

Abstract
Proteomics has revealed itself as a powerful tool in the identification and determination of proteins and their biological significance. More recently, several groups have taken advantage of the high-throughput nature of proteomics in order to gain a more in-depth understanding of the human brain. In turn, this information has provided researchers with invaluable insight into the potential pathways and mechanisms involved in the pathogenesis of several neurodegenerative disorders, e.g. Alzheimer and Parkinson disease. Furthermore, these findings likely will improve methods to diagnose disease and monitor disease progression as well as generate novel targets for therapeutic intervention. Despite these advances, comprehensive understanding of the human brain proteome remains challenging, and requires development of improved sample enrichment, better instrumentation, and innovative analytic techniques. In this review we will focus on the most recent progress related to identification of proteins in the human brain under normal as well as pathological conditions, mainly Alzheimer and Parkinson disease, their potential application in biomarker discovery, and discuss current advances in protein identification aimed at providing a more comprehensive understanding of the brain.
Keywords: Proteomics, laser dissection microscopy, Parkinson disease, Alzheimer disease, human brain, cerebrospinal fluid, biomarkers and mass spectrometry
Introduction
Recent advances in proteomics technologies provide powerful means to systematically profile the proteome of a complex biological system. The evolution of two-dimensional gel electrophoresis (2-DE) based approaches into liquid chromatography (LC) based high-resolution tandem mass spectrometry (MS) enables large-scale protein identification at high-throughput. The emerging technology of quantitative proteomics provides unique opportunities to reveal static or perturbation-induced changes in a protein profile. However, due to the enormous complexity of biological systems and the complex nature of proteins, an effective, in-depth protein profiling requires a multi-disciplinary approach to accomplish the goal of protein identification and quantification. Such a concerted approach typically includes several components, including sample preparation, protein or peptide separation, tandem MS (MS/MS) analysis and bioinformatics data processing, as well as independent confirmation and validation.
Advances in proteomics concept and technology have stimulated a great interest in applying this technology for studying neurodegenerative disorders. Neurodegenerative disorders, e.g. Alzheimer disease (AD), Parkinson disease (PD), and amyotrophic lateral sclerosis (ALS), represent one of the most important groups of diseases in the aging population that is fast growing in both developing and industrialized countries. The pathogeneses of these disorders remains unclear, and their clinical diagnoses, which are largely based on clinical observations currently, are unsatisfactory. State-of-the-art proteomics has revolutionized protein biomarker discovery critical to the disease diagnosis/monitoring of disease progression and has greatly facilitated studies to reveal the molecular events underlying neurodegenerative disease. In the past several years, numerous proteomic investigations using different proteomics approaches and sample types have been reported dealing with AD and PD. Thus, in this chapter, we will first briefly summarize a few proteomic techniques used in neuroproteomics, followed by discussion on proteomic characterization of proteins of normal and diseased human brain, especially in AD and PD, and the utility of identifying brain proteins in clinical biomarker discovery. Future directions of CNS proteomics will also be addressed briefly.
1. Common Proteomic Techniques
A variety of proteomic techniques have been developed for protein or peptide separation, an essential step in proteomic analysis of proteins of low abundance, prior to MS analysis: including one or 2-DE, size exclusion, ion chromatography, and reverse phase LC (RP-LC). Modern MS consists of three major modules: ion source, mass analyzer and detection unit. Based on the difference of ion source used, most of the mass spectrometers used in the field of proteomics can be generally categorized into two types, i.e. electrospray ionization (ESI) and matrix assisted laser desorption/ionization (MALDI) instruments. The most widely used mass analyzers include ion trap, triple quadrupole, time-of flight (TOF) and Fourier transform ion cyclotron resonance (FTICR). They are very different in their mechanism of ion separation, mass accuracy and resolution, and complementary in protein identification when used in concert. The interpretation of enormous amounts of mass spectrometric data relies on bioinformatics tools and protein database. The most widely used database search algorithms include SEQUEST, Mascot and Xtandem.
While identifying proteins is important in understanding any given biological process, a more informative step is to detect quantitative alterations of proteins when a system is perturbed. Traditionally, proteins are quantified by 2-DE, which is a labor intensive technique; thus, development of novel quantitative proteomics represents a milestone of proteomics technology for its applications in clinical studies, such as biomarker discovery. In the past several years, many MS based quantitative proteomics methods have been developed [1]. These methods include the use of chemical reactions to introduce isotopic tags at specific functional groups on peptides or proteins, such as ICAT (isotope coded affinity tags) [2] and iTRAQ (isobaric tags for relative and absolute quantitation) [3]; metabolic isotope labeling using heavy amino acids, such as SILAC (stable isotope labeling of amino acids in cell culture) [4]; and methods that introduce stable isotope tags via enzymatic reactions, such as enzymatic 18O incorporation [5]. Although different in sample preparation and other aspects, these methods share the use of stable isotope labeling to generate the mass signatures that identify sample origin and serve as the basis for accurate quantification, thus affording comparison of two or more proteomes simultaneously. More recently, label free quantitative proteomics is also being actively investigated exploring new alternatives for comparative proteomics studies. These techniques, though not being utilized in CNS proteomics, appear to provide excellent quantitative data, at least for abundant proteins [6, 7].
2. Human Brain Proteome
Characterization of the human brain proteome is the first step towards understanding its structure and function. In contrast to most organ systems in humans, the brain is highly spatially organized and specialized. Additionally, most neurodegenerative diseases are region-specific. For instance, AD is characterized by neuronal loss primarily in hippocampus and closely related regions as well as certain regions of cerebral cortex. In contrast, the neuronal degeneration that defines PD is largely focused in the brainstem (see Figure 1), before affecting multiple brain regions as the disease progresses.
Figure 1.
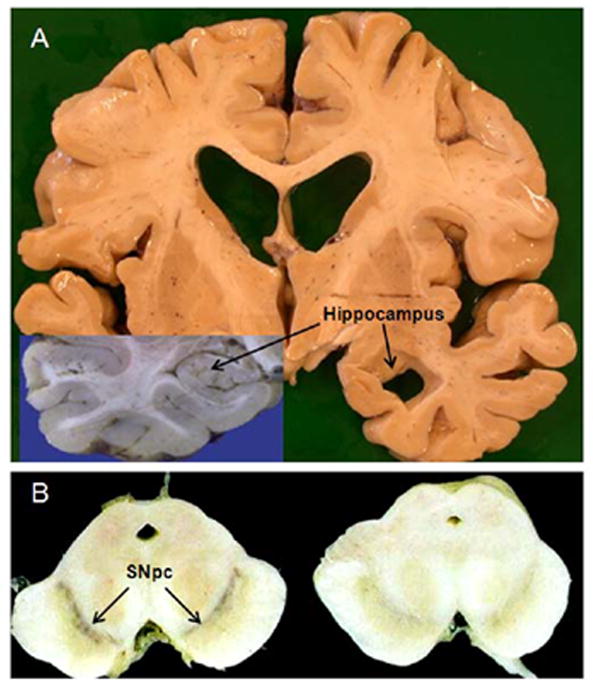
Regional neurodegeneration in patients with pathologically confirmed AD and PD. (A) Representative coronal section from patient with AD. Left side of section shows a superimposed picture of an intact hippocampus from a normal patient. Note the dramatic atrophy of the hippocampus in the AD patient (right side of figure). (B) Representative transverse section of the midbrain from a normal (left) and PD patient (right). A significant loss of the pigmented dopaminergic cell bodies in the SNpc (substantia nigra pars compacta) can be seen in the PD compared with the normal patient.
Thus, identification of proteins unique to each brain region, particularly those involved in neurodegenerative disease, could provide greater insight into the pathogenic nature of the disease as well as reveal novel therapeutic avenues aimed at attenuating or abrogating the disease process.
3. Proteomic analysis of the human cortex
3.1. Proteome of the human cortex under normal conditions
The initial large scale proteomics analysis of the human cortex has primarily focused upon the temporal lobe, a region commonly involved in the generation and propagation of some forms of epilepsy. Three independent studies by a consortium, HUPO (Human Proteome Organization), have utilized various proteomic platforms to examine tissue taken from the temporal lobe of epileptic patients in order to identify proteins localized to this specific brain region. One such study identified 1533 proteins through a multidimensional fractionation and separation approach [8]. Further analysis of the temporal lobe using 2-DE coupled with MALDI-TOF/TOF identified 375 proteins [9], while 209 proteins were identified following a shotgun proteomics approach [10]. Of note, shotgun proteomics is defined by John Yates and associates where proteins are separated by strong cation exchange (SCX) and RP-LC, followed by MS analysis [11].
Subsequently, the proteome of the human frontal cortex, a region associated with many neurodegenerative disorders, including AD, PD with dementia (PDD), dementia with Lewy body disease (DLB) and frontotemporal dementia (FTD) has been investigated. For example, in a recent shotgun proteomics-based study, tryptically digested peptides from four biochemical fractions were separated by SCX and RP-LC, followed by MALDI-TOF/TOF analysis [12]. This study identified 1782 non-redundant proteins; of those, 812 were reliably identified by 2 or more peptides. Interestingly, 463 of these proteins were also identified in the human temporal lobe [8–10]. The molecular functions of the proteins identified in the human cortex ranged from protein binding and catalytic activity to cellular transport and signal transduction. In addition, several proteins specific to the frontal cortex were identified that have previously been associated with neurodegenerative disease, such as apolipoprotein E and cathepsin D, which have been implicated in AD [13, 14], as well as neurofilament triple H protein, which has been implicated in amyotrophic lateral sclerosis (ALS) [15]. The caveat with respect to the regional specific proteins, however, is the characterization of any given regional proteome is incomplete. A more detailed discussion as to cortical regional specific proteins vs. those overlapped between the cortical regions or vs. other brain regions as well as the cerebrospinal fluid can be found in our recent publications [12, 16].
3.2. Proteomic identification of cortical proteins associated with pathological structures unique to neurodegenerative diseases
The precise cellular mechanism responsible for various neurodegenerative diseases is unclear; however, pathologically, most of them are associated with unique pathological structures in the cortex, including Lewy bodies (LBs) in PD, and neurofibrillary tangles (NFTs) and neuritic plaques in AD, respectively. The presence of several insoluble proteins, such as α-synuclein (SNCA), amyloid β(Aβ), and tau, all of which are critically involved in PD and AD, has been identified within many of these pathological inclusions. Thus, a better understanding of these inclusions and their contents may provide greater insight into the formation and pathogenesis of these disorders, as well as generate potential avenues for therapeutic intervention.
3.2.1. Cortical Lewy Bodies
Lewy body inclusions are one of the pathological hallmarks of PD, PDD, and DLB [17, 18]. Using conventional biochemical and/or histochemical methods, previous studies have identified approximately 70 molecular components of LBs, in both the brainstem and cortex, involved in a wide range of cellular functions [19], with SNCA being one of the major components. It is the presence of SNCA in LBs that has allowed a reliable means to identify and localize LB pathology in more advanced PD cases [20]. A recent study coupled laser capture microdissection (LCM) of cortical tissue immunostained for SNCA with LC/MS/MS of LBs from patients with a clinical and pathological diagnosis of DLB. This platform yielded the identification of 156 proteins (identified by 2 or more peptides) [21]. Interestingly, 17 of the proteins identified in this study had been previously demonstrated to be associated with cortical or brainstem LBs [21], which suggests that the majority of the proteins discovered are novel and have not been shown to associate with cortical LBs. One particular protein, heat shock cognate 71 (HSC70) associated previously only with brainstem LBs, has been implicated in endocytosis, oxidative stress, as well as apoptosis. It is anticipated that further characterization of proteins identified through this analysis could provide an improved understanding of the pathogenesis of PD as well as other disorders associated with LBs.
3.2.2. Neurofibrillary Tangles
Neurofibrillary tangles in the cortex and hippocampus are one of the pathologic hallmarks of AD [22], with the microtubule binding protein, tau, being one of the major proteins aggregated in NFTs [22]. While the mechanisms leading to the formation of NFTs and their subsequent pathology remain unclear, the identification of proteins that associate with NFTs may provide new insight into protein-protein interactions that might be important to AD. In particular, the mitochondrial molecular chaperone, peptidylprolyl-cis, transisomerase (Pin1) has been shown to play a role in modulating the phosphorylation state of tau, i.e. dephosphorylate hyperphosphorylated tau, thereby reducing the generation of NFTs [23–25]. Consistent with this argument, Pin1 has been shown to be oxidatively modified and reduced in AD, a process that should facilitate accumulation of hyperphosphorylated tau and NFTs [26, 27].
To take an unbiased approach to revealing novel proteins in NFTs, a recent study used LCM to isolate individual NFTs from hippocampal pyramidal cells in AD patients, followed by proteomic analysis of these accumulations by LC/MS/MS. With this technique, 72 proteins were identified, of which 63 proteins had no previous known association with NFTs [28]. Further analysis of one of these proteins, GAPDH was found to exist as a detergent-insoluble fraction from AD patients, in addition to colocalizing to NFTs. GAPDH has been shown to localize with other proteinaceous inclusions, including LBs [29], in addition to having an increased expression in the hippocampus of AD patients, compared with control [30]. Whether or not GAPDH is important to the generation of NFTs remains to be investigated.
3.2.3 Amyloid plaques
In addition to the presence of intraneuronal NFTs, another pathological characteristic of AD is the extracellular aggregation of amyloid plaques, composed primarily of insoluble Aβ [31, 32] in addition to other neuronal and glial components [33]. Until recently, the identification of proteins associated with plaques was largely by immunohistochemical staining methods. These proteins, though limited in number, have provided a general understanding of the composition of these inclusions and potential mechanisms involved in their formation. For instance, as mentioned above, Pin1 has been implicated in the modulation of tau phosphorylation, which affects the formation of NFTs [23–25]. Recent work has also identified Pin1 as playing a role in Aβ production, with a reduction in Pin1 resulting in an increase in Aβ [34]. Given the previous proteomic data demonstrating the oxidative modification and reduction in Pin1 in AD brains, this implies that Pin1 may be a specific protein involved in plaque formation, although the precise mechanism involved remains to be elucidated.
Similar to identification of proteins contained in LBs and NFTs, a recent study by Liao et al., (2004) [35], took a more nonbiased approach to characterize the protein composition of amyloid plaques taken from the cortex of AD patients. This investigation was able to identify 488 proteins, of which 26 were found to be unique to amyloid plaques, including well characterized plaque proteins as well as proteins previously unknown to exist as plaque components. It needs to be stressed that the results from this study confirm, for the most part, the presence of plaque-associated proteins previously identified by immunohistochemistry [33].
3.3. Quantitative proteomic analysis of neurodegenerative diseases
3.3.1. Lewy body progression
While the experiments discussed above have greatly facilitated the identification of particular proteins associated with specific neuropathological markers of disease, more recent work has focused on quantifying the changes in abundance of candidate proteins to allow for the progression of the disease to be followed. This line of research can be exemplified by proteomics analysis of LB progression, a concept defined by Braak et al. [20], who have suggested that as PD advances, it is invariably accompanied by spreading of LBs from the brainstem to the limbic system and eventually to the isocortex. Furthermore, the progression of these pathological inclusions appear to parallel the appearance of many of the non-motor features of PD [36]. Thus, identification of novel proteins associated with LBs and the changes in their relative abundance as the disease progresses from the brainstem to the isocortex could yield useful information concerning specific proteins that are imperative or mediate PD progression.
As the first step towards identifying proteins associated with LB progression, a recent investigation quantitatively compared tissue from control and PD patients at different stages [36]. Samples were labeled with specific iTRAQ reagents before being analyzed by MALDI-TOF/TOF. Through this analysis a total of 1864 non-redundant proteins were identified, of which 199 displayed significant changes in relative abundance between PD and control groups [37], thus providing the identification of many novel proteins that likely contribute to the progression of PD. Indeed, many of these proteins are involved in energy metabolism and neurotransmission. One particular protein that was further validated by an alternative means (Western blot) and whose relative levels were found to be decreased in all PD groups was mortalin. Mortalin has been demonstrated to have roles as a molecular chaperone, mitochondrial import and energy generation, as well as oxidative stress [38]. Although the precise mechanism by which mortalin produces its effects is still unknown, given its role in diverse biological functions it is likely that changes in levels of mortalin in cortical neurons may also be essential in PD progression. A more in depth discussion of mortalin and its biological significance can be found in the original publication [39].
3.3.2. Proteomic Analysis of Insoluble Proteins in Cortex
As mentioned earlier, the accumulation of insoluble proteins, forming unique pathological structures, is an important characteristic of several neurodegenerative diseases, including AD and PD [40, 41]. While the exact implications of these accumulations is not known, it has been previously demonstrated that the aberrant conversion of a protein to an insoluble form disrupts its endogenous functional characteristics and can lead to further damage within the cell [42] before formation of unique structures, e.g. LB and amyloid plaques/NFTs for PD and AD, respectively. Given the association of the accumulation of insoluble proteins with neurodegenerative disease, the identification of these proteins at different stages of disease progression, including those prior to developing unique pathological structures, could provide further insight into the pathogenic mechanisms involved in these diseases as well as their role in the progression of these disorders. These investigations are particularly informative as to identifying novel proteins critical to formation of LB and plaques/NFTs. Two examples of such studies are discussed in more detail below.
3.3.2.1. Proteomic analysis of insoluble proteins in AD
In an attempt to determine the extent of insoluble proteins that might be involved in the course of AD pathogenesis, the amount of accumulated insoluble proteins was determined in the cortex of patients with prodromal AD who had a mild reduction in cognitive function without dementia, late onset AD (LOAD), as well as patients with autosomal dominant forms of AD derived from mutations in the genes for presenilin 1 and 2 (PSEN1 and 2) [43]. Tissue from these groups was separated to yield a detergent insoluble fraction before undergoing proteomic analysis using LC/MS/MS. While a slight increase in insoluble proteins, such as 14-3-3, GAPDH, and HSC70 was demonstrated in the age-matched control samples, a much greater increase was seen in the LOAD group with an intermediate increase observed in the prodromal samples. Moreover, patients with presenilin mutations exhibited an increase in insoluble proteins comparable to that found in the LOAD patients. Remarkably, analysis of the deposition of insoluble Aβ and tau, the major components of amyloid plaques and NFTs, respectively, in each of these groups showed increases in insoluble Aβ in all groups. In contrast, insoluble tau was only seen in the LOAD samples as well as the dominant autosomal AD samples. These changes in tau are consistent with the amyloid hypothesis of AD that proposes pathologic changes in tau occur relatively late in the pathogenesis of AD. Additionally, the increased accumulation of insoluble Aβ from prodromal AD and LOAD may represent an alteration in the biochemical state of this protein that may contribute to the pathogenesis of AD. Of note is the observation that several of the insoluble proteins shown to increase in the prodromal, LOAD and PSEN1 and 2 samples have also been demonstrated to have differential protein levels in the hippocampus of AD patients compared with control [30]. Furthermore, several of these proteins have been shown to be oxidatively modified [30], which may affect their biochemical conformation and their propensity to accumulate.
3.3.2.2. Proteomic analysis of insoluble proteins in Parkinsonism-Dementia Complex of Guam
In addition to AD and PD, other neurodegenerative diseases are characterized by the pathological accumulation of insoluble proteins. One such disease is Parkinsonism-dementia complex (PDC) of Guam, which is classified histopathologically as a tauopathy, a group of neurodegenerative diseases, including AD, which shares in common abnormal tau-immunoreactive structures in different brain regions. Clinically, patients with PDC exhibit an akinetic-rigidity resembling PD, and are accompanied by dementia that is similar to AD. In cortical tissue taken from patients with PDC, AD, and age-matched controls, MALDI-TOF/TOF analysis, in conjunction with iTRAQ labeling, revealed 106 proteins, of which six, including Aβ-42, tau and ApoE, and ubiquitin were determined to have a higher abundance in the AD group and two, such as isoform A of microtubule associated protein (MAP) tau, as well as SNCA demonstrated a higher abundance in the PDC, compared with the control group [44]. These data appear to agree with previous studies that detected the presence of SNCA and tau positive pathology in PDC brain tissue [45–48]. Thus, these data robustly establish some and challenge other aspects of the proposed characterization of PDC. For instance, while establishing the presence of abnormal SNCA in relevant brain regions of PDC, these data do not support the characterization of PDC as a triple amyloidosis, meaning formation of brain amyloid from Aβ species, tau and SNCA [42], given the fact that no quantifiable alteration in levels of insoluble Aβ was observed.
4. Human Midbrain Proteome
Until recently, very little was known about the proteome of the human midbrain, whose degeneration has been linked to several neurodegenerative disorders with motor symptoms and signs, including PD, multiple system atrophy (MSA), and progressive supranuclear palsy (PSP). Indeed, it is the degeneration of dopamine (DA)-containing neurons in the SNpc that underlies the motor deficits that characterize most movement disorders [49]. Thus, a more thorough characterization of the human midbrain proteome is imperative to understanding the pathophysiology of PD as well as other neurodegenerative disorders associated with the midbrain.
4.1. Proteomic identification of proteins in the human midbrain
The first unbiased profiling work done in the human midbrain, where 44 proteins were identified, is based on 2-DE, followed by MALDI-TOF/MS [50]. A more extensive investigation of the human SNpc performed more recently identified 1263 non-redundant proteins, utilizing MALDI-TOF/TOF as well as LTQ MS platforms [16]. This study is part of a project aimed at identification of proteins in the midbrain that are unique to disease or confer regional specificity to human movement disorders. Proteins were identified and quantified by comparing samples of different brain regions from PD and control patients labeled with specific iTRAQ or ICAT reagents [2, 12, 51] before being analyzed by MALDI TOF/TOF and LTQ mass spectrometric platforms, respectively (see below for more detailed discussion about quantitative changes).
Among the proteins identified in the midbrain tissue, many are functionally or structurally involved in neurodegeneration. In particular, DJ-1, glutathione S-transferase omega1, manganese superoxide dismutase, and UCHL-1 are well-known to be important in PD pathogenesis, including mitochondrial function, oxidative stress, protein degradation, and neuroinflammation [52–55]. Two recent proteomic studies also demonstrated the oxidative modification and altered expression of DJ-1 and UCHL-1 in PD brains [56, 57]. While this study represents the most comprehensive survey of the human midbrain conducted, to date, further characterization of the SNpc proteome will significantly facilitate our understanding of the function and expression of proteins involved in PD and other midbrain-associated disorders.
4.2. Differential vulnerability of SNpc and VTA
The clinical features of PD, the most common degenerative disease involving the midbrain, are comprised of motor symptoms such as resting tremor, cogwheel rigidity, postural instability, and slowness of movement (bradykinesia) [58]. The manifestation of these clinical symptoms can be attributed to the progressive degeneration of the DA-containing neurons of the ventral midbrain, particularly the SNpc and the subsequent reduction of DA in the striatum [49, 59–61]. It has been hypothesized that DA containing neurons are especially vulnerable to neurodegeneration in PD, largely because of increased oxidative stress secondary to DA metabolism in these neurons [53, 62, 63].
Interestingly, while the SNpc and VTA are both DAergic, are in close proximity to each other and are derived from the same progenitor cells, they display a differential vulnerability to the DAergic degeneration seen in PD. For example, whereas upwards of 90% of DAergic neurons are lost in the SNpc, a 40% reduction has been recorded in the VTA [64–68]. A similar pattern of DAergic degeneration has been observed in several animal models of PD, including mice or monkeys treated with MPTP (1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine) and rats challenged by rotenone [69–74]. This suggests that the DAergic neurons of the SNpc possess an intrinsic property that renders them substantially more susceptible to the pathogenic mechanism(s) involved in PD.
To better comprehend the differences between these two DAergic regions, several groups have undertaken global proteomic and genomic studies of the ventral midbrain to examine the differential expression of genes and proteins in the VTA and SNpc in control, as well as parkinsonian brains. To date, the most comprehensive genomic analysis of differential gene expression between the VTA and SNpc utilized laser LCM and gene microarrays in the rat ventral midbrain to identify 3500 genes [75]. Of these, 144 genes showed a differential pattern of expression between VTA and SNpc, most notably, genes related to mitochondrial function were more prevalent in the SNpc. A similar study in mouse midbrain identified 42 genes that were differentially elevated in the SNpc, compared to 61 genes in the VTA [76]. In contrast, the results using human material are much less comprehensive. Work by Basso et al., [50], examined differences in protein expression in the SNpc of post mortem human control and PD subjects. Using 2-DE and MALDI-TOF analysis, 44 proteins were identified, of which 9 showed changes in expression between control and disease brains.
To extensively and quantitatively compare the protein profiles in the SNpc with that of VTA in human brain directly, we have recently taken advantage of shotgun proteomics, along with iTRAQ labeling [3, 77], to reveal the differentially expressed proteins in the VTA and SNpc of control as well as PD patients. This analysis discovered 722 proteins that exhibited a greater than 50% change in their relative abundance between these two regions. Of these proteins 33 were also identified by two or more peptides as well as consistently identified in two or more experimental replicates. A similar analysis of protein profiles between the SNpc of control and PD subjects identified 549 proteins that exhibited a greater than 50% change in their relative abundance. Of these 6 proteins were also identified by two or more peptides as well as in two or more experimental replicates. Validation of these proteins and their changes are currently underway in our lab. It is hoped that further analysis of these proteins will provide some insight into the differential variability of the SNpc and VTA as well as help identify proteins in the SNpc that may be involved in PD pathogenesis (a complete report on this dataset is under preparation as a separate manuscript).
5. Bioinformatics in proteomics
It cannot be overemphasized that proteomics only provides a list of proteins that are potentially involved in neurodegenerative diseases, i.e. proteins identified require further validation in order to determine their biological significance [78]. This is largely because proteins can be misidentified due to an incomplete human database, a limitation common to all current discovery types of proteomic studies. Another problem is that there is currently no method available for validating candidate proteins in a high-throughput manner. The challenge, then, becomes how to select hundreds, if not thousands, of candidate proteins revealed via discovery proteomics for further validation with conventional biochemical methods? Two approaches that are being used are discussed in more detail below:
5.1. Gene Ontology analysis
In order to identify relevant biological processes or functions from the protein expression data, many groups, including us, have take advantage of Gene Ontology (GO) analysis developed for microarray [12, 37, 79] that determine enhanced gene categories. This approach identifies GO categories by evidence of over-representation of significant proteins as well as allows the categorization of proteins that belong to a similar biological pathway or have similar biological functions. Thus, the biological function is expected to be more relevant based on the degree of over-representation of particular proteins within that biological category. This method leads to a concentrated evaluation of particular proteins identified in MS screens based on their perceived biological importance, eliminating much of the guess work that might be involved in choosing proteins of interest for further analysis and confirmation.
To further illustrate this approach, let’s examine the databases generated from the human midbrain proteome as well as LB progression studies that identified more than 892 proteins related to PD or LB progression. The top ranked or most significant GO categories include synaptic regulation and cellular respiration (Figure 2). Not surprisingly, several of the proteins identified in the top ranked GO categories have been associated with PD, including DJ-1 [80], SNCA [81], and the DA transporter (DAT) [82]. In addition to proteins known to be involved in PD, numerous other proteins were identified as having important roles in cellular functions of the CNS, such as synaptic vesicle glycoprotein 2A [83] and septin 9 [84], yet no known involvement in PD. Consequently, with this method, in addition to confirming the presence of significant proteins involved in PD, particularly those located in the midbrain or LBs, we have also generated a list of proteins for future analysis which may provide us with a greater insight into PD pathogenesis.
Figure 2.

The distribution of proteins identified from the human midbrain proteome as well as LB progression according to GO analysis and annotations. (A) Biological processes. (B) Cellular components.
5.2. Network analysis
Another advantage that can be taken off established gene array platforms is network analysis, which can greatly enhance our understanding of functional networks and pathways associated with the proteins of interest. Such an approach can expand a core list of key elements identified, such as a candidate protein list, toward a relevant interpretation of a biological condition at the level of reaction networks and pathways, providing the potential to boost our understanding of biological processes at a cellular or systems level. In a recent proteomics study of oxidative injury associated with the pathogenesis of neurodegenerative disease, pathways and network component analysis was applied to analyze the oxidatively modified proteins identified [85]. The results suggested that there are three major protein networks that could be potentially altered in a transgenic mouse model (PS1 + AβPP) of AD as a result of oxidative modifications. These pathways include inducible nitric oxide synthase (iNOS)-integrin signaling pathway, CRE/CBP (cAMP response element binding protein) transcription regulation and rab-lyst (Rab 4-lysosomal trafficking) vesicular trafficking. The development of bioinformatics and computational tools, in conjunction with the continuous increase in biological data should provide a more comprehensive approach to interpret the enormous complexity embedded in cellular reaction networks.
6. Overlap with CSF, potential biomarkers for disease diagnosis and monitoring disease progression
Currently, diagnoses of major neurodegenerative diseases are based, primarily on clinical criteria that are supported by laboratory investigations, and more recently, structural and functional imaging analysis. Even under the best circumstances these techniques allow for the accurate diagnosis of a particular neurodegenerative disorder 50–90% of the time (depending on the disease of interest). Additionally, in many cases the diagnosis remains uncertain until death. Thus, the discovery of disease-specific markers can assist with clinical diagnosis, monitoring of disease progression, and identification of new therapeutic targets. The search for consistent and reliable biomarkers for disease states is being actively pursued in the cerebral spinal fluid (CSF), plasma, urine, and saliva. CSF appears to hold the greatest promise for yielding reliable biomarkers. A major reason for the apparent superiority of CSF over plasma or any other body fluids lies in the proximity of the CSF to the region of disease, which more faithfully reflects the pathological state. In contrast, markers found in the plasma may not truly represent the specificity of the biomarker for the CNS diseases, largely because of confounding factors involved in plasma proteins (brain only accounts for ~4% of total body weight).
While the use of CSF appears to be the best suited vehicle for biomarker discover, it is not without its own challenges. These limitations are primarily centered on the large dynamic range of protein concentrations present in the CSF that limits the sensitivity of modern mass spectrometry for proteins of low abundance (the problem is even greater for proteomics of plasma; see Table 1 which summarizes pros and cons of biomarker discovery with CSF vs. plasma/serum samples).
Table 1.
Cerebral spinal fluid versus serum/plasma for biomarker discovery
| Advantages | Disadvantages | |
|---|---|---|
| Cerebral Spinal Fluid |
|
|
| Serum/Plasma |
|
|
Furthermore, of these proteins, 50% and 15% of the total protein content is composed of albumin and IgG, respectively, which can lead to the overshadowing of the lower abundance proteins. In addition, CSF proteins are extremely heterogeneous, as a result of PTMs, making the identification of specific proteins particularly difficult. Given these challenges associated with protein identification and biomarker discovery in the CSF, we have used integrated techniques over the last few years to establish a more comprehensive identification of proteins unique to neurodegenerative disorders. These include separation of trypsinized peptides employing orthogonal mechanisms, e.g. organic precipitation, SCX and RP-LC and the use of MS technologies with an increase in mass accuracy, high scan frequency, and complementary MS instrument. Finally, the analysis of a subproteome (glycoproteins), a topic discussed in more detail later, within the CSF has allowed for an increase in protein identification and an improved understanding of the CSF. As of today, a total of close to 3,000 CSF proteins have been identified when all studies are combined [86].
Although identification of a total of close to 3,000 proteins is quite significant in its own right, the proteins identified that are specific to the CNS are in the minority. In addition, the overlap between brain proteome and CSF proteome is quite low. For instance, we have compared our CSF dataset to the datasets acquired in our proteomic analysis of the human cortex and midbrain in order to determine the amount of overlap between particular proteins, only identifying, 198 and 140 overlapped with CSF in the midbrain and cortical proteome, respectively [12, 16]. Additionally, several proteins critical to AD and PD, e.g. tau and SNCA, are not seen in the CSF proteome. The huge dynamic range of the human brain and CSF proteome has been mainly blamed for the inability to achieve a more complete proteome identification. However, other issues including post-translational modification (PTM) may also be a major contributor, as the peptides with PTM will be ignored during profiling process. In our opinion, there are at least two strategies, namely identification of subproteomes and targeted proteomics, to circumvent this problem at least partially. Both of which will be discussed in the Future of Proteomics below.
7. Future of Proteomic Research for Neurodegenerative Diseases
While tremendous advances in the field of proteomics have been achieved in the last decade, providing researchers a more powerful tool with which to decipher larger and more complex proteomes, the future of proteomics research remains wide open. An area of proteomics that has been gaining considerable attention, both in terms of technical advancement and general research interest is the identification and quantification of the subproteome with unique PTMs. PTMs, e.g. phosphorylation, ubiquitination, and glycosylation, determine the activity, localization and signal transduction properties of proteins. Thus, given the transient and dynamic nature of these PTMs, a majority of recent research has focused on improved techniques that allow the reliable identification and quantification of these subproteomes. Analysis of subproteomes may also improve the dynamic range of proteomics as well as contribute to a greater understanding of molecular mechanisms underlying neurodegenerative disease.
7.1 Phosphorylation
Phosphorylation of proteins is broadly involved in cell signaling and cellular regulation. Phosphorylated proteins appear to play a prominent role in the formation of pathologic inclusions in both AD and PD. Hyperphosphorylation of the microtubule protein, tau, has been demonstrated to be a major mechanism in generation of NFTs, a major pathologic hallmark of AD [87]. Furthermore, the phosphorylation of SNCA at serine residue 129 has been demonstrated to be a contributor to the aberrant accumulation of SNCA as well as the formation of LBs [88]
Future research on proteins with phosphorylation lies in characterization of novel sites that are unique to each disease or disease progression. To this end, the sites of phosphorylation on a particular protein are typically identified through MS by mass shifts in the fragment ions. However, a common problem with identification of phosphorylation sites as well as other PTM sites is the instability of this modification during MS/MS fragmentation. Another limitation routinely encountered is the low abundance of phosphoproteins and phosphopeptides in addition to low detection sensitivity of these species. Thus, several techniques involving noncovalent as well as covalent properties have been introduced to enrich phosphoanalytes and improve detection sensitivity. A specific noncovalent strategy for phospho-enrichment, immobilized metal affinity chromatography (IMAC) purifies phosphoproteins and peptides by adsorbing them to chelated metal ions, such as Fe3+ and Ga3+ through metal-phosphate ion pair interactions [89]. Another noncovalent method to isolate phosphoproteins and phosphopeptides is the immunoprecipitation of phosphorylated species using phospho-specific antibodies followed by MS analysis [90–92]. Covalent techniques utilized for purification of phosphoanalytes involve mass tagging of the phosphor-species, either through stable isotope labeling, such as SILAC [4], or through the use of chemical derivatization to couple stable isotope labels to peptides using β-elimination or phosphoramidate coupling [93–95]. Furthermore, several of these techniques, most notably IMAC have been utilized to enrich and examine phosphorylated species in the brain, both in vitro and in vivo [96–98], demonstrating the utility of these strategies in neuroproteomics. It is expected that a nonbiased profiling of phosphorylated proteins in different brain regions that are unique to each disease or stages of a disease will significantly contribute to our understanding of these diseases. The recent advances in mass spectrometry for ion fragmentation techniques, such as electron capture dissociation (ECD) and electron transfer dissociation (ETD), has greatly enhance the capability of identifying phosphorylated peptides [99–101], which typically suffer from low sensitivity. It is anticipated that the combination of complementary phosphoprotein/peptide enrichment methods with an advanced tandem mass spectrometer equipped with ETD or ECD should provide a powerful tool to study PTMs, in particular phosphorylation, that are meaningful to neurodegenerative diseases.
7.2 Ubiquitination
Like phosphorylation, ubiquitination is involved in several aspects of cellular regulation, including protein stability, activity, localization and degradation. One of the most important functions of ubiquitin is its coupling to the proteasomal system, which is critical to the degradation of normal proteins as well as misfolded proteins. It is the accumulation of misfolded proteins that has been implicated in the pathogenesis of several neurodegenerative diseases [102]. Most proteins found in LBs, NFTs and amyloid plaques, including SNCA, tau and Aβ, have been found to be ubiquinated [103–105]. Ubiquitin is covalently coupled to lysine residues on target proteins through E3 ligases. This presents a unique challenge for MS-based identification of ubiquitinated proteins because of the trypsin proteolysis of ubiquitin and its target proteins at lysine residues, resulting in the generation of cleavage products that are easily missed during MS analysis. Similar to phosphorylation, these issues can be reduced by enrichment of the ubiquitin signal by affinity purification. Indeed, immunopurification techniques have proven successful in analyzing ubiquitinated proteins isolated from cortical neuron cultures following ischemic conditions [106]. In addition, more comprehensive databases of ubiquinated proteins and sites have become an important tool in the identification and quantification of this PTM. It is hoped that these advanced technologies will facilitate our understanding of aberrant molecular events that play a role in neurodegeneration.
7.3. Glycosylation
Protein glycosylation is one of the most common PTMs and can be categorized as being O-linked (carbohydrates linked to serine or threonine residues) or N-linked (linked to asparagine residues). Glycosylated proteins are especially prevalent in proteins destined for peripheral environments, such as plasma, or CSF, making them excellent candidates for biomarker discovery. However, several aspects of glycosylated proteins and peptides make identification of these species particularly difficult. For instance, glycoproteins generally exist as a heterogeneous complex of glycoforms consisting of varying degrees and sites of glycosylation. Furthermore, the low abundance of glycosylated proteins and peptides in biological samples, coupled with the low signal intensity of glycosylated peptides compared to non-glycosylated peptides, also contributes to the challenge of proteomics analysis of glycoproteins. Thus, as with phosphorylation and ubiquitination, the enrichment of the glycosylated species is imperative to a more robust identification of target proteins. Currently, two methods exist for glycoprotein isolation and enrichment, lectin affinity purification and hydrazide chemistry. These methods were recently applied during our analysis of the glycoprotein subproteome of human CSF and allowed for the reliable identification of several hundred non-redundant proteins that have potential use as biomarkers in neurodegenerative disease research [107]. Although extensive analysis of glycosylated proteins has not been done yet, glycosylation is clearly implicated in neurodegenerative diseases, e.g. the ubiquitination of glycosylated SNCA by parkin in PD [108]. Additionally, a few proposed markers of AD are also glycosylated proteins; these include: wheat germ agglutinin (WGA)-binding glycoprotein, reelin, and lysosome-associated membrane protein 1 (LAMP-1) [109–111].
In summary, analysis of PTMs of proteins with advancing technologies will be critical in the near future for better understanding of various neurodegenerative diseases. It also needs to be stressed that neurodegenerative diseases involve PTMs beyond just phosphorylation, ubiquitination and glycosylation. For example, oxidative stress-mediated modification of SNCA as well as other proteins has been demonstrated in several neurodegenerative diseases, including AD [53, 112–114].
7.4. Targeted quantitative proteomics
Targeted quantitative proteomics technology is another area of active research, which indicates a progression away from unbiased profiling and toward a multi-phase technology that allows key elements that uniquely represent a specific biological condition to be analyzed [115, 116]. The technology uses isotope dilution followed by MS analysis [116–120], in which test-samples are supplemented (spiked) with isotope-labeled synthetic peptides, which serve as the signature markers for the identification and quantification of native peptides (target) within each sample. To date, few investigations have been reported to utilize the concept of candidate-based targeted quantitative proteomics to study selected peptides/proteins for biomarker verification/validation via ESI or MALDI based platforms. For the ESI approach, a hybrid triple-quadrupole/ion trap mass spectrometer was used to identify and quantify a selected group of targeted proteins within human plasma [120]. Alternatively, an off-line LC MALDI-TOF/TOF platform was established to monitor a panel of targeted glycopeptides/glycoproteins in human serum, in conjunction with a sample preparation strategy that allowed for the extraction of deglycosylated N-linked glycopeptides from human serum [116]. A similar approach is also taken to validate candidate markers in human CSF [121]. Figure 3 depicts the identification of a targeted protein in CSF using a LC MALDI-TOF/TOF based platform. These early investigations have demonstrated the feasibility and advantages of the MS-based targeted quantitative proteomics to simultaneously identify and quantify a panel of selected peptides/proteins in a complex milieu, and consequently could be applied for biomarker verification/validation of AD and PD. For instance, proteins identified in human brain unique to AD, PD or disease progression that are not seen in human CSF can be tested via targeted proteomics, which significantly increases the sensitivity, as in this scenario, mass spectrometers will ignore irrelevant proteins/peptides.
Figure 3.
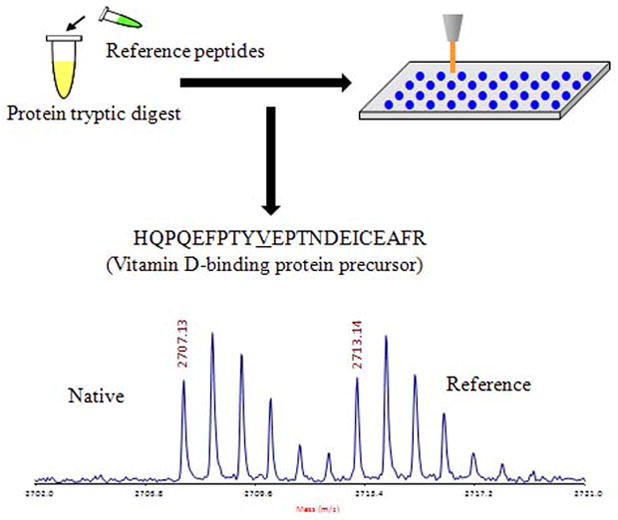
Identification of a targeted protein in CSF using a LC MALDI-TOF/TOF based platform. A signature peptide from a target protein was selected and synthesized to contain a stable isotope label and spiked into a tryptically-digested sample. The synthetic reference peptide serves as a signature marker as well as s standard for identification and subsequent quantification of the corresponding native peptides. The spiked sample is separated by LC and analyzed via MALDI-TOF/TOF targeting candidate peptides. The quantification of targeted proteins is determined by using the intensity ratio of the native and reference peptides and the known amount of spiked reference compound.
Concluding Remarks
The application of proteomics to the study of the human brain in the last decade has allowed us to make great strides in creating a comprehensive analysis of the human proteome in searching for biomarkers related to early diagnosis and therapy as well as identifying novel mechanisms underlying the pathogeneses of various neurodegenerative disorders. However, our knowledge is still far from complete. In our opinion, the following areas demand further attention. First, it is necessary to obtain a more comprehensive characterization of the human brain proteome before neurodegenerative markers can be analyzed more successfully. To this end, the existing Brain Proteome Project organized by HUPO (Human Proteome Organization) can contribute significantly by including more neuroscience oriented proteomics labs worldwide. Second, continued evaluation of the different brain subproteomes, e.g., phosphorylated and glycosylated proteins and peptides likely provides a platform from which to better evaluate the human brain proteome as well as further extend this knowledge to the diagnosis and treatment of neurodegenerative disease. Furthermore, the future of developing biomarkers for neurodegenerative disease or related neurological diseases rests in the continued development and implementation of advanced MS methodologies and analysis tools as well as elaboration of the existing proteomic databases. Finally, it is imperative to standardize methods for quality controls of sample processing as well as data interpretation and data sharing.
Acknowledgments
This work related to our own lab was supported by the National Institutes of Health Grants ES012703, AG025327, AG029808, AG05136, and NS060252 as well as grants from the Michael J. Fox Foundation, American Parkinson’s Disease Association, and the C.-M. Shaw Endowment.
Abbreviations
- AD
Alzheimer disease
- Aβ
Amyloid β
- CNS
Central Nervous System
- CSF
Cerebrospinal Fluid
- ESI
Electrospray Ionization
- FTICR
Fourier transform ion cyclotron resonance
- GO
Gene Ontology
- ICAT
isotope coded affinity tags
- iTRAQ
isobaric tags for relative and absolute quantitation
- LCM
laser capture microdissection
- LB
Lewy body
- MALDI TOF/TOF
Matrix Assisted Laser Desorption/Ionization Time-Of-Flight tandem mass spectrometer
- MS
Mass spectrometry
- NFT
neurofibrillary tangle
- PD
Parkinson disease
- RP LC
reverse phase liquid chromatography
- SCX
strong cation exchange chromatography
- SILAC
stable isotope labeling with amino acids in cell culture
- SNpc
Substantia nigra pars compacta
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature biotechnology. 1999;17:994–9. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 3.Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 5.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–42. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 6.Finney GL, Blackler AR, Hoopmann MR, Canterbury JD, et al. Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high-resolution muLC-MS data. Analytical chemistry. 2008;80:961–71. doi: 10.1021/ac701649e. [DOI] [PubMed] [Google Scholar]
- 7.Cutillas PR, Vanhaesebroeck B. Quantitative profile of five murine core proteomes using label-free functional proteomics. Mol Cell Proteomics. 2007;6:1560–73. doi: 10.1074/mcp.M700037-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Park YM, Kim JY, Kwon KH, Lee SK, et al. Profiling human brain proteome by multi-dimensional separations coupled with MS. Proteomics. 2006;6:4978–86. doi: 10.1002/pmic.200600098. [DOI] [PubMed] [Google Scholar]
- 9.He S, Wang Q, He J, Pu H, et al. Proteomic analysis and comparison of the biopsy and autopsy specimen of human brain temporal lobe. Proteomics. 2006;6:4987–96. doi: 10.1002/pmic.200600078. [DOI] [PubMed] [Google Scholar]
- 10.Dumont D, Noben JP, Verhaert P, Stinissen P, Robben J. Gel-free analysis of the human brain proteome: application of liquid chromatography and mass spectrometry on biopsy and autopsy samples. Proteomics. 2006;6:4967–77. doi: 10.1002/pmic.200600080. [DOI] [PubMed] [Google Scholar]
- 11.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683–90. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 12.Pan S, Shi M, Jin J, Albin RL, et al. Proteomics identification of proteins in human cortex using multidimensional separations and MALDI tandem mass spectrometer. Mol Cell Proteomics. 2007;6:1818–23. doi: 10.1074/mcp.M700158-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Bizzarro A, Marra C, Acciarri A, Valenza A, et al. Apolipoprotein E epsilon4 allele differentiates the clinical response to donepezil in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2005;20:254–61. doi: 10.1159/000087371. [DOI] [PubMed] [Google Scholar]
- 14.Papassotiropoulos A, Bagli M, Kurz A, Kornhuber J, et al. A genetic variation of cathepsin D is a major risk factor for Alzheimer’s disease. Ann Neurol. 2000;47:399–403. [PubMed] [Google Scholar]
- 15.Julien JP. A role for neurofilaments in the pathogenesis of amyotrophic lateral sclerosis. Biochem Cell Biol. 1995;73:593–7. doi: 10.1139/o95-064. [DOI] [PubMed] [Google Scholar]
- 16.Kitsou E, Pan S, Zhang JP, Shi M, Zabeti A, Dickson DW, Albin R, Gearing M, Kashima DT, Wang Y, Beyer RP, Zhou Y, Pan C, Caudle WM, Zhang J. Identification of proteins in human substantia nigra. Proteomics Clinical Applications. 2008;2:776–782. doi: 10.1002/prca.200800028. [DOI] [PubMed] [Google Scholar]
- 17.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–34. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 18.Kosaka K, Iseki E. Dementia with Lewy bodies. Curr Opin Neurol. 1996;9:271–5. doi: 10.1097/00019052-199608000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. 2007;27:494–506. doi: 10.1111/j.1440-1789.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- 20.Braak H, Del Tredici K, Rub U, de Vos RA, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 21.Leverenz JB, Umar I, Wang Q, Montine TJ, et al. Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 2007;17:139–45. doi: 10.1111/j.1750-3639.2007.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braak H, Braak E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- 23.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–8. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 24.Holzer M, Gartner U, Stobe A, Hartig W, et al. Inverse association of Pin1 and tau accumulation in Alzheimer’s disease hippocampus. Acta Neuropathol (Berl) 2002;104:471–81. doi: 10.1007/s00401-002-0581-1. [DOI] [PubMed] [Google Scholar]
- 25.Ramakrishnan P, Dickson DW, Davies P. Pin1 colocalization with phosphorylated tau in Alzheimer’s disease and other tauopathies. Neurobiology of disease. 2003;14:251–64. doi: 10.1016/s0969-9961(03)00109-8. [DOI] [PubMed] [Google Scholar]
- 26.Sultana R, Boyd-Kimball D, Poon HF, Cai J, et al. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol Aging. 2006;27:918–25. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Butterfield DA, Abdul HM, Opii W, Newman SF, et al. Pin1 in Alzheimer’s disease. Journal of neurochemistry. 2006;98:1697–706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Woltjer RL, Cimino PJ, Pan C, et al. Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. Faseb J. 2005;19:869–71. doi: 10.1096/fj.04-3210fje. [DOI] [PubMed] [Google Scholar]
- 29.Olah J, Tokesi N, Vincze O, Horvath I, et al. Interaction of TPPP/p25 protein with glyceraldehyde-3-phosphate dehydrogenase and their co-localization in Lewy bodies. FEBS Lett. 2006;580:5807–14. doi: 10.1016/j.febslet.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 30.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, et al. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–64. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 31.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and biophysical research communications. 1984;120:885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 32.Masters CL, Simms G, Weinman NA, Multhaup G, et al. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atwood CS, Martins RN, Smith MA, Perry G. Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides. 2002;23:1343–50. doi: 10.1016/s0196-9781(02)00070-0. [DOI] [PubMed] [Google Scholar]
- 34.Pastorino L, Sun A, Lu PJ, Zhou XZ, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–34. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 35.Liao L, Cheng D, Wang J, Duong DM, et al. Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J Biol Chem. 2004;279:37061–8. doi: 10.1074/jbc.M403672200. [DOI] [PubMed] [Google Scholar]
- 36.Braak H, Rub U, Del Tredici K. Cognitive decline correlates with neuropathological stage in Parkinson’s disease. J Neurol Sci. 2006;248:255–8. doi: 10.1016/j.jns.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 37.Shi M, Jin J, Wang Y, Beyer RP, et al. Mortalin: a protein associated with progression of Parkinson disease? Journal of neuropathology and experimental neurology. 2008;67:117–24. doi: 10.1097/nen.0b013e318163354a. [DOI] [PubMed] [Google Scholar]
- 38.Kaul SC, Deocaris CC, Wadhwa R. Three faces of mortalin: A housekeeper, guardian and killer. Exp Gerontol. 2007;42:263–74. doi: 10.1016/j.exger.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 39.Jin J, Hulette C, Wang Y, Zhang T, et al. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol Cell Proteomics. 2006;5:1193–204. doi: 10.1074/mcp.M500382-MCP200. [DOI] [PubMed] [Google Scholar]
- 40.Baba M, Nakajo S, Tu PH, Tomita T, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–84. [PMC free article] [PubMed] [Google Scholar]
- 41.Trojanowski JQ, Lee VM. Brain degeneration linked to “fatal attractions” of proteins in Alzheimer’s disease and related disorders. J Alzheimers Dis. 2001;3:117–119. doi: 10.3233/jad-2001-3116. [DOI] [PubMed] [Google Scholar]
- 42.Skovronsky DM, Lee VM, Trojanowski JQ. Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu Rev Pathol. 2006;1:151–70. doi: 10.1146/annurev.pathol.1.110304.100113. [DOI] [PubMed] [Google Scholar]
- 43.Woltjer RL, Cimino PJ, Boutte AM, Schantz AM, et al. Proteomic determination of widespread detergent-insolubility including Abeta but not tau early in the pathogenesis of Alzheimer’s disease. Faseb J. 2005;19:1923–5. doi: 10.1096/fj.05-4263fje. [DOI] [PubMed] [Google Scholar]
- 44.Yang W, Woltjer RL, Sokal I, Pan C, et al. Quantitative proteomics identifies surfactant-resistant alpha-synuclein in cerebral cortex of Parkinsonism-dementia complex of Guam but not Alzheimer’s disease or progressive supranuclear palsy. Am J Pathol. 2007;171:993–1002. doi: 10.2353/ajpath.2007.070015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winton MJ, Joyce S, Zhukareva V, Practico D, et al. Characterization of tau pathologies in gray and white matter of Guam parkinsonism-dementia complex. Acta Neuropathol (Berl) 2006;111:401–12. doi: 10.1007/s00401-006-0053-0. [DOI] [PubMed] [Google Scholar]
- 46.Sebeo J, Hof PR, Perl DP. Occurrence of alpha-synuclein pathology in the cerebellum of Guamanian patients with parkinsonism-dementia complex. Acta Neuropathol. 2004;107:497–503. doi: 10.1007/s00401-004-0840-4. [DOI] [PubMed] [Google Scholar]
- 47.Forman MS, Schmidt ML, Kasturi S, Perl DP, et al. Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol. 2002;160:1725–31. doi: 10.1016/s0002-9440(10)61119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamazaki M, Arai Y, Baba M, Iwatsubo T, et al. Alpha-synuclein inclusions in amygdala in the brains of patients with the parkinsonism-dementia complex of Guam. J Neuropathol Exp Neurol. 2000;59:585–91. doi: 10.1093/jnen/59.7.585. [DOI] [PubMed] [Google Scholar]
- 49.Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr. 1960;38:1236–9. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- 50.Basso M, Giraudo S, Corpillo D, Bergamasco B, et al. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics. 2004;4:3943–52. doi: 10.1002/pmic.200400848. [DOI] [PubMed] [Google Scholar]
- 51.Abdi F, Quinn JF, Jankovic J, McIntosh M, et al. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis. 2006;9:293–348. doi: 10.3233/jad-2006-9309. [DOI] [PubMed] [Google Scholar]
- 52.Schapira AH, Mann VM, Cooper JM, Krige D, et al. Mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann Neurol. 1992;32(Suppl):S116–24. doi: 10.1002/ana.410320720. [DOI] [PubMed] [Google Scholar]
- 53.Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S26–36. doi: 10.1002/ana.10483. discussion S36–8. [DOI] [PubMed] [Google Scholar]
- 54.McNaught KS, Jackson T, JnoBaptiste R, Kapustin A, Olanow CW. Proteasomal dysfunction in sporadic Parkinson’s disease. Neurology. 2006;66:S37–49. doi: 10.1212/wnl.66.10_suppl_4.s37. [DOI] [PubMed] [Google Scholar]
- 55.McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord. 2004;10(Suppl 1):S3–7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 56.Choi J, Levey AI, Weintraub ST, Rees HD, et al. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem. 2004;279:13256–64. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 57.Choi J, Sullards MC, Olzmann JA, Rees HD, et al. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem. 2006;281:10816–24. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 59.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–75. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 60.Crossman AR. Neural mechanisms in disorders of movement. Comp Biochem Physiol A. 1989;93:141–9. doi: 10.1016/0300-9629(89)90201-6. [DOI] [PubMed] [Google Scholar]
- 61.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–5. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- 62.Cohen G. Oxy-radical toxicity in catecholamine neurons. Neurotoxicology. 1984;5:77–82. [PubMed] [Google Scholar]
- 63.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of neurochemistry. 2006;97:1634–58. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 64.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain. 1999;122(Pt 8):1437–48. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 65.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 66.Pakkenberg B, Moller A, Gundersen HJ, Mouritzen Dam A, Pakkenberg H. The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson’s disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry. 1991;54:30–3. doi: 10.1136/jnnp.54.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24:574–6. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- 68.McRitchie DA, Cartwright HR, Halliday GM. Specific A10 dopaminergic nuclei in the midbrain degenerate in Parkinson’s disease. Exp Neurol. 1997;144:202–13. doi: 10.1006/exnr.1997.6418. [DOI] [PubMed] [Google Scholar]
- 69.German DC, Dubach M, Askari S, Speciale SG, Bowden DM. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonian syndrome in Macaca fascicularis: which midbrain dopaminergic neurons are lost? Neuroscience. 1988;24:161–74. doi: 10.1016/0306-4522(88)90320-x. [DOI] [PubMed] [Google Scholar]
- 70.Varastet M, Riche D, Maziere M, Hantraye P. Chronic MPTP treatment reproduces in baboons the differential vulnerability of mesencephalic dopaminergic neurons observed in Parkinson’s disease. Neuroscience. 1994;63:47–56. doi: 10.1016/0306-4522(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 71.Waters CM, Hunt SP, Jenner P, Marsden CD. An immunohistochemical study of the acute and long-term effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the marmoset. Neuroscience. 1987;23:1025–39. doi: 10.1016/0306-4522(87)90178-3. [DOI] [PubMed] [Google Scholar]
- 72.German DC, Nelson EL, Liang CL, Speciale SG, et al. The neurotoxin MPTP causes degeneration of specific nucleus A8, A9 and A10 dopaminergic neurons in the mouse. Neurodegeneration. 1996;5:299–312. doi: 10.1006/neur.1996.0041. [DOI] [PubMed] [Google Scholar]
- 73.Rodriguez M, Barroso-Chinea P, Abdala P, Obeso J, Gonzalez-Hernandez T. Dopamine cell degeneration induced by intraventricular administration of 6-hydroxydopamine in the rat: similarities with cell loss in parkinson’s disease. Exp Neurol. 2001;169:163–81. doi: 10.1006/exnr.2000.7624. [DOI] [PubMed] [Google Scholar]
- 74.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 75.Greene JG, Dingledine R, Greenamyre JT. Gene expression profiling of rat midbrain dopamine neurons: implications for selective vulnerability in parkinsonism. Neurobiology of disease. 2005;18:19–31. doi: 10.1016/j.nbd.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 76.Chung CY, Seo H, Sonntag KC, Brooks A, et al. Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum Mol Genet. 2005;14:1709–25. doi: 10.1093/hmg/ddi178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Link AJ, Eng J, Schieltz DM, Carmack E, et al. Direct analysis of protein complexes using mass spectrometry. Nature biotechnology. 1999;17:676–82. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 78.Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–7. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 79.Zougman A, Pilch B, Podtelejnikov A, Kiehntopf M, et al. Integrated Analysis of the Cerebrospinal Fluid Peptidome and Proteome. Journal of proteome research. 2008;7:386–399. doi: 10.1021/pr070501k. [DOI] [PubMed] [Google Scholar]
- 80.Bonifati V, Rizzu P, van Baren MJ, Schaap O, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 81.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 82.Sossi V, de la Fuente-Fernandez R, Schulzer M, Troiano AR, et al. Dopamine transporter relation to dopamine turnover in Parkinson’s disease: a positron emission tomography study. Ann Neurol. 2007;62:468–74. doi: 10.1002/ana.21204. [DOI] [PubMed] [Google Scholar]
- 83.Crowder KM, Gunther JM, Jones TA, Hale BD, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A) Proceedings of the National Academy of Sciences of the United States of America. 1999;96:15268–73. doi: 10.1073/pnas.96.26.15268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kuhlenbaumer G, Hannibal MC, Nelis E, Schirmacher A, et al. Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat Genet. 2005;37:1044–6. doi: 10.1038/ng1649. [DOI] [PubMed] [Google Scholar]
- 85.Soreghan BA, Lu BW, Thomas SN, Duff K, et al. Using proteomics and network analysis to elucidate the consequences of synaptic protein oxidation in a PS1 + AbetaPP mouse model of Alzheimer’s disease. J Alzheimers Dis. 2005;8:227–41. doi: 10.3233/jad-2005-8302. [DOI] [PubMed] [Google Scholar]
- 86.Pan S, Zhu D, Quinn JF, Peskind ER, et al. A combined dataset of human cerebrospinal fluid proteins identified by multi-dimensional chromatography and tandem mass spectrometry. Proteomics. 2007;7:469–73. doi: 10.1002/pmic.200600756. [DOI] [PubMed] [Google Scholar]
- 87.Trojanowski JQ, Lee VM. The role of tau in Alzheimer’s disease. Med Clin North Am. 2002;86:615–27. doi: 10.1016/s0025-7125(02)00002-0. [DOI] [PubMed] [Google Scholar]
- 88.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–18. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 89.Andersson L, Porath J. Isolation of phosphoproteins by immobilized metal (Fe3+) affinity chromatography. Analytical biochemistry. 1986;154:250–4. doi: 10.1016/0003-2697(86)90523-3. [DOI] [PubMed] [Google Scholar]
- 90.Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1488–93. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Wolf-Yadlin A, Ross PL, Pappin DJ, et al. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol Cell Proteomics. 2005;4:1240–50. doi: 10.1074/mcp.M500089-MCP200. [DOI] [PubMed] [Google Scholar]
- 92.Rush J, Moritz A, Lee KA, Guo A, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nature biotechnology. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- 93.Oda Y, Nagasu T, Chait BT. Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome. Nature biotechnology. 2001;19:379–82. doi: 10.1038/86783. [DOI] [PubMed] [Google Scholar]
- 94.Steen H, Mann M. A new derivatization strategy for the analysis of phosphopeptides by precursor ion scanning in positive ion mode. Journal of the American Society for Mass Spectrometry. 2002;13:996–1003. doi: 10.1016/S1044-0305(02)00415-4. [DOI] [PubMed] [Google Scholar]
- 95.Zhou H, Watts JD, Aebersold R. A systematic approach to the analysis of protein phosphorylation. Nature biotechnology. 2001;19:375–8. doi: 10.1038/86777. [DOI] [PubMed] [Google Scholar]
- 96.Kang TH, Bae KH, Yu MJ, Kim WK, et al. Phosphoproteomic analysis of neuronal cell death by glutamate-induced oxidative stress. Proteomics. 2007;7:2624–35. doi: 10.1002/pmic.200601028. [DOI] [PubMed] [Google Scholar]
- 97.Ishihama Y, Wei FY, Aoshima K, Sato T, et al. Enhancement of the efficiency of phosphoproteomic identification by removing phosphates after phosphopeptide enrichment. Journal of proteome research. 2007;6:1139–44. doi: 10.1021/pr060452w. [DOI] [PubMed] [Google Scholar]
- 98.DeGiorgis JA, Jaffe H, Moreira JE, Carlotti CG, Jr, et al. Phosphoproteomic analysis of synaptosomes from human cerebral cortex. Journal of proteome research. 2005;4:306–15. doi: 10.1021/pr0498436. [DOI] [PubMed] [Google Scholar]
- 99.Chi A, Huttenhower C, Geer LY, Coon JJ, et al. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2193–8. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mikesh LM, Ueberheide B, Chi A, Coon JJ, et al. The utility of ETD mass spectrometry in proteomic analysis. Biochimica et biophysica acta. 2006;1764:1811–22. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zubarev RA. Electron-capture dissociation tandem mass spectrometry. Current opinion in biotechnology. 2004;15:12–6. doi: 10.1016/j.copbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 102.Ross CA, Pickart CM. The ubiquitin-proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol. 2004;14:703–11. doi: 10.1016/j.tcb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 103.Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, et al. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem. 2002;277:49071–6. doi: 10.1074/jbc.M208046200. [DOI] [PubMed] [Google Scholar]
- 104.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, et al. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–60. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 105.McKeith IG, Ince P, Jaros EB, Fairbairn A, et al. What are the relations between Lewy body disease and AD? J Neural Transm Suppl. 1998;54:107–16. doi: 10.1007/978-3-7091-7508-8_10. [DOI] [PubMed] [Google Scholar]
- 106.Meller R, Thompson SJ, Lusardi TA, Ordonez AN, et al. Ubiquitin proteasome-mediated synaptic reorganization: a novel mechanism underlying rapid ischemic tolerance. J Neurosci. 2008;28:50–9. doi: 10.1523/JNEUROSCI.3474-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pan S, Wang Y, Quinn JF, Peskind ER, et al. Identification of glycoproteins in human cerebrospinal fluid with a complementary proteomic approach. Journal of proteome research. 2006;5:2769–79. doi: 10.1021/pr060251s. [DOI] [PubMed] [Google Scholar]
- 108.Shimura H, Hattori N, Kubo S, Mizuno Y, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–5. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 109.Fodero LR, Saez-Valero J, Barquero MS, Marcos A, et al. Wheat germ agglutinin-binding glycoproteins are decreased in Alzheimer’s disease cerebrospinal fluid. Journal of neurochemistry. 2001;79:1022–6. doi: 10.1046/j.1471-4159.2001.00640.x. [DOI] [PubMed] [Google Scholar]
- 110.Botella-Lopez A, Burgaya F, Gavin R, Garcia-Ayllon MS, et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5573–8. doi: 10.1073/pnas.0601279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barrachina M, Maes T, Buesa C, Ferrer I. Lysosome-associated membrane protein 1 (LAMP-1) in Alzheimer’s disease. Neuropathology and applied neurobiology. 2006;32:505–16. doi: 10.1111/j.1365-2990.2006.00756.x. [DOI] [PubMed] [Google Scholar]
- 112.Butterfield DA. Proteomics: a new approach to investigate oxidative stress in Alzheimer’s disease brain. Brain research. 2004;1000:1–7. doi: 10.1016/j.brainres.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 113.Perluigi M, Poon HF, Maragos W, Pierce WM, et al. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005;4:1849–61. doi: 10.1074/mcp.M500090-MCP200. [DOI] [PubMed] [Google Scholar]
- 114.Poon HF, Frasier M, Shreve N, Calabrese V, et al. Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice--a model of familial Parkinson’s disease. Neurobiology of disease. 2005;18:492–8. doi: 10.1016/j.nbd.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 115.Aebersold R. Constellations in a cellular universe. Nature. 2003;422:115–6. doi: 10.1038/422115a. [DOI] [PubMed] [Google Scholar]
- 116.Pan S, Zhang H, Rush J, Eng J, et al. High throughput proteome screening for biomarker detection. Mol Cell Proteomics. 2005;4:182–90. doi: 10.1074/mcp.M400161-MCP200. [DOI] [PubMed] [Google Scholar]
- 117.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100:6940–5. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Anderson NL, Anderson NG, Haines LR, Hardie DB, et al. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA) J Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 119.Anderson L. Candidate-based proteomics in the search for biomarkers of cardiovascular disease. J Physiol. 2005;563:23–60. doi: 10.1113/jphysiol.2004.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 121.Pan S, Rush J, Peskind ER, Galasko D, et al. Application of targeted quantitative proteomics analysis in human cerebrospinal fluid using a liquid chromatography matrix-assisted laser desorption/ionization time-of-flight tandem mass spectrometer (LC MALDI TOF/TOF) platform. Journal of proteome research. 2008;7:720–30. doi: 10.1021/pr700630x. [DOI] [PubMed] [Google Scholar]