting the presence of maternal transcripts in embryos (6, 41, 42). Unexpectedly, we found expression at earlier developmental stages more tightly correlated with binding at later stages, rather than RNA Pol II–binding anticipating RNA production (Fig. 2B). Specifically, the correlation is low initially, reaches a maximum at the matching stage, and then remains high for later stages. This can be interpreted as RNA Pol II binding to genes at the same developmental stage at which they are initially expressed, and Pol II then remaining bound in later stages, even if expression drops. The initial round of transcription may affect the accessibility of the promoter, which may then remain unaltered in later stages for nondividing cells. Alternatively, this result may reflect paused RNA Pol II at genes with reduced expression at later stages. We have found several examples of genes in which RNA Pol II binding remains high in later stages but gene expression is low [such as isl-1 and pgp-2 (fig. S31)], which is consistent with RNA Pol II stalling.
Overall, we have shown how the analysis of relatively few TFs allows the construction of a fairly elaborate network. To improve these networks in the future, we will need to identify the precise cells and stages in which the TFs and miRNAs are expressed.
Chromatin Organization and Its Implications
One modENCODE goal is to identify elements that control chromosome behavior and regulate the function of DNA elements. C. elegans chromosomes have several distinctive features. Instead of having centromeres embedded in highly repeated sequences, its chromosomes are holocentric, with microtubule attachment sites distributed along their length. In hermaphrodites (XX), gene expression from both X chromosomes is down-regulated in somatic cells by a dosage compensation mechanism and so better match expression in males, which have one X chromosome (XO) (43). Furthermore, the entire X chromosome is under-expressed relative to the autosomes in the germline cells of both hermaphrodites and males (44). C. elegans autosomes have distinct domains—a central region flanked by two distal “arms” that together comprise more than half of the chromosome. Compared with the centers, the arms have higher meiotic recombination rates, lower gene density, and higher repeat content (5, 45, 46). Arms are not as sharply defined on the X chromosome.
Chromosome-scale domains of histone modification
The distribution of 19 histone modifications and two key histone variants (C. elegans homologs of H2A.z and H3.3) revealed striking, broad domains of histone modification states on the autosomes, with relatively sharp boundaries between the central region of each autosome and the arms (Fig. 5, A to C) (47–49). Modifications traditionally associated with gene activity and euchromatin such as acetylation and H3K4 and H3K36 methylation are enriched in the central regions of the chromosomes. In contrast, H3K9 mono-, di-, and trimethylation marks associated with transcriptional repression and heterochromatin formation are relatively depleted from the central regions and enriched on the arms of the autosomes (Fig. 5A). These megabase-scale chromosomal domains are not homogeneous; there are small zones of repressive marks within the generally active central regions and active marks within the generally repressed arms. The chromosome-scale domains of histone modification do not vary substantially in composition or position between embryos and L3 larvae. Despite the biased distribution of repressive marks, the arms of the chromosomes do not appear heterochromatic through 4′,6′-diamidino-2-phenylindole (DAPI) staining or classical banding techniques (50). Although our samples did not include appreciable meiotic tissue, the broad domains of histone modifications correspond to regions defined by differences in recombination rate, with the boundaries located at the recombination rate inflection points (Fig. 5A) (5, 46). On each chromosome, one arm contains a meiotic pairing center that mediates homologous pairing and synapsis (50, 51). As previously reported, H3K9me3 is more highly enriched on that arm (Fig. 5A) (52). However, methylation is not particularly enriched within the pairing center regions themselves (53). H3K9me3 is also highly enriched on silent genes on arms, and all forms of H3K9 methylation are enriched in repetitive elements, which are more prevalent on chromosome arms (fig. S32).
Fig. 5.
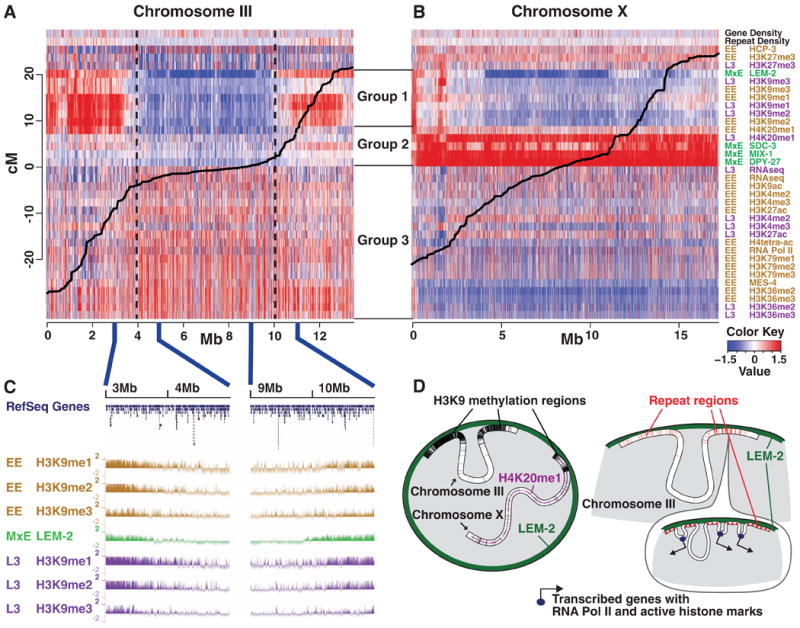
Chromosome-scale domains of chromatin organization. (A and B) Whole-genome ChIP-chip data for various histone modifications and chromatin-associated proteins, along with relevant genome annotations, were normalized, placed into 10-kb bins, and displayed as a heat map. Red indicates a stronger signal, and blue indicates a weaker signal. The continuous black line plots the relationship between physical (x axis) and genetic (y axis) distance. Three major groups were identified by hierarchical clustering. Group 1 contains H3K9 methylation marks and LEM-2, which tend to be enriched at distal autosomal regions, and correlate with repetitive DNA and a high recombination rate. Group 2 contains dosage compensation complex members and H4K20me1, which are highly enriched on X. Group 3 contains marks associated with active chromatin. Generally, signals for active marks are weaker on the X chromosome than the autosomes. This megabase-scale chromatin organization persists through all stages examined. (A) Chromosome III is representative of autosomes. (B) X has a distinct chromatin configuration. (C) H3K9me1, - 2, and -3 signals decrease gradually at the boundaries between the central and distal domains, whereas the boundaries defined by LEM-2 are relatively sharp. (D) A schematic representation of key findings.
The X chromosome
Gene density, recombination rates, and repeat content are more uniformly distributed along the X chromosome than autosomes (5). Consistent with this, chromatin marks on the X are more uniformly distributed. A high density of repressive marks, similar to that seen throughout the autosome arms, is associated with only two narrow ∼300-kb regions at the left end of the X that flank the meiotic pairing center (Fig. 5B). The genomic distribution of DPY-26, DPY-27, DPY-28, and SDC-3, proteins mediating dosage compensation, is highly enriched on the X chromosome (Fig. 5B) (25, 54, 55). H4K20me1, a modification linked in mammals to chromosome maturation and X-chromosome inactivation (56), is also enriched on the X. This X-enrichment is detectable in early embryo populations, when some embryos have initiated dosage compensation, and becomes more pronounced in L3 animals, when dosage compensation is fully established.
Chromosomes and nuclear envelope interactions
Interactions between the genome and the nuclear envelope were determined by means of ChIP of LEM-2, a transmembrane protein associated with the nuclear lamina (57). In embryos, LEM-2 interacts with the repeat-rich, H3K9-methylated arms of the autosomes but not with the autosome centers (Fig. 5, A and D). Similar to H3K9 methylation, the transition between LEM-2–enriched arms and the central chromosomal regions is relatively sharp, coinciding with the transition between regions of high and low meiotic recombination rate (Fig. 5B). Within the arm regions, LEM-2 enrichment exhibits a complex underlying subdomain structure (57). On the X chromosome, LEM-2 interacts with only the small regions on the left end that harbor repressive chromatin marks (Fig. 5B). This suggests a particular organization for the X chromosome within the nucleus (Fig. 5D).
Histone mono-methylation
We plotted the distribution of each chromatin mark relative to transcript starts and ends and further subdivided these plots by the expression level of the associated gene on autosomes versus the X chromosome (Fig. 6 and fig. S34). Overall, the results are consistent with the known distributions and functions of chromatin marks in other eukaryotes (58). However, the distribution of several mono-methyl marks—including H4K20me1, H3K9me1, and H3K27me1—are associated more with the bodies of highly transcribed genes on the X chromosome than with similarly expressed genes on autosomes. Further, H3K36me1 is confined sharply to gene bodies on X, in contrast to broader enrichment that spans promoters and 3′ UTRs on autosomal genes. Conversely, H3K36me3 and H3K36me2 are more associated with autosomal genes than with X-linked ones (Fig. 6 and fig. S34). Differences in several marks are observed between early embryogenesis and more differentiated L3 animals—most notably a redistribution of H3K27me1 and H3K27me3 (Fig. 6 and fig. S34, bottom row).
Fig. 6.

Chromatin patterns around genes. Average gene profiles around the TSS and TTS of various histone marks displayed for the (red) X chromosome and (blue) autosomes. Genes were further stratified according to their expression level, with the top 20% of expressed genes shown in darker shade and the bottom 20% of expressed genes shown in lighter color. Marks typically associated with active or repressed transcription are labeled on the left.
Nucleosome organization
Consistent with micrococcal nuclease (MNase) nucleosome-mapping experiments (52, 59, 60), both X and autosomal genes exhibit a typical nucleosome-depleted region upstream of TSSs, a well-positioned +1 nucleosome, and nucleosome depletion at the 3′ ends. However, we observed that the average nucleosome occupancy immediately upstream of the +1 nucleosome on the X chromosome was 1.6-fold higher than that of genes on autosomes (at −300 to +200 bp relative to the TSS; P < 2.2e−16, Wilcoxon rank-sum test) (61). Relative to autosomal genes, promoters of X-linked genes have higher GC content, which is predictive of high nucleosome occupancy in vitro (fig. S33) (61–63). We observed a similar difference between X and autosomal promoters when naked DNA was digested with MNase, although this result was expected because the known DNA sequence preferences of MNase are similar to the sequence preferences of linker DNA (64, 65). DNA sequences associated with nucleosome occupancy evolve according to expression requirements (66, 67), suggesting that the higher GC content on X promoters may relate to mechanisms of X-specific gene regulation in the soma and germline.
Epigenetic transmission of chromatin state to progeny
The activity of the C. elegans protein MES-4—a histone H3K36 methyltransferase required for the survival of nascent germ cells in developing animals—mediates the transmission of information about the pattern of germline gene expression from mother to progeny. Similar to other H3K36 methyltransferases, MES-4 is associated with gene bodies. However, in contrast to previously studied H3K36 methyltransferases (68) MES-4 is able to associate with genes in an RNA Pol II–independent manner (69). In the embryo, MES-4 is preferentially bound to genes that were highly expressed in the maternal germline but may no longer be expressed in embryos (69). Conversely, MES-4 is not associated with genes expressed specifically in early embryos, despite recruitment of RNA Pol II to those genes (69). Therefore, RNA Pol II association with genes is neither necessary nor sufficient to recruit MES-4 in embryos (69). These findings suggest that MES-4, which is required for fertility, functions as a maintenance histone methyltransferase and propagates the memory of gene expression from the maternal germline to the cells of the next generation (69).
Models relating chromatin to TF binding
To integrate chromatin with other types of modENCODE data, we sought to relate the patterns of histone marks with the observed TF-binding sites. Across the whole genome, we observed only weak direct correlations between the two (fig. S38A). However, the relationship between chromatin and TFs may involve complex, nonlinear relationships. To probe these, we built machine-learning models to identify TF-binding peaks from chromatin features (fig. S39). Investigating the association of individual histone marks with TF-binding sites, we found some that discriminate TF-binding sites from the genomic background with reasonable accuracy (Fig. 7A). Often, this is connected with their actual presence at binding sites; for example, when comparing the background to binding peaks, on average, some marks have stronger signals, where-as others have weaker ones [such as H3K4me3 versus H3K9me3 (fig. S41)]. Individual chromatin marks and RNA Pol II–binding signals could also distinguish HOT regions from the genomic background, highlighting the association with active transcription in these regions.
Fig. 7.
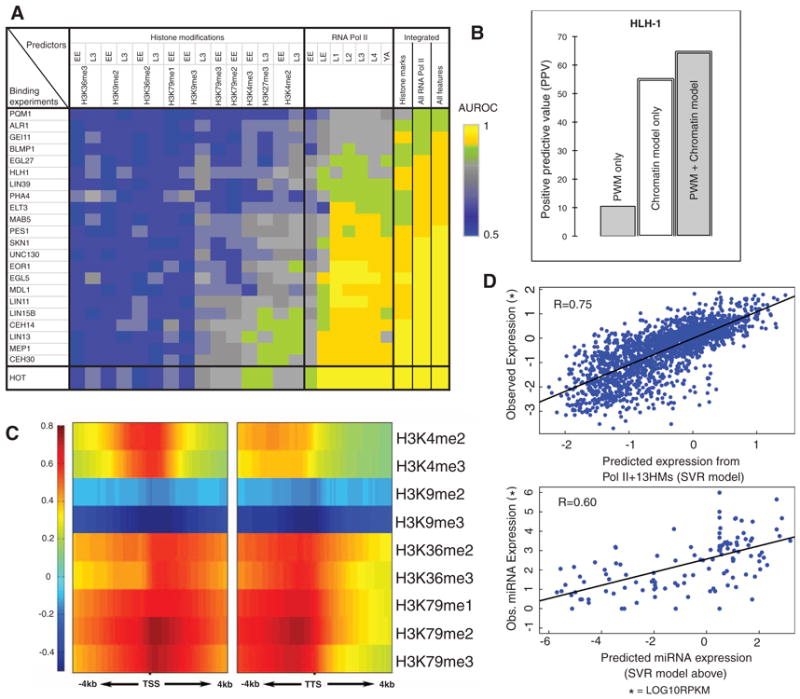
Statistical models predicting TF-binding and gene expression from chromatin features. (A) Modeling TF-binding sites with chromatin features. The color of each cell represents the accuracy of a statistical model in which a chromatin feature or a set of features acts as predictor for TF binding or HOT regions. (B) An example of combining chromatin and sequence features. Potential binding sites of HLH-1 were predicted by using only sequence motifs, only chromatin features, or both. (C) Correlation pattern for a number of chromatin features in 100-bp bins around the TSS (± 4 kb) and TTS (± 4 kb) of transcripts at the early embryo (EE) stage. The Spearman correlation coefficient of each chromatin feature with gene-expression levels was calculated for each bin. (D) Chromatin features can predict expression levels for both protein-coding genes and miRNAs. (Top) A model involving all chromatin features. (Bottom) The model for protein-coding genes can also be used to predict accurately miRNA expression levels.
Because chromatin features work in combination to influence binding-site selection (70), we combined all the histone marks together in a classifier. The resulting models could identify binding sites better than those based on any individual mark (Fig. 7A and figs. S38B and S40A).
We further observed that chromatin features are particularly good at identifying the binding peaks of some specific TFs. For example, H3K4me2 and H3K4me3, which are usually enriched in promoters, identified the binding peaks of a group of five factors (CEH-14, CEH-30, LIN-13, LIN-15B, and MEP-1) better than the other TFs. This association is specifically due to a relative enrichment of these H3K4me2 and H3K4me3 at the binding peaks of this group of five TFs (fig. S41). It further suggests that the chromatin features can be useful in discriminating not only binding sites from the genomic background but also the sites of specific TFs in comparison with other TFs. Indeed, we were able to build integrated models to do this with reasonable accuracy (fig. S40B). The same approach was also successful in discriminating HOT regions from all TF-binding regions (fig. S40B). Our models perform best when chromatin features are measured at the same stage as the TFs, suggesting a dynamic relationship between chromatin and binding sites across developmental stages (fig. S42).
To provide additional predictive power, we incorporated into our models the information from the specific sequence motif recognized by a TF, summarized by a position-weight matrix. The combined models with both chromatin and sequence information were more accurate than were models involving either type of information alone (Fig. 7B and fig. S43). Thus, chromatin features enable one to predict TF-accessible regions and broad classes of binding sites, and motifs provide additional information on the exact sites bound by particular factors, chosen from these broad classes.
Models relating chromatin to gene expression
Next, we developed a model to relate chromatin marks to gene expression levels. We divided the regions around each TSS and transcript termination site (TTS) into small (100 bp) bins and calculated the average signal of each chromatin feature and RNA Pol II (13 features in total) in a set of 160 bins up to 4 kb upstream and downstream of these two anchors (to include even long-range effects). Then at each bin, we correlated the chromatin signals with the stage-matched gene expression value (Fig. 7C). There is clear variation across the bins in this correlation, with the effect of making activating marks more sensitive than are repressive ones to their exact positioning relative to the TSS or TTS.
By combining all features at each of the 160 bins, we built a model for gene expression, predicting the quantitative expression levels of transcripts with support vector regression (SVR) (6). Predicted expression levels were highly correlated with measured ones [correlation coefficient (r) = 0.75, cross-validated]. As an overall benchmark, we compared our chromatin model with one based on the level of RNA Pol II–binding alone (r = 0.37); our model achieves better prediction accuracy for expression levels.
To find the relative importance for gene expression of the 160 possible bin locations, we divided genes into highly and lowly expressed classes and predicted the class of each gene from each bin. The best predictions were obtained from bins immediately after the TSS and just before the TTS. With increasing distance upstream of the TSS, predictive power decreased smoothly. Intriguingly, the predictive capability of chromatin features extended as much as 4 kb upstream of the TSS and 4 kb downstream of the TTS, even when we restricted the analysis to widely separated genes with distant neighbors. This may indicate a long-range influence of chromatin on gene expression.
In contrast to protein-coding genes, the association between histone modifications and miRNA expression has not been explored in detail. Because protein-coding and miRNA genes are both transcribed by RNA Pol II, we applied the above chromatin model, derived from protein-coding genes, to the regions around candidate pre-miRNAs. We then predicted expression levels for 162 microRNAs, for which genomic locations are provided by miRBase (71), and compared these predictions to the measurements in the modENCODE small RNA-seq data set. We found a correlation of 0.60 (r = 0.62 for just miRNAs far from known genes) (Fig. 7D). That expression of miRNAs can be predicted accurately by using a chromatin model trained on protein-coding genes is consistent with miRNAs and protein-coding gene regulation sharing similar mechanistic connections to histone marks.
Conservation Analysis
Because mutations are constantly accumulating over evolutionary time, purifying selection slows the rate of divergence of functional relative to nonfunctional sequences (72). For this reason, evolutionarily constrained regions can assist in identifying functional elements (73). Although some functional sequences may not be conserved, are conserved in a way that we are unable to detect, or are under positive selection (resulting in accelerated divergence), the coverage of constrained bases by identified functional elements is a valuable measure of the completeness of our understanding of the genome. We characterized regions of the C. elegans genome under evolutionary constraint by constructing a multiple alignment among the nematodes C. elegans, C. remanei, C. briggsae, C. brenneri, C. japonica, and Pristionchus pacificus using methods previously developed (1). We then calculated conservation scores with PhastCons (6, 74). These procedures identified 59,504 constrained blocks that cover 29.6% of the C. elegans genome as a whole and range from 27.4% of chromosome IV to 31.9% of chromosome X. The single largest constrained block was 3558 bp on chromosome V, but conserved blocks were typically much smaller (mean 49 ± 58.6 bp).
These conserved regions are highly correlated with functional elements. We first examined the proportion of evolutionarily constrained regions that overlap experimentally annotated portions of the genome (Fig. 8A and fig. S44). In the last WormBase freeze before the incorporation of modENCODE data (6), 50.8% of the constrained regions were covered by annotations supported by direct experimental evidence. Adding modENCODE protein-coding gene evidence increased the coverage of constrained bases to 58.3%. Other modENCODE increases came from the 7k-set of ncRNAs (1.9%), TF-binding sites, (5.9%), dosage compensation (9.3%), and other chromatin-associated factors (2.8%). Thus, modENCODE explains an additional 27.4% (8.1 Mb) of the constrained portion of the genome; together with remaining unconfirmed WormBase gene predictions (0.7%) and pseudogenes (0.6%), coverage now totals 79.5% of constrained bases.
Fig. 8.

Relative proportion of annotations among constrained sequences. (A) Relative proportion of constrained and unconstrained bases in the C. elegans genome. Within the constrained region, the stacked bar chart shows the cumulative proportion covered by various classes of annotated genomic elements. (B) Fraction of element classes covering (red) constrained and (gray) unconstrained bases. The error bars show the 95% confidence interval for random placement of elements calculated with GSC. If the ends of the columns are outside the confidence interval, then it is unlikely that the fraction of the element class overlapping constrained and/or unconstrained bases could have occurred by chance. (C) Constraint profiles of broad categories of elements. The x axis indicates the PhastCons score of bases covered by the element ranging from 0 (no conservation) to 1.0 (perfect conservation). The y axis indicates the log ratio of the number of bases with the given score covered, relative to what would be expected by random element placement (dotted line) (fig. S45 shows more detail).
We then estimated the extent of constraint on different functional elements by plotting the distribution of the PhastCons conservation scores for each type of element (Fig. 8, B and C, and fig. S45). The most constrained elements were ncRNAs (both known and the 7k-set), presumably reflecting the fact that conservation was a criterion used to identify them. Next came protein-coding elements, followed by miRNAs, TF-binding sites, and other chromatin factor–binding sites. Pseudogenes, introns, and regions of the genome not covered by modENCODE data sets all have low levels of conservation. We then used the genome structure correction (GSC) statistic (1, 75) to calculate confidence intervals on the degree of overlap between evolutionarily constrained bases and functional elements defined by modENCODE and other sources. This demonstrated that coding regions, ncRNAs, TF-binding sites, and other chromatin factor–binding sites are significantly more constrained than would be expected by chance, whereas regions covered by pseudogenes, introns, and unannotated regions are significantly depleted in constrained regions relative to chance.
Roughly 20.5% of the constrained genome remains uncovered by known functional elements, but a portion of this sequence directly abuts known functional elements. If the borders of transcribed regions and chromatin-associated protein-binding sites are extended across all constrained blocks that neighbor them, ∼ 4.1 Mb (14%) in isolated constrained blocks remains. These residual constrained bases are highly enriched in introns and intragenic regions (table S14), are moderately enriched in the 1-kb regions upstream of TSSs, and are depleted in the 1-kb regions downstream of TTSs. One potential explanation for the residual constrained bases is that they correspond to the binding sites of untested TFs. Indeed, a plot of coverage of constrained sequence against numbers of TF experiments shows that the relatively small numbers of TFs studied here are far from saturating constrained bases (fig. S47), implying that additional TFs may explain part of the remaining constrained bases in these regions. Other explanations for the residual constrained regions include other intronic regulatory sites, transcribed regions that are expressed only under rare circumstances, and possibly as-yet unknown classes of functional elements.
Discussion
Our analysis illustrates patterns at multiple genomic scales: individual gene, chromosomal domain, and whole-chromosome. At the first scale, in addition to improving annotation of protein-coding genes, we identified transcribed pseudogenes and many previously unidentified ncRNAs, mapped binding sites of TFs, built regulatory networks, and constructed models predicting binding location and expression levels from chromatin marks. On a larger scale, we found chromosomal domains—characterized by repressive marks and interactions with the nuclear envelope on the autosome arms—and noted how the boundaries in these domains align with changes in recombination frequency. We also identified additional properties of the entire X chromosome, including the preferential accumulation of multiple mono-methylated histone marks. Our large-scale approach also discovered unexpected biological phenomena that would be difficult to uncover in conventional studies. In particular, upon profiling the binding sites of 23 factors we identified regions of clustered binding (HOT regions).
One limitation of the modENCODE strategy is that we cannot readily distinguish low levels of biochemical noise, such as a rare nonfunctional transcription splice form, from biologically important phenomena. The presence of such noise may be an unavoidable part of the cell regulatory machinery (76) and will only be distinguished from biologically important signals through careful follow-up experimentation. Another limitation is that almost all experiments were performed in populations of whole animals composed of multiple tissues. Future studies will increase the tissue-specific resolution of the data.
Model organisms such as C. elegans have long served as key experimental systems for developing technology and providing fundamental insights into human biology. Comparing our modENCODE results with the ENCODE pilot, which assessed functional elements in 1% of the human genome, we can already begin to see commonalities (6). For instance, for some aggregated binding signals (such as for RNA Pol II) the overall shape of the signal distributions relative to the TSS are quite similar between human and C. elegans. Likewise, the overall amount (per base pair) of transcription and binding by TFs is comparable (fig. S49 and tables S15 and S16). However, there are differences in the shape of the aggregated signal distributions for a few matched histone modifications (Fig. 6 versus fig. S50). Moreover, the relative proportion of constrained genome covered by experimental annotation is quite different in human and nematode, perhaps reflecting evolutionary pressures for a compact genome in the latter (fig. S48). A more comprehensive comparison, including the Drosophila genome data presented in the accompanying article, must await genome-wide analysis of human cells—an effort currently underway in the ENCODE project.
The modENCODE data sets are intended as an enduring resource for the genomics community. All raw and analyzed data, metadata, and interpreted results are available at www.modencode.org, where they can be searched, displayed, and downloaded. Raw sequencing reads and microarray data are archived at the Short-read Archive and the Gene Expression Omnibus, and higher-order results are being incorporated into WormBase (77). In addition, we have assembled a compact guide to the data sets used (at www.modencode.org/publications/integrative_worm_2010) (table S1) (6) and have populated a community cloud-computing resource with the data and analysis tools to facilitate further investigation by interested researchers (6). We expect that analyses of these data sets in the coming years will provide additional insights into general principles of genome organization and function, which will ultimately aid in annotating and deciphering the human genome.
Supplementary Material
Acknowledgments
Funding for this work came from the NHGRI of the NIH as part of the modENCODE project, NIH (grant R01GM088565), Muscular Dystrophy Association, and the Pew Charitable Trusts (J.K.K.); the Helmholtz-Alliance on Systems Biology (Max Delbrück Centrum Systems Biology Network) (S.D.M.); the Wellcome Trust (J.A.); the William H. Gates III Endowed Chair of Biomedical Sciences (R.H.W.); and the A. L. Williams Professorship (M.B.G.). M. Snyder has an advisory role with DNANexus, a DNA sequence storage and analysis company. Transfer of GFP-tagged fosmids requires a Materials Transfer Agreement with the Max Planck Institute of Molecular Cell Biology and Genetics. Raw microarray data are available from the Gene Expression Omnibus archive, and raw sequencing data are available from the SRA archive (accessions are in table S18). We appreciate help from S. Anthony, K. Bell, C. Davis, C. Dieterich, Y. Field, A. S. Hammonds, J. Jo, N. Kaplan, A. Manrai, B. Mathey-Prevot, R. McWhirter, S. Mohr, S. Von Stetina, J. Watson, K. Watkins, C. Xue, and Y. Zhang, and B. Carpenter. We thank C. Jan and D. Bartel for sharing data on poly(A) sites before publication, WormBase curator G. Williams for assistance in quality checking and preparing the transcriptomics data sets for publication, as well as his fellow curator P. Davis for reviewing and hand-checking the list of pseudogenes.
Footnotes
Supporting Online Material: www.sciencemag.org/cgi/content/science.1196914/DC1, Materials and Methods, Figs. S1 to S50, Tables S1 to S18, Refrences
References and Notes
- 1.Birney E, et al. ENCODE Project Consortium. NISC Comparative Sequencing Program; Baylor College of Medicine Human Genome Sequencing Center; Washington University Genome Sequencing Center; Broad Institute; Children's Hospital Oakland Research Institute. Nature. 2007;447:799. [Google Scholar]
- 2.Celniker SE, et al. Nature. 2009;459:927. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sulston JE, Schierenberg E, White JG, Thomson JN. Dev Biol. 1983;100:64. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 4.White JG, Southgate E, Thomson JN, Brenner S. Philos Trans R Soc London B Biol Sci. 1986;314:1. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 5.C. elegans Sequencing Consortium. Science. 1998;282:2012. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- 6.Materials and methods are available as supporting material on Science Online.
- 7.Reboul J, et al. Nat Genet. 2003;34:35. doi: 10.1038/ng1140. [DOI] [PubMed] [Google Scholar]
- 8.Lamesch P, et al. Genome Res. 2004;14(10B):2064. doi: 10.1101/gr.2496804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Nat Methods. 2008;5:621. doi: 10.1038/nmeth.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hillier LW, et al. Genome Res. 2009;19:657. doi: 10.1101/gr.088112.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mangone M, et al. Science. 2010;329:432. doi: 10.1126/science.1191244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jan CH, Friedman RC, Ruby JG, Burge CB, Bartel DP. Nature. doi: 10.1038/nature09616. published online 17 November 2010. [DOI] [Google Scholar]
- 13.Allen MA, Hillier LW, Waterston RH, Blumenthal T. Genome Res. doi: 10.1101/gr.113811.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal A, et al. BMC Genomics. 2010;11:383. doi: 10.1186/1471-2164-11-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spence WC, et al. Genome Res. doi: 10.1101/gr.114595.110. [DOI] [Google Scholar]
- 16.Harrison PM, Gerstein M. J Mol Biol. 2002;318:1155. doi: 10.1016/s0022-2836(02)00109-2. [DOI] [PubMed] [Google Scholar]
- 17.Sasidharan R, Gerstein M. Nature. 2008;453:729. doi: 10.1038/453729a. [DOI] [PubMed] [Google Scholar]
- 18.Mattick JS. Science. 2005;309:1527. doi: 10.1126/science.1117806. [DOI] [PubMed] [Google Scholar]
- 19.Kato M, de Lencastre A, Pincus Z, Slack FJ. Genome Biol. 2009;10:R54. doi: 10.1186/gb-2009-10-5-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoeckius M, et al. Nat Methods. 2009;6:745. doi: 10.1038/nmeth.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruby JG, Jan CH, Bartel DP. Nature. 2007;448:83. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung W, et al. Genome Res. doi: 10.1101/gr.113050.110. [DOI] [Google Scholar]
- 23.Ruby JG, et al. Cell. 2006;127:1193. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 24.Lu ZJ, et al. Genome Res. doi: 10.1101/gr.110189.110. [DOI] [Google Scholar]
- 25.Ercan S, et al. Nat Genet. 2007;39:403. doi: 10.1038/ng1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu W, et al. Genome Res. doi: 10.1101/gr.114587.110. [DOI] [Google Scholar]
- 27.Fukushige T, Krause M. Development. 2005;132:1795. doi: 10.1242/dev.01774. [DOI] [PubMed] [Google Scholar]
- 28.Grove CA, et al. Cell. 2009;138:314. doi: 10.1016/j.cell.2009.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy PJ, Stuart JM, Lund J, Kim SK. Nature. 2002;418:975. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, et al. Cell. 2009;139:623. doi: 10.1016/j.cell.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamath RS, et al. Nature. 2003;421:231. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 32.Ashburner M, et al. Nat Genet. 2000;25:25. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carr A, Biggin MD. EMBO J. 1999;18:1598. doi: 10.1093/emboj/18.6.1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moorman C, et al. Proc Natl Acad Sci U S A. 2006;103:12027. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacArthur S, et al. Genome Biol. 2009;10:R80. doi: 10.1186/gb-2009-10-7-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alon U. Nat Rev Genet. 2007;8:450. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 37.Yu HY, Gerstein M. Proc Natl Acad Sci U S A. 2006;103:14724. doi: 10.1073/pnas.0508637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simonis N, et al. Nat Methods. 2009;6:47. doi: 10.1038/nmeth.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez NJ, et al. Genes Dev. 2008;22:2535. doi: 10.1101/gad.1678608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hornstein E, Shomron N. Nat Genet. 2006;38(suppl):S20. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- 41.Edgar LG, Wolf N, Wood WB. Development. 1994;120:443. doi: 10.1242/dev.120.2.443. [DOI] [PubMed] [Google Scholar]
- 42.Seydoux G, Fire A. Development. 1994;120:2823. doi: 10.1242/dev.120.10.2823. [DOI] [PubMed] [Google Scholar]
- 43.Meyer BJ. X-Chromosome Dosage Compensation. In: The C. elegans Research Community, editor. WormBook. WormBook; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelly WG, et al. Development. 2002;129:479. doi: 10.1242/dev.129.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barnes TM, Kohara Y, Coulson A, Hekimi S. Genetics. 1995;141:159. doi: 10.1093/genetics/141.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rockman MV, Kruglyak L, Przeworski M. PLoS Genet. 2009;5:e1000419. doi: 10.1371/journal.pgen.1000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu T, et al. Genome Res. doi: 10.1101/gr.115519.110. [DOI] [Google Scholar]
- 48.Ooi SL, Henikoff JG, Henikoff S. Nucleic Acids Res. 2010;38:e26. doi: 10.1093/nar/gkp1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egelhofer TA, et al. Nat Struct Mol Biol. doi: 10.1038/nsmb.1972. [DOI] [Google Scholar]
- 50.Albertson DG, Rose AM, Villeneuve AM. In: C elegans II. Riddle DL, Blumenthal T, Meyer BJ, Preiss JR, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. pp. 47–78. [PubMed] [Google Scholar]
- 51.MacQueen AJ, et al. Cell. 2005;123:1037. doi: 10.1016/j.cell.2005.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gu SG, Fire A. Chromosoma. 2010;119:73. doi: 10.1007/s00412-009-0235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phillips CM, et al. Nat Cell Biol. 2009;11:934. doi: 10.1038/ncb1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ercan S, Dick LL, Lieb JD. Curr Biol. 2009;19:1777. doi: 10.1016/j.cub.2009.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jans J, et al. Genes Dev. 2009;23:602. doi: 10.1101/gad.1751109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kohlmaier A, et al. PLoS Biol. 2004;2:E171. doi: 10.1371/journal.pbio.0020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ikegami K, Egelhofer TA, Strome S, Lieb JD. Genome Res. doi: 10.1186/gb-2010-11-12-r120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kouzarides T. Cell. 2007;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 59.Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ. Genome Res. 2006;16:1505. doi: 10.1101/gr.5560806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valouev A, et al. Genome Res. 2008;18:1051. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ercan S, Lubling Y, Segal E, Lieb JD. Genome Res. doi: 10.1101/gr.115931.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaplan N, et al. Nature. 2009;458:362. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tillo D, et al. PLoS ONE. 2010;5:e9129. doi: 10.1371/journal.pone.0009129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hörz W, Altenburger W. Nucleic Acids Res. 1981;9:2643. doi: 10.1093/nar/9.12.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Segal E, Widom J. Curr Opin Struct Biol. 2009;19:65. doi: 10.1016/j.sbi.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Field Y, et al. PLoS Comput Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsankov AM, et al. PLoS Biol. 2010;8:e1000414. doi: 10.1371/journal.pbio.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kizer KO, et al. Mol Cell Biol. 2005;25:3305. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rechtsteiner A, et al. PLoS Genet. 2010;6:e1001091. doi: 10.1371/journal.pgen.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berger SL. Nature. 2007;447:407. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 71.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. Nucleic Acids Res. 2008;36(Database issue):D154. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cooper GM, et al. Genome Res. 2004;14:539. doi: 10.1101/gr.2034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moses AM, et al. PLOS Comput Biol. 2006;2:e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siepel A, et al. Genome Res. 2005;15:1034. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bickel PJ, Boley N, Brown JB, Huang H, Zhang N. Annals Appl Stat. 2010;0:1. [Google Scholar]
- 76.Eldar A, Elowitz MB. Nature. 2010;467:167. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harris TW, et al. Nucleic Acids Res. 2010;38(Database issue):D463. doi: 10.1093/nar/gkp952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.