

Abstract
Activated platelets have been implicated in playing a major role in transfusion-related acute lung injury (TRALI), as platelets can trigger neutrophils, resulting in vascular damage. We hypothesized that binding of platelet CD40 ligand (CD40L) to endothelial CD40 is essential in the onset of TRALI. Mice were challenged with monoclonal major histocompatibility complex (MHC)-1 antibody which induced TRALI, evidenced by pulmonary oedema, accompanied by significantly elevated bronchoalveolar fluid (BALF) levels of total protein and elevated plasma levels of keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-2 (MIP-2) compared to infusion of isotype antibody (all Ps < 0·05). Treatment with ciglitazone, which inhibits platelet CD40L expression, had no effect on pulmonary and systemic inflammation compared to controls. In addition, treatment with anti-CD40L antibody, which antagonizes all CD40–CD40L interactions, also did not abrogate the TRALI reaction. Furthermore, levels of soluble CD40L were measured in a cohort of cardiac surgery patients, who were followed prospectively for the onset of TRALI after transfusion. Plasma levels of sCD40L at baseline and at time of developing TRALI did not differ between TRALI patients and controls (transfused cardiac surgery patients not developing acute lung injury) (275 ± 192 versus 258 ± 346 and 93 ± 82 versus 93 ± 123 pg/ml, respectively, not significant). In conclusion, these results do not support the idea that the CD40–CD40L interaction is involved in mediating TRALI.
Keywords: cardiac surgery, CD40 ligand, MHC-1 antibody, TRALI, transfusion
Introduction
Transfusion-related acute lung injury (TRALI) is a major cause of transfusion-related morbidity and mortality [1]. Activation of lung endothelium induced by the underlying condition of the patient (e.g. cardiac surgery or sepsis) primes polymorphic neutrophils (PMNs) in the lung. Transfusion of blood products can result in activation of these primed PMNs and enhance microvascular permeability with subsequent pulmonary leakage [2,3]. The pathogenesis of activation of PMNs during TRALI is not understood fully. Donor-derived leucocyte antibodies appear to be a key stimulus of PMNs in TRALI. Of these, leucocyte antibodies directed against the human leucocyte antigen class (HLA) II, human neutrophil alloantigen-3a and HLA-A2 antigens are associated with the most severe TRALI cases [4]. Other factors that have been implicated in TRALI are bioactive substances which accumulate during storage of blood products [5,6].
Recently, platelets were identified as important mediators of vascular damage in TRALI [7,8]. Both platelet depletion and aspirin protected against lung injury and reduced mortality in a two-event mouse model of TRALI and aspirin was coined as a novel therapy [9]. However, the molecular mechanism of the protective effect of blocking platelet activation in TRALI still needs to be unravelled.
CD40 ligand (CD40L; CD154) is a proinflammatory mediator found in soluble (sCD40L) and cell-associated forms [10]. CD40L is primarily platelet-derived, but is also expressed on T cells and binds to CD40, expressed on endothelial and epithelial cells, neutrophils, B cells, dendritic cells and macrophages [11–13]. The CD40–CD40L system plays a pivotal role in inflammation and thrombosis in various clinical settings, including cancer [14], autoimmune diseases [15] and atherothrombosis [16,17]. Recombinant sCD40L was found to serve as the second event in an in vitro model of PMN-mediated endothelial damage [13,18]. In line with this, blocking of CD40–CD40L has been found to be protective in various models of acute lung injury (ALI) [19–21].
In TRALI, the CD40–CD40L system is also postulated to play a role. sCD40L accumulates during storage of red blood cells and platelet concentrates which are implicated in TRALI reactions [22]. In addition, increased post-transfusion levels of sCD40L were found in patients with TRALI [13]. Thereby, CD40L on platelets may induce lung injury through binding of CD40 with the endothelium in the lung, or with neutrophils or other immune cells involved in TRALI.
In this paper, we investigated the role of CD40L as mediator of lung injury in a murine model of antibody-mediated TRALI. Furthermore, plasma sCD40L levels were measured in patients who developed TRALI after cardiac surgery. Cardiac surgery patients were chosen because cardiac surgery is a risk factor for TRALI; these patients are often transfused and sCD40L is released during cardiopulmonary bypass [23].
Materials and methods
Experiments were performed with healthy male BALB/c mice (Charles River, Someren, the Netherlands), aged 10–12 weeks and weighing 22–25 g, assigned randomly to six groups (n = 8 per group). Animal studies were approved by the Animal Care and Use Committee of the Academic Medical Center at the University of Amsterdam, the Netherlands (no. 102033). Animal procedures were carried out in compliance with Institutional Standards for Human Care and Use of Laboratory Animals.
Interventions
Two interventions were performed with appropriate control groups. First, 24 h before induction of TRALI, mice were pretreated intraperitoneally (i.p.) with ciglitazone {ciglitazone 5-[4-(1-methylcyclohexylmethoxy) benzyl]-thiazolidine-2,4-dione} (5 mg/kg) (Enzo Life Science, Zandhoven, Belgium), as described previously [24]. Ciglitazone, an anti-diabetic drug with anti-inflammatory capacity, inhibits the expression of CD40L on platelets [25] and lowers the serum level of sCD40L [26]. Controls received vehicle (2·5% ethanol in 200 µl saline) i.p. Secondly, immediately prior to infusion of TRALI-inducing antibodies, mice were pretreated with anti-CD40L antibody i.p. (10 mg/kg diluted in phosphate-buffered saline (PBS) in a volume–weight-dependent dose of 180–220 µl). Controls received isotype antibody (hamster–anti-rat CD40L, both from Bioceros, Utrecht, the Netherlands). Anti-CD40L antibody is capable of antagonizing all CD40–CD40L interactions, thereby making no distinction between CD40L from platelets and T cells [27,28].
Experimental study protocol
After prehydration with 1 ml NaCl 0·9% i.p., mice were anaesthetized i.p. with 0·075 ml/10 g of a mix containing ketamine (EurovetAnimal Health BV, Bladel, the Netherlands), medetomidine (Pfizer Animal Health BV, Capell a/d Ijssel, the Netherlands) and atropine (Pharmachemie, Haarlem, the Netherlands) in a ratio of 1·26 ml 100 mg/ml ketamine, 0·2 ml 1 mg/ml medetomidine and 1 ml 0·5 mg/ml atropine in 5 ml NaCl 0·9%. Then, mice were placed supine on a warming blanket and the jugular vein was isolated. Using a 30-gauge sterile needle attached to polyethylene tubing, venous blood was aspirated from the jugular vein to verify intravascular placement of the needle. Mice were infused with either MHC-1 antibody [immunoglobulin (Ig)G2a, κ, 4·5 mg/kg], which has been shown previously to induce TRALI [8,29] or matched isotype antibody (IgG2a, CRl-1908) (both from the American Type Culture Collection). The skin was closed with prolene 5–0. The mice were kept under a heating lamp until recovery from anaesthesia and then placed back into their cages. After 2 h, mice were exsanguinated by drawing blood from the carotic artery. The left lung was ligated and the right lung was lavaged three times with 0·5 ml of normal saline. Approximately 1·0 ml of lavage fluid was retrieved per mouse. Left lungs were weighed and homogenized in 4× lung weight (mg) in 0·9% saline using a tissue homogenizer (Biospec Products, Bartlesville, OK, USA) and diluted 1:1 with Greenberger lysis buffer. Supernatant was stored at −20°C for total protein level and cytokine measurement. The left lung was used to calculate wet lung to body weight ratio.
Clinical study
Blood samples were derived from a larger trial on TRALI incidence performed in the mixed medical–surgical intensive care unit of a university hospital in the Netherlands [30]. The study was approved by the Institutional Review Board (06/201 no. 06·17·1506). Prior to valvular and/or coronary artery surgery, informed consent was requested from patients aged 18 years or older for participation in the study. Exclusion criteria were off-pump surgery, emergency surgery and use of immunosuppressive drugs. Patients were followed prospectively for the development of TRALI using the consensus definition (new-onset hypoxaemia or deterioration demonstrated by a PaO2/FiO2 ratio < 300, occurring within 6 h after transfusion, with bilateral pulmonary changes on the chest radiograph and a pulmonary arterial occlusion pressure of ≤ 18 mmHg) [31]. Cardiogenic pulmonary oedema was identified when pulmonary arterial occlusion pressure was > 18 mmHg, or by the presence of at least two of the following: central venous pressure > 15 mmHg, a history of heart failure or valve dysfunction, ejection fraction < 45% as estimated by echocardiogram and a positive fluid balance [32]. Sixteen cardiac surgery patients were identified as having suspected TRALI. Cases were matched randomly with controls in a 1:2 ratio. Controls were transfused cardiac surgery patients not developing acute lung injury. All transfused red blood cells (RBCs) were leucoreduced [buffy coat was removed and the erythrocyte suspension was filtered to remove the leucocytes (< 1 × 106)], which is the standard of practice in the Netherlands. Blood for analysis was drawn before and 6 h after surgery.
Assays
Total protein levels (Bradford Protein Assay Kit, OZ Bioscience, Marseille, France) were measured in bronchoalveolar lavage fluid (BALF). Keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-2 (MIP-2) were measured in BALF and plasma using enzyme-linked immunosorbent assay (ELISA), according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA). We chose to measure KC and MIP-2 because CD40L on activated platelets triggers an inflammatory reaction of endothelial cells [33]. Human sCD40L/TNFSF5 was measured in plasma according to the instructions of the manufacturer using an ELISA (Quantikine; R&D Systems). The detection limit was 4·2 pg/ml. To minimize post-venapuncture CD40L hydrolysis, ethylenediamine tetraacetic acid (EDTA) anti-coagulation was used [23].
Donor antibody analysis
Donor antibody analysis was performed as described previously [30]. In short, in donor samples of platelets (PLTs) and fresh frozen plasma (FFP) products, leucocyte-reactive antibodies were examined using a standard complement-dependent cytotoxicity (CDC) assay with an HLA-typed donor panel (to detect complement-fixing antibodies to HLA classes I and II) and a Luminex screening assay. Leucocyte agglutinating antibodies were examined using a leucocyte agglutination technique. Granulocyte-reactive antibodies were examined by the granulocyte immunofluorescence test.
Statistical analysis
Data were expressed as mean ± standard deviation (s.d.) or median [interquartile range (IQR)] when appropriate. Comparisons between experimental groups were performed using Student's t-test or Mann–Whitney U-test depending on data distribution. On clinical data, we performed a secondary analysis using patients from a case–control study. For comparison of sCD40L levels, a Mann–Whitney U-test was used. A P-value < 0·05 was considered statistically significant. Statistical analyses were performed with spss version 17·0 (SPSS Inc., Chicago, IL, USA) and Prism version 5·0 (GraphPad Software, San Diego, CA, USA).
Results
All mice in the isotype MHC-1 antibody control group survived. In contrast, 26% of the mice challenged with TRALI antibodies were killed because of respiratory distress. All mice were used for analysis.
Induction of TRALI with MHC-1 antibody
Infusion with MHC-1 antibodies resulted in an increase in lung-to-body weight ratio and total protein concentration in the BALF compared to controls, indicating a decrease in alveolar fluid clearance and an increased lung vascular permeability (Fig. 1), with a non-significant elevation of the level of KC in the BALF (Fig. 2). The level of MIP-2 in BALF did not differ between TRALI and control mice. Plasma levels of KC and MIP-2 were elevated after MHC-1 antibody injection compared to controls (Fig. 2).
Fig. 1.
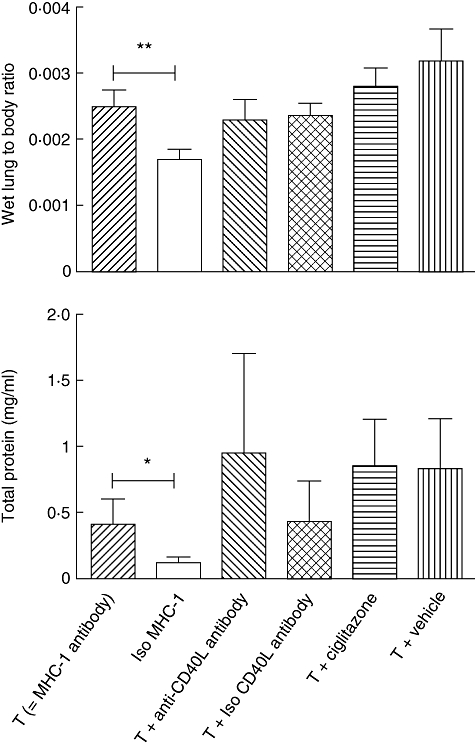
Pulmonary oedema and protein leakage after induction of transfusion-related acute lung injury and treatment with anti-CD40 ligand antibody or ciglitazone and controls. *P < 0·05; **P < 0·01.
Fig. 2.
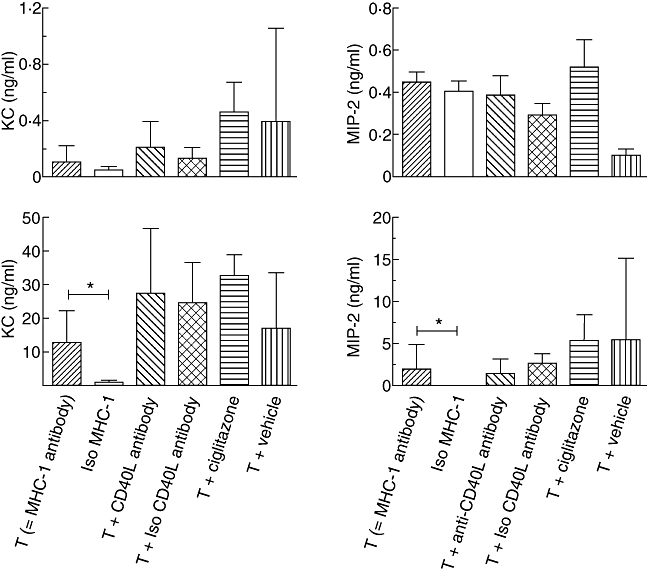
Levels of chemoattractants in broncoalveolar lavage fluid (BALF; upper panels) and plasma (lower panels) after induction of transfusion-related acute lung injury and treatment with anti-CD40 ligand antibody or ciglitazone and controls. KC: keratinocyte-derived chemokine; MIP-2: macrophage-inflammatory protein-2.
The effect of blocking CD40L in a TRALI model
Pretreatment with ciglitazone did not reduce pulmonary oedema nor BALF protein concentration compared to the TRALI group who had received vehicle (Fig. 1). Also, ciglitazone did not modulate local or systemic inflammation compared to the TRALI group and vehicle controls (Fig. 2).
To investigate the effect of antagonizing CD40L on all cell types, mice were treated i.p. with anti-CD40L antibody. Antagonizing of CD40L did not reduce lung oedema or protein leakage compared to the TRALI group (Fig. 1). Also, local and systemic levels of chemoattractants were not different between the anti-CD40L antibody-treated group and controls (Fig. 2).
Levels of sCD40L in TRALI patients and controls before and after cardiac surgery
Patients who had developed TRALI after cardiac surgery were compared to transfused cardiac surgery patients who did not develop ALI [30]. Baseline characteristics are presented in Table 1. There was no difference in factors associated with an increase in sCD40L levels, including diabetes mellitus (DM) and vascular disease, or a decrease in sCD40L levels, such as use of statins and aspirin in type 2 DM [34]. None of the diabetic patients used a thiazolidinedione derivate. Patients developing TRALI had received more RBCs, FFP and PLTs than controls (Table 1). In 63% of these patients, a leucocyte antibody was detected in an associated blood product. Patients developing TRALI received significantly more blood products containing HLA-I, HLA-II and human nuclear antigen (HNA) antibodies compared to controls (38 versus 3%, 44 versus 6% and 19 versus 3%, respectively, P = 0·005). Of the HLA/HNA antibody-positive products, 80% originated from female donors [30].
Table 1.
Baseline characteristics, transfusion data and preoperative medication of patients developing transfusion-related acute lung injury (TRALI) compared to transfused control subjects
| TRALI (n = 16) | Controls (n = 32) | P-value | |
|---|---|---|---|
| Age‡ | 74 (29) | 68 (13) | 0·19 |
| Gender, male | 12 (75%) | 20 (63%) | 0·52 |
| EuroSCORE‡ | 6·0 (5·5) | 5·0 (3·8) | 0·94 |
| Myocardial infarction | 2 (13%) | 11 (34%) | 0·17 |
| Hypertension | 8 (50%) | 24 (75%) | 0·18 |
| PVD | 3 (19%) | 10 (31%) | 0·20 |
| Diabetes | 4 (25%) | 12 (63%) | 0·40 |
| RBCs, units† | 3·2 (±2·3) | 2·0 (±1·5) | 0·02 |
| Fresh frozen plasma, units† | 3·3 (±3·5) | 1·2 (±1·8) | 0·02 |
| Platelets, units† | 0·8 (±0·8) | 0·3 (±0·5) | 0·03 |
| Thiazolidinedione | 0 | 0 | – |
| Aspirin | 9 (56%) | 22 (69%) | 0·52 |
| Statin | 11 (69%) | 26 (81%) | 0·49 |
ALI, acute lung injury; EuroSCORE, European System for Cardiac Operative Risk Evaluation; PVD, peripheral vascular disease; RBCs, red blood cells. Data are presented as number (%)
mean (± standard deviation)
median (interquartile range).
Prior to surgery, baseline levels of sCD40L did not differ between TRALI patients and controls. Post-surgery, at onset of TRALI, levels of sCD40L were significantly lower in both groups compared to baseline values (P ≤ 0·01 to both), but there were no differences between cases and controls (Fig. 3). Because a drop in platelet counts may account for lower sCD40L concentrations, levels of platelets between groups were compared. Platelet counts correlated with sCD40L levels, showing a decrease in platelet count after surgery in both groups (Fig. 3), but no difference was observed between TRALI and controls.
Fig. 3.
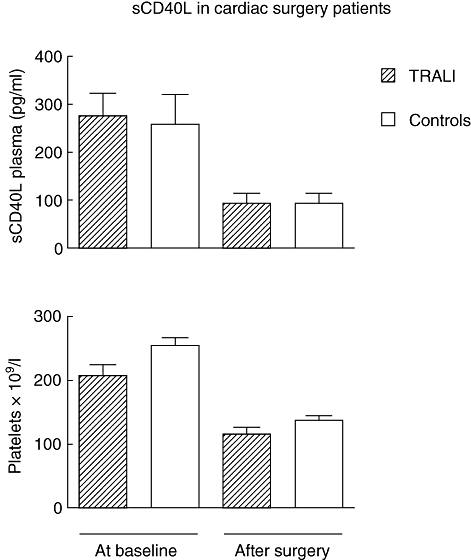
Plasma levels of sCD40 ligand and platelet count before and after cardiac surgery in transfusion-related acute lung injury (TRALI) patients and transfused controls.
Discussion
In this study on the role of CD40L in TRALI, we found that blocking of CD40L–CD40 is not protective in an antibody-mediated murine model of TRALI, suggesting that there is no role for CD40L in the onset of immune-mediated TRALI. Furthermore, and confirming this observation, the levels of sCD40L in patients developing TRALI were not different from transfused controls.
Infusion of MHC-1 antibodies resulted in a decrease in alveolar fluid clearance and an increased lung vascular permeability, as found previously [8]. The CD40–CD40L interaction was inhibited in two different ways. As platelets were found to be pivotal in mediating lung injury in this TRALI model, ciglitazone was given, which inhibits CD40L expression on platelets. A dose was used which was found previously to ameliorate lung injury during murine pneumonia [24]. However, ciglitazone did not reduce lung injury in our model. As immune cells other than platelets may also express CD40L and contribute to the development of TRALI, animals were then infused with anti-CD40L antibody, which antagonizes binding of CD40L with all immune cells and with the endothelium [35], and completely blocks any CD40–CD40L interaction [27,28]. In this model, however, anti-CD40L antibody also had no effect on pulmonary leakage and inflammation. Therefore, these results indicate that CD40–CD40L interaction does not play a major role in this model of TRALI.
A possible explanation for contrasting results with previous experimental findings may be a difference in the models used. In a previous study, sCD40L has been implicated in a ‘two-event’in vitro TRALI model after lipopolysaccharide (LPS) priming [13], whereas an immune-mediated antibody model was used in this study. As sCD40L accumulates during storage of blood [13,22], it can be hypothesized that blocking of the CD40–CD40L interaction may be protective in transfusion models using stored blood products. However, in a rat transfusion model of lung injury induced by stored PLT products after LPS priming, we found previously that pulmonary expression of CD40L was not enhanced [36].
To examine further the role of the CD40–CD40L pathway in TRALI, sCD40L was measured in transfused cardiac surgery patients developing TRALI. Cardiac surgery is recognized as a risk factor for TRALI [3,32], due possibly to PMN priming during cardiopulmonary bypass [37,38]. As recombinant sCD40L was found to activate primed PMNs in vitro[13], we hypothesized that sCD40L activates primed PMNs following cardiac surgery, increasing susceptibility for a TRALI reaction. However, levels of sCD40L in TRALI cases and controls in this study were not different, suggesting that sCD40L is not implicated in the onset of TRALI in cardiac surgery patients.
These findings do not accord with previous observations suggesting that sCD40L is implicated in TRALI [13]. However, the finding of an accumulation of sCD40L in stored platelets is indirect evidence, and may not have a functional consequence for a mediating role of sCD40L. In accordance, the increase in sCD40L found in platelet products implicated in transfusion reactions was accompanied by a concomitant increase in levels of several other inflammatory mediators, suggesting that any single mediator is unlikely to account for a transfusion reaction [39]. Of note, the increase in sCD40L found previously was not univocal, occurring in only eight of 12 patients with TRALI, and was not statistically different from sCD40L concentrations in the pretransfusion samples [13]. Therefore, increased levels of sCD40L may have been associated, but not causal in TRALI.
An alternative explanation for an absence of elevated levels of sCD40L in TRALI patients may have been the timing of sampling. However, as levels of sCD40L were measured before and at the onset of TRALI, it is less likely that the timing of sCD40L measurement was too late in the pathophysiological process of TRALI, allowing for CD40 to internalize or ligate sCD40L [40,41]. Moreover, it should be noted that transfused RBCs in this clinical study were leucoreduced, which has been shown previously to result in lower sCD40L levels compared to non-leucoreduced RBCs, due possibly to a reduction in contaminating platelets [13]. However, TRALI also continues to occur after the introduction of leucoreduction [42,43]. In line with this, 16 TRALI cases were detected in our prospective study using leucoreduced blood [44].
Of note, sCD40L levels were evidently lower in both groups after cardiac surgery. This drop in sCD40L probably reflects the concomitant decrease in platelet counts in both groups, as a tight correlation between sCD40L and platelet count has been found previously [23,45]. We cannot exclude that the decrease in platelets observed after cardiac surgery may have influenced results. Therefore, our clinical data cannot be generalized to a non-cardiac surgery population.
The molecular mechanism by which aspirin was found to protect against TRALI in a murine model is still not known. Of note, the use of aspirin did not differ between TRALI patients and controls in this study, which does not support the findings in the animal model [9]. However, patient numbers may have been too small in this study to comment on the effect of aspirin use.
In conclusion, antagonizing CD40–CD40L does not ameliorate lung injury in a murine model of antibody-mediated TRALI. Furthermore, in the reported clinical setting, TRALI is not associated with increased sCD40L levels compared to transfused controls. Therefore, these results do not underline an important role for CD40–CD40L as a mediating pathway in TRALI.
Disclosure
None.
References
- 1.Goldman M, Webert KE, Arnold DM, Freedman J, Hannon J, Blajchman MA. Proceedings of a consensus conference: towards an understanding of TRALI. Transfus Med Rev. 2005;19:2–31. doi: 10.1016/j.tmrv.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Silliman CC. The two-event model of transfusion-related acute lung injury. Crit Care Med. 2006;34:S124–S131. doi: 10.1097/01.CCM.0000214292.62276.8E. [DOI] [PubMed] [Google Scholar]
- 3.Silliman CC, Boshkov LK, Mehdizadehkashi Z, et al. Transfusion-related acute lung injury: epidemiology and a prospective analysis of etiologic factors. Blood. 2003;101:454–62. doi: 10.1182/blood-2002-03-0958. [DOI] [PubMed] [Google Scholar]
- 4.Bux J. Antibody-mediated (immune) transfusion-related acute lung injury. Vox Sang. 2011;100:122–8. doi: 10.1111/j.1423-0410.2010.01392.x. [DOI] [PubMed] [Google Scholar]
- 5.Silliman CC, Paterson AJ, Dickey WO, et al. The association of biologically active lipids with the development of transfusion-related acute lung injury: a retrospective study. Transfusion. 1997;37:719–26. doi: 10.1046/j.1537-2995.1997.37797369448.x. [DOI] [PubMed] [Google Scholar]
- 6.Silliman CC, Bjornsen AJ, Wyman TH, et al. Plasma and lipids from stored platelets cause acute lung injury in an animal model. Transfusion. 2003;43:633–40. doi: 10.1046/j.1537-2995.2003.00385.x. [DOI] [PubMed] [Google Scholar]
- 7.Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15:384–91. doi: 10.1038/nm.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fc gamma receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest. 2006;116:1615–23. doi: 10.1172/JCI27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119:3450–61. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res. 2003;92:1041–8. doi: 10.1161/01.RES.0000070111.98158.6C. [DOI] [PubMed] [Google Scholar]
- 11.van Kooten C, Banchereau J. CD40–CD40 ligand. J Leukoc Biol. 2000;67:2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- 12.Phipps RP. Atherosclerosis: the emerging role of inflammation and the CD40–CD40 ligand system. Proc Natl Acad Sci USA. 2000;97:6930–2. doi: 10.1073/pnas.97.13.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan SY, Kelher MR, Heal JM, et al. Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood. 2006;108:2455–62. doi: 10.1182/blood-2006-04-017251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roselli M, Mineo TC, Basili S, et al. Soluble CD40 ligand plasma levels in lung cancer. Clin Cancer Res. 2004;10:610–14. doi: 10.1158/1078-0432.ccr-0348-03. [DOI] [PubMed] [Google Scholar]
- 15.Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009;21:293–300. doi: 10.1016/j.smim.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novo S, Basili S, Tantillo R, et al. Soluble CD40L and cardiovascular risk in asymptomatic low-grade carotid stenosis. Stroke. 2005;36:673–5. doi: 10.1161/01.STR.0000154878.58398.14. [DOI] [PubMed] [Google Scholar]
- 17.Aukrust P, Muller F, Ueland T, et al. Enhanced levels of soluble and membrane-bound CD40 ligand in patients with unstable angina. Possible reflection of T lymphocyte and platelet involvement in the pathogenesis of acute coronary syndromes. Circulation. 1999;100:614–20. doi: 10.1161/01.cir.100.6.614. [DOI] [PubMed] [Google Scholar]
- 18.Wyman TH, Bjornsen AJ, Elzi DJ, et al. A two-insult in vitro model of PMN-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol. 2002;283:C1592–C1603. doi: 10.1152/ajpcell.00540.2001. [DOI] [PubMed] [Google Scholar]
- 19.Adawi A, Zhang Y, Baggs R, Finkelstein J, Phipps RP. Disruption of the CD40-CD40 ligand system prevents an oxygen-induced respiratory distress syndrome. Am J Pathol. 1998;152:651–7. [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto N, Kawabe T, Imaizumi K, et al. CD40 plays a crucial role in lipopolysaccharide-induced acute lung injury. Am J Respir Cell Mol Biol. 2004;30:808–15. doi: 10.1165/rcmb.2003-0197OC. [DOI] [PubMed] [Google Scholar]
- 21.Adawi A, Zhang Y, Baggs R, et al. Blockade of CD40–CD40 ligand interactions protects against radiation-induced pulmonary inflammation and fibrosis. Clin Immunol Immunopathol. 1998;89:222–30. doi: 10.1006/clin.1998.4606. [DOI] [PubMed] [Google Scholar]
- 22.Kaufman J, Spinelli SL, Schultz E, Blumberg N, Phipps RP. Release of biologically active CD154 during collection and storage of platelet concentrates prepared for transfusion. J Thromb Haemost. 2007;5:788–96. doi: 10.1111/j.1538-7836.2007.02412.x. [DOI] [PubMed] [Google Scholar]
- 23.Nannizzi-Alaimo L, Rubenstein MH, Alves VL, Leong GY, Phillips DR, Gold HK. Cardiopulmonary bypass induces release of soluble CD40 ligand. Circulation. 2002;105:2849–54. doi: 10.1161/01.cir.0000019068.32280.b3. [DOI] [PubMed] [Google Scholar]
- 24.Stegenga ME, Florquin S, de Vos AF, van der Poll T. The thiazolidinedione ciglitazone reduces bacterial outgrowth and early inflammation during Streptococcus pneumoniae pneumonia in mice. Crit Care Med. 2009;37:614–18. doi: 10.1097/CCM.0b013e31819599b6. [DOI] [PubMed] [Google Scholar]
- 25.Akbiyik F, Ray DM, Gettings KF, Blumberg N, Francis CW, Phipps RP. Human bone marrow megakaryocytes and platelets express PPARgamma, and PPARgamma agonists blunt platelet release of CD40 ligand and thromboxanes. Blood. 2004;104:1361–8. doi: 10.1182/blood-2004-03-0926. [DOI] [PubMed] [Google Scholar]
- 26.Marx N, Imhof A, Froehlich J, et al. Effect of rosiglitazone treatment on soluble CD40L in patients with type 2 diabetes and coronary artery disease. Circulation. 2003;107:1954–7. doi: 10.1161/01.CIR.0000069272.06194.91. [DOI] [PubMed] [Google Scholar]
- 27.Foy TM, Shepherd DM, Durie FH, Aruffo A, Ledbetter JA, Noelle RJ. In vivo CD40-gp39 interactions are essential for thymus-dependent humoral immunity. II. Prolonged suppression of the humoral immune response by an antibody to the ligand for CD40, gp39. J Exp Med. 1993;178:1567–75. doi: 10.1084/jem.178.5.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stax AM, Gelderman KA, Kamerling SW, van der Geest R, Schlagwein N, van KC. Generation and characterization of a novel anti-rat CD40L antibody with inhibitory activities in vitro and in vivo. J Immunol Methods. 2008;335:46–52. doi: 10.1016/j.jim.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Vlaar AP, Wolthuis EK, Hofstra JJ, et al. Mechanical ventilation aggravates transfusion-related acute lung injury induced by MHC-I class antibodies. Intens Care Med. 2010;36:879–87. doi: 10.1007/s00134-010-1802-z. [DOI] [PubMed] [Google Scholar]
- 30.Vlaar AP, Hofstra JJ, Determann RM, et al. The incidence, risk factors and outcome of transfusion-related acute lung injury in a cohort of cardiac surgery patients: a prospective nested case control study. Blood. 2011;117:4218–25. doi: 10.1182/blood-2010-10-313973. [DOI] [PubMed] [Google Scholar]
- 31.Toy P, Popovsky MA, Abraham E, et al. Transfusion-related acute lung injury: definition and review. Crit Care Med. 2005;33:721–6. doi: 10.1097/01.ccm.0000159849.94750.51. [DOI] [PubMed] [Google Scholar]
- 32.Vlaar AP, Binnekade JM, Prins D, et al. Risk factors and outcome of transfusion-related acute lung injury in the critically ill: a nested case–control study. Crit Care Med. 2010;38:771–8. doi: 10.1097/CCM.0b013e3181cc4d4b. [DOI] [PubMed] [Google Scholar]
- 33.Henn V, Slupsky JR, Grafe M, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–4. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 34.Santilli F, Basili S, Ferroni P, Davi G. CD40/CD40L system and vascular disease. Intern Emerg Med. 2007;2:256–68. doi: 10.1007/s11739-007-0076-0. [DOI] [PubMed] [Google Scholar]
- 35.Nierkens S, van HP, Bol M, et al. Selective requirement for CD40-CD154 in drug-induced type 1 versus type 2 responses to trinitrophenyl-ovalbumin. J Immunol. 2002;168:3747–54. doi: 10.4049/jimmunol.168.8.3747. [DOI] [PubMed] [Google Scholar]
- 36.Vlaar AP, Hofstra JJ, Kulik W, et al. Supernatant of stored platelets causes lung inflammation and coagulopathy in a novel in vivo transfusion model. Blood. 2010;116:1360–8. doi: 10.1182/blood-2009-10-248732. [DOI] [PubMed] [Google Scholar]
- 37.Cameron D. Initiation of white cell activation during cardiopulmonary bypass: cytokines and receptors. J Cardiovasc Pharmacol. 1996;27(Suppl 1):S1–5. doi: 10.1097/00005344-199600001-00004. [DOI] [PubMed] [Google Scholar]
- 38.Gillinov AM, Bator JM, Zehr KJ, et al. Neutrophil adhesion molecule expression during cardiopulmonary bypass with bubble and membrane oxygenators. Ann Thorac Surg. 1993;56:847–53. doi: 10.1016/0003-4975(93)90342-f. [DOI] [PubMed] [Google Scholar]
- 39.Blumberg N, Gettings KF, Turner C, Heal JM, Phipps RP. An association of soluble CD40 ligand (CD154) with adverse reactions to platelet transfusions. Transfusion. 2006;46:1813–21. doi: 10.1111/j.1537-2995.2006.00979.x. [DOI] [PubMed] [Google Scholar]
- 40.Dongari-Bagtzoglou AI, Thienel U, Yellin MJ. CD40 ligation triggers COX-2 expression in endothelial cells: evidence that CD40-mediated IL-6 synthesis is COX-2-dependent. Inflamm Res. 2003;52:18–25. doi: 10.1007/s000110300009. [DOI] [PubMed] [Google Scholar]
- 41.Hakkinen T, Karkola K, Yla-Herttuala S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions. Colocalization with epitopes of oxidized low-density lipoprotein, scavenger receptor, and CD16 (Fc gammaRIII) Virchows Arch. 2000;437:396–405. doi: 10.1007/s004280000239. [DOI] [PubMed] [Google Scholar]
- 42.Uhlmann EJ, Isgriggs E, Wallhermfechtel M, Goodnough LT. Prestorage universal WBC reduction of RBC units does not affect the incidence of transfusion reactions. Transfusion. 2001;41:997–1000. doi: 10.1046/j.1537-2995.2001.41080997.x. [DOI] [PubMed] [Google Scholar]
- 43.Hebert PC, Fergusson D, Blajchman MA, et al. Clinical outcomes following institution of the Canadian universal leukoreduction program for red blood cell transfusions. JAMA. 2003;289:1941–9. doi: 10.1001/jama.289.15.1941. [DOI] [PubMed] [Google Scholar]
- 44.Vlaar AP, Hofstra JJ, Determann RM, et al. Transfusion-related acute lung injury in cardiac surgery patients is hemerus characterized by systemic inflammation prior to onset of disease, followed by pulmonary activation of inflammation and coagulopathy. Transfusion. 2010;50(Suppl 2):P4–020A. [Google Scholar]
- 45.Danese S, Katz JA, Saibeni S, et al. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut. 2003;52:1435–41. doi: 10.1136/gut.52.10.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]