

Abstract
Although metastasis accounts for >90% of cancer-related deaths, no therapeutic that targets this process has yet been approved. Because the chemokine receptor CXCR4 is one of the targets closely linked with tumor metastasis, inhibitors of this receptor have the potential to abrogate metastasis. In the current report, we demonstrate that celastrol can downregulate the CXCR4 expression on breast cancer MCF-7 cells stably transfected with HER2, an oncogene known to induce the chemokine receptor. Downregulation of CXCR4 by the triterpenoid was not cell type-specific as downregulation occurred in colon cancer, squamous cell carcinoma, and pancreatic cancer cells. Decrease in CXCR4 expression was not due to proteolysis as neither proteasome inhibitors nor lysosomal stabilization had any effect. Quantitative reverse transcription polymerase chain reaction analysis revealed that downregulation of CXCR4 messenger RNA (mRNA) by celastrol occurred at the translational level. Chromatin immunoprecipitation analysis revealed regulation at the transcriptional level as well. Abrogation of the chemokine receptor by celastrol or by gene-silencing was accompanied by suppression of invasiveness of colon cancer cells induced by CXCL12, the ligand for CXCR4. This effect was not cell type-specific as celastrol also abolished invasiveness of pancreatic tumor cells, and this effect again correlated with the disappearance of both the CXCR4 mRNA and CXCR4 protein. Other triterpenes, such as withaferin A and gedunin, which are known to inhibit Hsp90, did not downregulate CXCR4 expression, indicating that the effects were specific to celastrol. Overall, these results show that celastrol has potential in suppressing invasion and metastasis of cancer cells by down-modulation of CXCR4 expression.
Keywords: CXCR4, CXCL12, Colon cancer, NF-κB
Introduction
While most cancer therapeutic approaches focus on survival and growth of primary tumors, it is the tumor metastasis that is responsible for >90% deaths due to cancer [1]. No drug that targets tumor metastasis has yet been approved. Metastasis or the spread of the tumor that normally occurs to highly vital organs such as brain, lung, liver, bone, or lymph nodes is a highly complex process that is not fully understood. The role of a wide variety of molecules in metastasis has been implicated, including tumor necrosis factor-α (TNF-α) [2], tumor growth factor-β [3], vascular endothelial growth factor (VEGF) [4], and the chemokine receptor, CXCR4 [5]. While VEGF is thought to be a key mediator of angiogenesis and metastasis, antibodies against VEGF (called Avastin), although approved as a treatment, has little effect on tumor metastasis. In fact, there are reports that Avastin may upregulate CXCR4 and its ligand in tumors from patients with rectal cancer [6], thus leading to accelerated metastasis [7].
Among all other factors that have been linked with tumor metastasis, CXCR4 may be the most studied. Although different cancers preferentially metastasize to different organs, production of the chemokine by the organ is responsible for migration to that organ. The involvement of this receptor in tumor metastasis was first documented in breast cancer [5], but has now been linked with metastasis of a wide variety of cancers, including ovarian [8], colorectal [9], pancreatic [10], and prostate cancers [11]. It is now known that CXCR4 is closely linked to the metastasis of breast cancer, its expression is regulated by HER2 [12], and it is a predictor of recurrence [13]. CXCR4 binds to its ligand CXCL12 (also called stromal cell-derived factor-1 [SDF-1]) expressed in the organ to which the tumor migrates and activates the pathway that promotes invasion by the tumor. Thus, agents that can interrupt the CXCR4–CXCL12 cell signaling pathway have a potential to suppress tumor metastasis.
We decided to search for inhibitors of the CXCR4–CXCL12 cell signaling pathway among the traditional medicines. For over 20 years, our laboratory has worked on deciphering the mechanism of action of traditional medicines because most drugs used for treatment of cancer today have their origin from natural sources and they have been used for thousands of years. Celastrol, a triterpene, is one such compound, that was identified from the traditional Chinese medicine “God of Thunder Vine” or Tripterygium wilfordii Hook F. almost three decades ago and used for the treatment of cancer and other inflammatory diseases [14]. Various studies have indicated that this triterpene exhibits anticancer potential [15, 16] and eradicates leukemia stem cells [17]. It has been shown to suppress the production of inflammatory cytokines such as interleukin-1 (IL-1), TNF-α, IL-6, and IL-8 [18], induce heat shock response [19], and disrupt heat shock protein 90 (Hsp90) [20], possibly through its interaction with cdc37 [21] and co-chaperone p23 [22]. Gene expression signature-based analysis has also revealed that celastrol is a Hsp90 inhibitor [16]. Molecular docking studies have indicated that celastrol is a potent proteasome inhibitor [15]. This triterpene was actually described as a regulator of protein homeostasis [23].
In the present report, we investigated whether celastrol can modulate the expression of CXCR4 and thus inhibit tumor cell invasion. Our results show that this triterpene can downregulate CXCR4 expression induced by HER2 oncogenes and in various tumor cells that overexpress this chemokine receptor. This downregulation occurred at the transcriptional level and also at the translational level and led to inhibition of CXCL12-induced invasion by colon and pancreatic tumor cells.
Materials and methods
Reagents
A 10-mol/L solution of celastrol (Cayman Chemicals, Ann Arbor, MI), gedunin, and withaferin A (Tocris Bioscience, Ellisville, MO) were prepared in 100% dimethyl sulfoxide, stored as small aliquots at −20°C, and then diluted as needed in cell culture medium. RPMI 1640, Dulbecco’s modified Eagle’s medium (DMEM)/F12, Iscove’s modified Dulbecco’s medium (IMDM), DMEM, fetal bovine serum (FBS), 0.4% trypan blue vital stain, and an antibiotic–antimycotic mixture were obtained from Invitrogen. Rabbit polyclonal antibody to CXCR4 was obtained from Abcam. Lactacystin was obtained from Calbiochem. Small interfering RNA (siRNA; ON-TARGETplus SMARTpool) of CXCR4 (GenBank accession no. NM_003467) was purchased from Dharmacon (Lafayette, CO).
Cell lines, cell culture and DNA constructs
Breast cancer cell lines that express different levels of HER2, including stably transfected MCF-7/HER2 and their vector control, were kindly provided by Dr. D. Yu of MD Anderson Cancer Center. PANC-28 (human pancreatic carcinoma) cell lines were provided by Dr. S. Reddy of MD Anderson Cancer Center. The KBM-5 (human chronic myeloid leukemia) was kindly provided by Dr. N. Donato. The rest of the cell lines like SCC-4 (human squamous cell carcinoma), Caco-2 (human colorectal adenocarcinoma), HCT116 (human colorectal carcinoma), MDA-MB-231 (human breast adenocarcinoma), BxPC-3 (human pancreatic adenocarcinoma), MIA PaCa-2 (human pancreatic carcinoma), AsPC-1 (human pancreatic adenocarcinoma), and A293 (human embryonic kidney cells) were obtained from American Type Culture Collection. MCF-7/HER2 and its control cells were cultured in DMEM/F12 supplemented with 10% FBS, Caco-2, PANC-28, and MIA PaCa-2 cells cultured in RPMI 1640 with 10% FBS. KBM-5 cells were cultured in IMDM with 15% FBS. HCT116, AsPC-1, BxPC-3, A293, and MDA-MB-231 cells were cultured in DMEM with 10% FBS. SCC-4 cells were cultured in DMEM containing 10% FBS, 100 μmol/L nonessential amino acids, 1 mmol/L pyruvate, 6 mmol/L L-glutamine, and 1× vitamins. Culture media were also supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin except media for MCF-7/HER2 and its control. Cells were maintained at 37 °C in an atmosphere of 5% CO2–95% air. HA-CXCR4 construct was kindly provided by Dr. Benovic (Thomas Jefferson University, PA) [24] and anti-HA polyclonal antibodies were purchased from Cell Signaling.
Western blotting
For the detection of CXCR4, celastrol-treated whole-cell extracts were lysed in lysis buffer (20 mmol/L Tris (pH 7.4), 250 mmol/L NaCl, 2 mmol/L EDTA (pH 8.0), 0.1% Triton X-100, 0.01 mg/mL aprotinin, 0.005 mg/mL leupeptin, 0.4 mmol/L phenylmethylsulfonyl fluoride, and 4 mmol/L NaVO4). Lysates were then spun at 14,000 rpm for 10 min to remove insoluble material and resolved on a 10% sodium dodecyl sulfate gel. After electrophoresis, the proteins were electrotransferred onto a nitrocellulose membrane, blocked with 5% nonfat milk, and probed with anti-CXCR4 antibodies (1:3,000) overnight at 4°C. The blot was washed, exposed to horseradish peroxidase-conjugated secondary antibodies for 2 h, and finally examined by chemiluminescence (GE Healthcare).
Electrophoretic mobility shift assay
To assess NF-κB activation, we isolated nuclei from cells and carried out electrophoretic mobility shift assays (EMSAs) essentially as previously described. In brief, nuclear extracts prepared from cancer cells (1×106/mL) were incubated with 32P-end-labeled 45-mer double-stranded NF-κB oligonucleotide (4 μg of protein with 16 fmol of DNA) from the HIV long terminal repeat (5′-TTGTTACAAGGGACTTTC CGCTG GGGACTTTC CAGGGA GGCGT GG-3′; italicized letters indicate NF-κB binding sites) for 15 min at 37 °C. The resulting DNA–protein complex was separated from free oligonucleotides on 6.6% native polyacrylamide gels. The dried gels were visualized and radioactive bands were quantitated using Phosphorimager (GE Healthcare) and ImageQuant software.
RNA analysis and RT-PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). One microgram of total RNA was converted to complementary DNA (cDNA) by Superscript reverse transcriptase and then amplified by Platinum Taq polymerase using Superscript One-Step reverse transcription polymerase chain reaction (RT-PCR) kit (Invitrogen). The relative expression of CXCR4 and CXCR7 was analyzed by quantitative RT-PCR with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. The following pairs of forward and reverse primer sets were used: CXCR4, 5′-GAAGCTGTTGGCTGAAAAGG-3′ and 5′-GAGTCG ATGCTGATCCCAAT-3′ (PCR product size, 345 bp; Gen-Bank accession no. NM_003467); CXCR7, 5′-CT CACGTGCAAAGTCACACA-3′ and 5′-CGATAATGGAG AAGGGAACG-3′ (PCR product size, 343 bp; GenBank accession no. NM_001047841). The RT-PCR mixture contained 12.5 μL of 2× reaction buffer, 10 μL each of cDNA, 0.5 μL each of forward and reverse primers, and 1 μL of RT-Platinum Taq in a final volume of 50 μL. The reaction was done at 50 °C for 30 min, 94 °C for 2 min, and then 30 cycles of denaturation at 94°C for 15 s, annealing at 54 °C for 30 s, and extension 72 °C for 1 min. The final extension was done at 72 °C for 10 min. PCR products were run on 2% agarose gel and then stained with ethidium bromide. Stained bands were visualized under UV light and photographed.
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) following the manufacturer’s protocol. The messenger RNA (mRNA) expression of CXCR4 in HCT116 cells was determined using real-time PCR. For mRNA quantification, cDNA was synthesized using 3 μg RNA through a RT reaction (iScriptTM cDNA Synthesis Kit, Bio-Rad). Using SYBR Green/Fluorescein PCR Master Mix (SuperArray Bioscience Corporation), cDNA was amplified using real-time PCR with a Bio-Rad MyiQ thermocycler and the SYBR green detection system (Bio-Rad). Samples were run in triplicate to ensure amplification integrity. Manufacturer-supplied (SuperArray Bioscience Corporation) primer pairs were used to measure the mRNA levels of CXCR4. The standard PCR conditions were as follows: 95 °C for 15 min, then 40 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, as recommended by the primer’s manufacturer. The expression levels of genes were normalized to the expression level of GAPDH mRNA in each sample. The threshold for positivity of real-time PCR was determined based on negative controls. For mRNA analysis, the calculations for determining the relative level of gene expression were made using the cycle threshold (Ct) method. The mean Ct values from duplicate measurements were used to calculate the expression of the target gene with normalization to a housekeeping gene used as an internal control (GAPDH) and using the 2−ΔCt formula.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was done as previously described [25] with some modifications. HCT116 cells (2×107) were incubated with or without 3 μmol/L celastrol for the indicated times and immunoprecipitated with anti-p65 antibody. PCR analyses were carried out for 39 cycles with primers 5′-TCGAAAGCTTATT GCCGCCTACT-3′ (forward) and 5′-TCGAGGATCCCC AACAAACTGAAGTTTCTG-3′ (backward) for CXCR4, the amplified DNA fragment (−417 to +1), which contains two NF-κB binding sites [26].
Transfection with small interfering RNA
HCT116 cells (0.25×106 cells per well) were plated in six-well plates, allowed to adhere for 12 h, and transfected with siRNAs following the protocol given by the manufacturer. Briefly, 12 μL HiPerFect transfection reagent (Qiagen) were added to 50 nmol/L siRNAs in a final volume of 100 μL culture medium. To determine the effect of siRNA transfection, the transfected cells were collected after 48 h and the protein levels of CXCR4 were measured by Western blot.
Invasion assay
In vitro invasion assay was done using the BD Bio-Coat Matrigel invasion assay system (BD Biosciences) according to the manufacturer’s instructions. Cancer cells (2×105) were suspended in medium (10% FBS–RPMI 1640 for AsPC-1, 10% FBS–DMEM for HCT116, and 12% FBS–DMEM for MIA PaCa-2) and seeded into the Matrigel-precoated Transwell chambers with polycarbonate membranes of 8-μm pore size. After preincubation with or without celastrol (3 μmol/L) for 6 h, Transwell chambers were then placed into 24-well plates in which was added the basal medium only or basal medium containing 100 ng/mL CXCL12. After incubation, the upper surface of Transwell chambers was wiped off with a cotton swab and invading cells were fixed and stained with a Diff-Quick stain. The invading cell numbers were counted in five randomly selected microscope fields (×200).
Statistical analysis
The experiments were performed in triplicate and repeated twice. The P value was obtained after ANOVA and Student–Newman–Keul tests.
Results
The present study was designed to determine the effect of celastrol on the expression of CXCR4 that is induced by the oncogene HER2 and on the constitutive expression of CXCR4 by various tumor cells. The mechanism by which celastrol modulates the chemokine receptor was also examined. Furthermore, we determined the effect of the celastrol on colon and pancreatic tumor cell invasion induced by CXCL12.
Celastrol suppresses the expression of CXCR4 protein in HER2-overexpressing breast cancer cells
HER2 overexpression has been linked with metastasis of breast cancer. Furthermore, HER2 has been shown to induce the expression of CXCR4 on breast cancer cells [12]. As shown in Fig. 1a, expression of HER2 in MCF-7 cells indeed induced the expression of CXCR4. This experiments showed that celastrol suppressed CXCR4 expression and only partially affected HER2 expression (Fig. 1b), thus suggesting that celastrol can downregulate the CXCR4 expression. Furthermore, our results also suggest that downregulation of CXCR4 by celastrol is not due to modulation of HER2 expression.
Fig. 1.
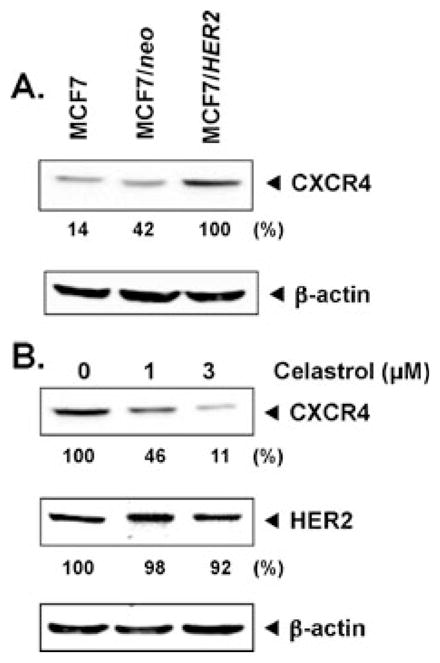
Celastrol suppresses CXCR4 in MCF-7/HER2 cells. a Western blot analysis of CXCR4 expression. Whole-cell extracts of MCF-7, MCF-7/neo, and MCF-7/HER2 (40 μg) were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and probed with anti-CXCR4 antibody. As a loading control, stripped membrane was probed with β-actin antibodies. The results shown are representative of three independent experiments. b Celastrol inhibits CXCR4 and HER2 expression. MCF-7/HER2 cells were incubated with the indicated concentrations of celastrol for 24 h. Whole-cell extracts were prepared and analyzed by Western blot analysis with antibodies against HER2 and CXCR4. The results shown are representative of three independent experiments
Celastrol-induced down-modulation of CXCR4 is not cell type-specific
Irrespective of HER2 expression, CXCR4 is overexpressed on a wide variety of tumor cells. Thus, we investigated whether celastrol downregulates expression of CXCR4 in leukemia (KBM-5), head and neck squamous cell carcinoma (SCC-4), kidney (A293), colon (HCT116, Caco-2), and pancreatic (AsPC-1, BxPC-3, MIA PaCa-2, and PANC-28) cancer cell lines. For this, cells were treated with 3 μmol/L celastrol for 24 h and then examined for CXCR4 expression. Figure 2a shows that celastrol downregulated CXCR4 in most of the cell lines, but most dramatically in colon cancer (HCT116), pancreatic cancer cells (AsPC-1), and embryonic kidney cells (A293 cells). The effect of celastrol on CXCR4 expression was minimal on pancreatic cancer BxPC-3 and MIA PaCa-2. Celastrol has no effect on the expression of CXCR4 in Caco-2 cells. Thus, these results suggest that CXCR4 downregulation by celastrol may involve cell type-specific mechanism.
Fig. 2.

Celastrol downregulates CXCR4 in different cell types. a Different cells were incubated with 3 μmol/L celastrol for 24 h. Whole-cell extracts were prepared and analyzed by Western blot analysis with antibodies against CXCR4. The same blots were stripped and reprobed with β-actin antibody to show equal protein loading. The results shown are representative of three independent experiments. b Celastrol suppresses CXCR4 levels in a dose-dependent manner. HCT116 cells (1×106) were treated with the indicated concentrations of celastrol for 24 h. Whole-cell extracts were then prepared, and 40 μg of protein was resolved on SDS-PAGE, electrotransferred onto nitrocellulose membranes, and probed for CXCR4. The results shown are representative of three independent experiments. c Celastrol suppresses CXCR4 levels in a time-dependent manner. HCT116 cells (1×106) were treated with 3 μmol/L celastrol for the indicated times, after which Western blotting was done as described above. The same blots were stripped and reprobed with β-actin antibody to show equal protein loading. The results shown are representative of three independent experiments
Whether the effect of celastrol on CXCR4 expression is dose-dependent and time-dependent was examined using HCT116, a colorectal cancer cell line. When these cells were incubated with different concentrations of celastrol for 24 h, celastrol suppressed the expression of CXCR4 in a dose-dependent manner (Fig. 2b). Celastrol-induced suppression could be observed at the lowest concentration tested, 2 μmol/L. We also found that celastrol at 3 μmol/L suppressed the expression of CXCR4 in a time-dependent manner (Fig. 2c) and minimum time required was 3 h. This downregulation was not due to a decrease in cell viability because >90% cells were viable under these conditions (data not shown).
Downregulation of CXCR4 by celastrol is not mediated through its degradation
CXCR4 has been shown to undergo ubiquitination at its lysine residue, which in turn leads to CXCR4 degradation [10, 27]. To determine whether celastrol induces down-regulation of CXCR4 through proteasomal degradation, we treated HCT116 cells with lactacystin, a proteasome inhibitor, 1 h before celastrol treatment. As shown in Fig. 3a (top right panel), lactacystin had no effect on celastrol-induced down-modulation of CXCR4, suggesting that proteasomal degradation is an unlikely mechanism by which celastrol downregulated CXCR4. Under these conditions, lactacystin inhibited the TNF-induced inhibitor of κBα (IκBα) degradation, indicating its ability to suppress proteasomal degradation (top left panel).
Fig. 3.

Celastrol suppresses CXCR4 through mRNA level. a Celastrol does not suppress CXCR4 through lysosomal and proteasomal degradation. Cells were treated with the indicated concentration of lactacystin (upper right panel) or chloroquine (lower right panel) for 1 h at 37 °C, followed by treatment with 3 μmol/L celastrol for 24 h. As a positive control for the effect of lactacystin on proteasomal degradation, TNF-induced IκBα was used (upper left panel). Similarly, as a positive control for chloroquine effect, TNF-induced p60 (TNFR1) was used (lower left panel). Whole-cell extracts were prepared and analyzed by Western blot analysis with antibodies against CXCR4. The same blots were stripped and reprobed with β-actin antibody to show equal protein loading. The results shown are representative of three independent experiments. b Celastrol suppresses the expression of CXCR4 mRNA. Cells were treated with 3 μmol/L celastrol for the indicated times. Total RNA was isolated and analyzed by RT-PCR assay as described in the “Materials and methods” section. GAPDH was used to show equal loading of total RNA. The results shown are representative of three independent experiments. c The result of mRNA expression of CXCR4 by quantitative real-time PCR is presented after normalization to GAPDH using the Ct method. Significantly less vs. control (0 vs. 24 h, *P<0.05, n=3)
There are studies showed that CXCR4 could undergo ligand-dependent lysosomal degradation [27]. Therefore, we treated cells with chloroquine, a lysosomal inhibitor, for 1 h before exposing them to celastrol. At a 200-μmol/L concentration, chloroquine did not prevent the down-modulation of CXCR4 (Fig. 3a, lower right panel), indicating that lysosomal degradation was also not the pathway involved in the suppression of expression of CXCR4. Under these conditions, ligand-induced degradation of its receptor TNFR1 (p60) was reversed by chloroquine (Fig. 3a, top left panel).
Celastrol downregulates CXCR4 by multiple mechanisms
Because celastrol did not downregulate CXCR4 expression by enhancing its degradation, we investigated whether suppression occurred at the transcriptional level using RT-PCR and also by quantitative PCR (real-time PCR). Cells were treated with celastrol for different times and then examined for steady-state mRNA level of CXCR4. As shown in Fig. 3b, celastrol induced the downregulation of CXCR4 mRNA. Major inhibition of CXCR4 mRNA occurred at 12 h of celastrol treatment. The kinetics of decrease in CXCR4 protein (Fig. 2c) was seen at 3 h, suggesting that down-modulation involved translational regulation. Interestingly, celastrol had no effect on the mRNA of another chemokine receptor, CXCR7 (Fig. 3b, left panel), thus indicating that the effects were specific for CXCR4 in our study.
Celastrol suppresses constitutive activation of NF-κB in HCT116 and TNF-inducible NF-κB Caco-2 in cells
The promoter of CXCR4 is known to contain several NF-κB binding sites. Moreover, celastrol has been shown to inhibit NF-κB activation in human chronic myeloid leukemia cells and also in wide variety of solid tumor cell lines [28–30]. In addition, HER2 oncogene has been shown to activate NF-κB [31] in breast cancer cells. Thus, it is possible that celastrol manifests its effect on CXCR4 by suppressing NF-κB activation. In a DNA-binding assay, celastrol inhibited constitutive NF-κB activation in HCT116 and TNF-induced NF-κB activation in Caco-2 cells, which occurred in a dose-dependent manner (Fig. 4a, left panel for HCT116 and right panel for Caco-2). Our results show that, although Caco-2 cells do not express constitutive NF-κB, they do express constitutive CXCR4, thus indicating that expression of NF-κB may not be linked to CXCR4 expression.
Fig. 4.
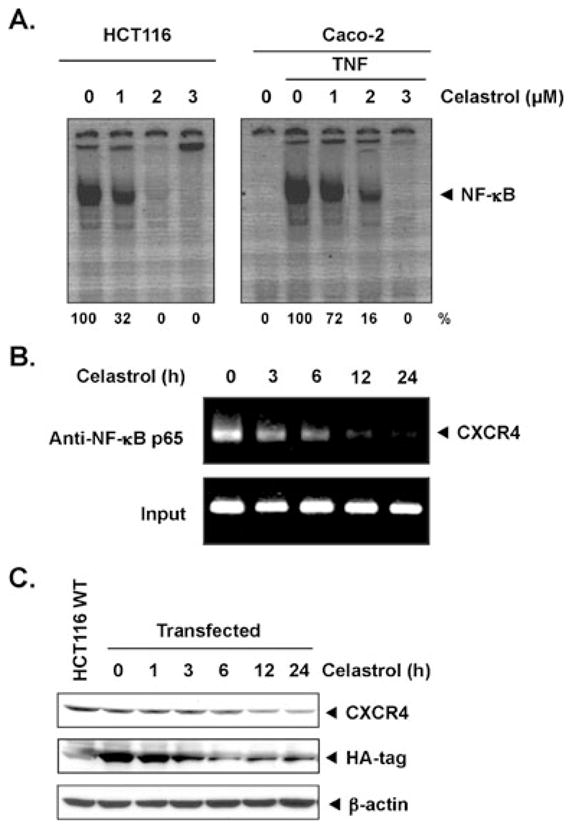
Celastrol suppresses NF-κB and CXCR4 through ChIP assay. a Celastrol inhibits HCT116 (upper left) constitutive NF-κB and Caco-2 (upper right) TNF-induced NF-κB activation in colon cancer cells. HCT116 and Caco-2 cells were incubated with celastrol for 24 h. The nuclear extracts were assayed for NF-κB activation by EMSA. The results shown are representative of three independent experiments. b Celastrol inhibits binding of NF-κB to the CXCR4 promoter. HCT116 cells were pretreated with 3 μmol/L celastrol for 3, 6, 12, and 24 h, and the proteins were cross-linked with DNA with formaldehyde and then subjected to ChIP assay with an anti-p65 antibody. PCR on the immunoprecipitate was performed using primers spanning −417 to +1 of the CXCR4 promoter. Reaction products were resolved by electrophoresis. c Celastrol suppresses CXCR4 expression in a NF-κB-independent manner—HCT116 cells were transfected with HA-tagged CXCR4 expression plasmid where CXCR4 expression was under the control of constitutive promoter (CMV). Transfected HCT116 cells (1×106) were treated with 3 μmol/L celastrol for the indicated times, after which Western blotting was done using anti-HA and anti-CXCR4 antibodies as described above. The same blots were stripped and reprobed with β-actin antibody to show equal protein loading
Celastrol inhibits CXCR4 expression by both NF-κB-dependent and NF-κB-independent manners
Since, as a late event, celastrol inhibited the steady-state mRNA expression of CXCR4 in HCT116 cells and it also inhibited NF-κB activation, ChIP assay using p65 antibodies was performed to find out whether downregulation of CXCR4 by celastrol in HCT116 cells was due to reduction of NF-κB at the CXCR4 promoter. As shown in Fig. 4b, celastrol reduced the NF-κB occupancy at the CXCR4 promoter. The reduction was substantial by 6 h and almost no NF-κB was found at the CXCR4 promoter after 12 h. The kinetics of reduction of NF-κB occupancy at the CXCR4 promoter preceded the down-modulation of CXCR4 mRNA (Fig. 3b). Overall, these results suggest that celastrol inhibits CXCR4 mRNA expression by suppressing binding of NF-κB to the CXCR4 promoter.
We also examined the effect of celastrol in HCT116 cells transfected with HA-tagged CXCR4 expression plasmid where CXCR4 expression was under the control of a constitutive promoter. Figure 4c shows that celastrol inhibited constitutive expression of HA-CXCR4 in a time-dependent manner, and the inhibition was effective as early as 3 h of celastrol treatment. This suggests that celastrol also inhibits CXCR4 expression in a NF-κB-independent manner.
CXCR4 is essential for CXCL12-induced invasion
Disruption of CXCR4 and CXCL12 interaction by selective antagonists or anti-CXCR4 antibody blocks cancer metastasis, suggesting an essential role for CXCR4. Therefore, when HCT116 cells were transfected with siRNA specific for CXCR4, it efficiently inhibited CXCL12-mediated invasion by 38% (Fig. 5a, b). Indeed, the CXCR4-specific siRNA reduced CXCR4 protein expression (Fig. 5c).
Fig. 5.

Celastrol suppresses invasion in colon cancer cells. a HCT116 cells (0.25×106 cells per well) were transfected with siRNAs and the transfected cells were collected after 48 h. After transfection, cells were seeded in the top chamber of Matrigel. Transwell chambers were then placed into 24-well plates in which either the basal medium was added or 100 ng/mL CXCL12 in the basal medium. After the incubation, invasion assay was done as described in the “Materials and methods” section. The results shown are representative of three independent experiments. b Histogram of data obtained from invasion assay in Fig. 4a. c Western blot for CXCR4 showing its down-modulation by siRNA. d HCT116 cells (2×105; 10% FBS–DMEM/F12) were seeded in the top chamber of Matrigel. After preincubation with or without celastrol (3 μmol/L) for 6 h, Transwell chambers were then placed into 24-well plates in which either the basal medium was added or 100 ng/mL CXCL12 in basal medium. After incubation, invasion assay was done as described in the “Materials and methods” section. The results shown are representative of three independent experiments. e Columns mean number of invaded cells, bars SE. *P<0.05
Celastrol suppresses CXCL12-induced colon cancer cell invasion
Several lines of evidence implicate CXCR4 in colon cancer metastasis and it was found that the motility and migration of colon cancer cells could be induced by exposure to CXCL12 [32]. In addition, colon cancer metastasis can be inhibited by silencing CXCR4 [10]. In an in vitro invasion assay, we found that CXCL12 induced the invasion of colon cancer cells and that celastrol effectively abrogated the invasion (Fig. 5d, e).
Celastrol inhibits CXCL12-induced pancreatic cancer cell invasion
CXCR4–CXCL12 signaling has also been shown to play a critical role in pancreatic cancer metastasis [10]. In a cell invasion assay (Fig. 6a), we found that treatment with celastrol suppressed CXCL12-induced invasion of pancreatic cancer AsPC-1 cells. We found that celastrol down-regulated expression occurred at both the mRNA (Fig. 6b) and protein levels (Fig. 6c) for CXCR4. No invasion was found in case of MIA PaCa-2 (Fig. 6d), which indicates that this effect was only for selected cell lines.
Fig. 6.

Celastrol suppresses CXCR4 and invasion in pancreatic cancer cells. a Right panel AsPC-1 cells (2×105; 2% FBS–DMEM) were seeded in the top chamber of Matrigel. After preincubation with or without celastrol (3 μmol/L) for 6 h, Transwell chambers were then placed into 24-well plates in which either the basal medium was added or 100 ng/mL CXCL12 in basal medium. After incubation, invasion assay was done as described in the “Materials and methods” section. The results shown are representative of three independent experiments. Left panel histogram of data obtained from invasion assay in Fig. 5a, right panel, SE. *P<0.01. b Celastrol suppresses expression of CXCR4 mRNA. AsPC-1 cells were treated with 3 μmol/L celastrol for the indicated times. Total RNA was isolated and analyzed by RT-PCR assay as described in the “Materials and methods” section. GAPDH was used to show equal loading of total RNA. The results shown are representative of three independent experiments. c Cells were incubated with 3 μmol/L celastrol for 24 h. Whole-cell extracts were prepared and analyzed by Western blot analysis with antibodies against CXCR4. The same blots were stripped and reprobed with β-actin antibody to show equal protein loading. The results shown are representative of three independent experiments. d Left panel MIA PaCa-2 cells (2×105; 2% FBS–DMEM) were seeded in the top chamber of Matrigel and invasion assay was done as describe above. The results shown are representative of three independent experiments. Right panel histogram of data obtained from invasion assay in Fig. 5d, left panel
Other terpenes that inhibit Hsp90 do not suppress CXCR4
Celastrol has been shown to disrupt Hsp90 and cdc37 interaction [20]. Molecular docking studies have indicated that celastrol is a potent inhibitor of Hsp90 [20] as shown to have gene expression signature-based analyses [16]. Besides celastrol, other terpenes such as withaferin A and gedunin have also been shown to block Hsp90 and regulate protein homeostasis (Fig. 7a) [23]; whether withaferin A and gedunin can also affect CXCR4 expression was examined. As shown in Fig. 7b, withaferin A and gedunin had no effect on CXCR4 expression, whereas celastrol down-modulated CXCR4 expression under similar conditions.
Fig. 7.
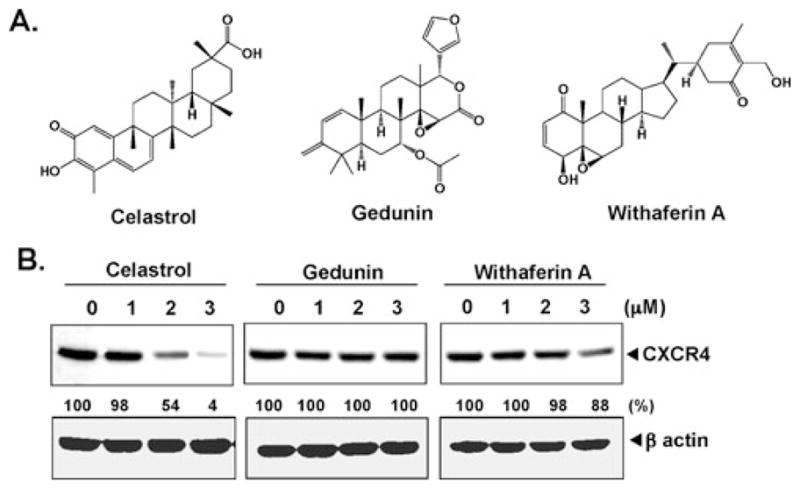
Except celastrol, no other Hsp90 inhibitor suppresses CXCR4. a Structures of celastrol, gedunin, and withaferin A. b HCT116 colon cancer cells were incubated with indicated concentration of celastrol (left panel), gedunin (middle panel), and withaferin A (right panel) for 24 h. Whole-cell extracts were prepared and analyzed by Western blot analysis with antibodies against CXCR4. The same blots were stripped and reprobed with β-actin antibody to show equal protein loading. The results shown are representative of three independent experiments
Discussion
A number of studies have suggested that the CXCR4–CXCL12 axis plays a pivotal role in triggering tumor metastasis. The goal of the present study was to determine whether celastrol could suppress the expression of CXCR4, a chemokine receptor that has been closely linked with cancer cell growth, invasion, angiogenesis, and metastasis. We have shown for the first time that celastrol abolished the expression of HER2-induced CXCR4 expression in breast cancer cells. We also showed that celastrol inhibited CXCR4 expression in a variety of different cancer cell types. It has been already published that celastrol has mild proteolytic activity, but our results showed that down-regulation of CXCR4 did not occur through proteolytic degradation of the receptor but rather through down-regulation of transcription. Furthermore, suppression of receptor expression led to reduced invasion, whether induced by tumor cells or CXCL12.
CXCR4, the chemokine receptor, has been reported to be overexpressed in a variety of different tumors and promotes metastasis of cancer [32]. Various reports suggested that expression of CXCR4 might be increased by inflammatory cytokines such as TNF [33] and VEGF [34]. Reports also suggested that transient transfection of NF-κB expression plasmid (p65 subunit) upregulated CXCR4 expression in human prostate cancer PC-3 cells [35]. It has also been found that anti-HER2 antibody in breast cancer down-regulated CXCR4 expression by suppressing CXCR4 translation and by allowing its degradation [34].
Many studies have documented the ligand-dependent downregulation of the CXCR4 expression by lysosomal degradation [36], which involves atrophin-interacting protein 4-mediated ubiquitination and degradation [27]. Our results, however, suggest that downregulation of CXCR4 by celastrol is induced not through lysosomal degradation or through proteasomal degradation as inhibitors of this pathway had no effect. We found, however, that down-regulation of CXCR4 by the triterpene occurs at both the transcriptional and translational levels.
The transcription factor NF-κB [37] has been implicated in the regulation of CXCR4, and celastrol has been shown to downregulate NF-κB activation. Therefore, it is possible that downregulation of CXCR4 mRNA occurs through downregulation of NF-κB activation. Indeed, the occupancy of NF-κB at the CXCR4 promoter was reduced by celastrol, suggesting that down-modulation of CXCR4 mRNA by celastrol involves regulation at the transcription initiation level. Our results are in agreement with those of Muller et al. [5] who reported that the transcription factor NF-κB regulates CXCR4 expression in breast cancer cells. NF-κB regulates the motility of breast cancer cells by direct upregulation of CXCR4. Overexpression of the IκB in breast cancer cells with constitutive NF-κB activity resulted in reduced expression of CXCR4 and a corresponding loss of CXCL12-mediated migration of breast cancer cells in vitro [37]. The possibility that mechanisms other than suppression of NF-κB activation are involved in the downregulation of CXCR4 by celastrol cannot be ruled out. Indeed, our results suggest that early event of regulation of CXCR4 expression by celastrol involves a NF-κB-independent mechanism, possibly a translational regulation. In addition, the regulation of CXCR4 by celastrol is cell type-specific. What the mechanism of NF-κB-independent regulation and cell type specificity of CXCR4 regulation is not clear at present.
Along with CXCR4, the activation of NF-κB also induces expression of various adhesion molecules including intracellular adhesion molecule-1, vascular cell adhesion molecule-1, and endothelial leukocyte adhesion molecule-1, which are also linked with cancer cell metastasis to other organs. Because celastrol can inhibit both inducible and constitutively activated NF-κB in a wide variety of tumor cell lines [28–30], it is possible that celastrol can suppress the expression of these adhesion molecules as well.
We also found that celastrol suppressed the ligand-induced invasion of both colorectal and pancreatic cancers, and this correlated with the downregulation of CXCR4, thus suggesting that this triterpene has a potential to suppress tumor metastasis through its action on CXCR4. Elevated levels of CXCR4 have been demonstrated in nodal metastasis of various human cancers [38].
Overexpression of mutant IκBα super-repressor suppressed CXCR4 gene expression in PC-3 cells [35]. This was supported by the findings that transient transfection with NF-κB expression plasmid (p65 subunit) upregulated CXCR4 expression in PC-3 cells and that CXCL12 enhances transcriptional activity of NF-κB in an IκBα-dependent manner, which in turn would upregulate expression of CXCR4, an effect that was reversed by a dominant-negative mutant IκBα [35].
Numerous molecular targets of celastrol have been identified [39], including IKK-α, IKK-β [30], cdc37 [21], p23 [22], heat shock factor 1 [19], and proteasomes [15]. The interaction with most of these targets is probably through cysteine residues in these proteins with quinone methide present in celastrol. We found that the quinone group of celastrol is critical for its activity, as at the same dose, gedunin and withaferin A, which lacks this group, had no activity. Celastrol has been shown to exhibit antiproliferative activity against a variety of tumor cells, including leukemia [40] and prostate cancer [15]. It also modulates the expression of proinflammatory cytokines [18], inducible nitric oxide synthase, adhesion molecules in endothelial cells [41], proteasome activity [15], topoiso-merase II [40], and heat shock response [19]. It is possible that some of these antitumor effects of celastrol are also mediated through CXCR4 regulation. We recently showed that celastrol could inhibit angiogenesis-mediated tumor growth. When given subcutaneously to mice bearing human prostate cancer xenografts, this triterpene significantly reduced tumor growth and decreased tumor angiogenesis [42]. This correlated with inhibition of VEGF-induced proliferation, migration, invasion, and capillary-like structure formation by primary cultured human umbilical vascular endothelial cells, suppressed the VEGF-induced activation of AKT, mammalian target of rapamycin, and ribosomal protein S6 kinase.
Taken together, our data suggest that celastrol can downregulate the expression of CXCR4, a key receptor involved in the cross-talk between tumor cells and its microenvironment, which contributes to its anti-invasive activity. Further in vivo studies are planned to show the relevance of these observations to cancer treatment.
Acknowledgments
Dr. Aggarwal is the Ransom Horne, Jr., Professor of Cancer Research. This work was supported by a program project grant from National Institutes of Health (NIH CA-124787-01A2) and a grant from Clayton Foundation for Research, USA. We would like to thank Dr. J. L. Benovic, Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA and Dr. M.C. Hung, Department of Molecular and Cellular Oncology, MD Anderson Cancer Center for HA-CXCR4 plasmid constructs and also Mr. Walter Pagel for his careful reading of the manuscript.
Footnotes
Conflict of interest The authors declare no conflict of interest.
Contributor Information
Vivek R. Yadav, Cytokine Research Laboratory, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
Bokyung Sung, Cytokine Research Laboratory, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Sahdeo Prasad, Cytokine Research Laboratory, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Ramaswamy Kannappan, Cytokine Research Laboratory, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Sung-Gook Cho, Institute of Biosciences and Technology, Department of Molecular and Cellular Medicine, Texas A&M University Health Science Center, Houston, TX 77030, USA.
Mingyao Liu, Institute of Biosciences and Technology, Department of Molecular and Cellular Medicine, Texas A&M University Health Science Center, Houston, TX 77030, USA.
Madan M. Chaturvedi, Cytokine Research Laboratory, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
Bharat B. Aggarwal, Email: aggarwal@mdanderson.org, Cytokine Research Laboratory, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
References
- 1.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orosz P, Echtenacher B, Falk W, Ruschoff J, Weber D, Mannel DN. Enhancement of experimental metastasis by tumor necrosis factor. J Exp Med. 1993;177:1391–1398. doi: 10.1084/jem.177.5.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leivonen SK, Kahari VM. Transforming growth factor-beta signaling in cancer invasion and metastasis. Int J Cancer. 2007;121:2119–2124. doi: 10.1002/ijc.23113. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 5.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 6.Xu L, Duda DG, di Tomaso E, Ancukiewicz M, Chung DC, Lauwers GY, Samuel R, Shellito P, Czito BG, Lin PC, Poleski M, Bentley R, Clark JW, Willett CG, Jain RK. Direct evidence that bevacizumab, an anti-VEGF antibody, up-regulates SDF1alpha, CXCR4, CXCL6, and neuropilin 1 in tumors from patients with rectal cancer. Cancer Res. 2009;69:7905–7910. doi: 10.1158/0008-5472.CAN-09-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porcile C, Bajetto A, Barbero S, Pirani P, Schettini G. CXCR4 activation induces epidermal growth factor receptor transactivation in an ovarian cancer cell line. Ann NY Acad Sci. 2004;1030:162–169. doi: 10.1196/annals.1329.021. [DOI] [PubMed] [Google Scholar]
- 9.Zeelenberg IS, Ruuls-Van Stalle L, Roos E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res. 2003;63:3833–3839. [PubMed] [Google Scholar]
- 10.Marchesi F, Monti P, Leone BE, Zerbi A, Vecchi A, Piemonti L, Mantovani A, Allavena P. Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 2004;64:8420–8427. doi: 10.1158/0008-5472.CAN-04-1343. [DOI] [PubMed] [Google Scholar]
- 11.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–1837. [PubMed] [Google Scholar]
- 12.Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 13.Holm NT, Byrnes K, Li BD, Turnage RH, Abreo F, Mathis JM, Chu QD. Elevated levels of chemokine receptor CXCR4 in HER-2 negative breast cancer specimens predict recurrence. J Surg Res. 2007;141:53–59. doi: 10.1016/j.jss.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Calixto JB, Campos MM, Otuki MF, Santos AR. Anti-inflammatory compounds of plant origin. Part II. Modulation of pro-inflammatory cytokines, chemokines and adhesion molecules. Planta Med. 2004;70:93–103. doi: 10.1055/s-2004-815483. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 16.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Hassane DC, Guzman ML, Corbett C, Li X, Abboud R, Young F, Liesveld JL, Carroll M, Jordan CT. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood. 2008;111:5654–5662. doi: 10.1182/blood-2007-11-126003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He W, Huang FC, Gavai A, Chan WK, Amato G, Yu KT, Zilberstein A. Novel cytokine release inhibitors. Part III: truncated analogs of tripterine. Bioorg Med Chem Lett. 1998;8:3659–3664. doi: 10.1016/s0960-894x(98)00671-4. [DOI] [PubMed] [Google Scholar]
- 19.Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem. 2009;284:35381–35389. doi: 10.1074/jbc.M109.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sreeramulu S, Gande SL, Gobel M, Schwalbe H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl. 2009;48:5853–5855. doi: 10.1002/anie.200900929. [DOI] [PubMed] [Google Scholar]
- 22.Chadli A, Felts SJ, Wang Q, Sullivan WP, Botuyan MV, Fauq A, Ramirez-Alvarado M, Mer G. Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone p23. J Biol Chem. 2010;285:4224–4231. doi: 10.1074/jbc.M109.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L, Kelly JW. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell. 2003;5:709–722. doi: 10.1016/s1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- 25.Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 26.Maroni P, Bendinelli P, Matteucci E, Desiderio MA. HGF induces CXCR4 and CXCL12-mediated tumor invasion through Ets1 and NF-kappaB. Carcinogenesis. 2007;28:267–279. doi: 10.1093/carcin/bgl129. [DOI] [PubMed] [Google Scholar]
- 27.Bhandari D, Trejo J, Benovic JL, Marchese A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J Biol Chem. 2007;282:36971–36979. doi: 10.1074/jbc.M705085200. [DOI] [PubMed] [Google Scholar]
- 28.Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood. 2007;109:2727–2735. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 29.Idris AI, Libouban H, Nyangoga H, Landao-Bassonga E, Chappard D, Ralston SH. Pharmacologic inhibitors of IkappaB kinase suppress growth and migration of mammary carcinosarcoma cells in vitro and prevent osteolytic bone metastasis in vivo. Mol Cancer Ther. 2009;8:2339–2347. doi: 10.1158/1535-7163.MCT-09-0133. [DOI] [PubMed] [Google Scholar]
- 30.Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, Hong YS, Lee JJ. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Biswas DK, Iglehart JD. Linkage between EGFR family receptors and nuclear factor kappaB (NF-kappaB) signaling in breast cancer. J Cell Physiol. 2006;209:645–652. doi: 10.1002/jcp.20785. [DOI] [PubMed] [Google Scholar]
- 32.Matsusue R, Kubo H, Hisamori S, Okoshi K, Takagi H, Hida K, Nakano K, Itami A, Kawada K, Nagayama S, Sakai Y. Hepatic stellate cells promote liver metastasis of colon cancer cells by the action of SDF-1/CXCR4 axis. Ann Surg Oncol. 2009;16:2645–2653. doi: 10.1245/s10434-009-0599-x. [DOI] [PubMed] [Google Scholar]
- 33.Kulbe H, Hagemann T, Szlosarek PW, Balkwill FR, Wilson JL. The inflammatory cytokine tumor necrosis factor-alpha regulates chemokine receptor expression on ovarian cancer cells. Cancer Res. 2005;65:10355–10362. doi: 10.1158/0008-5472.CAN-05-0957. [DOI] [PubMed] [Google Scholar]
- 34.Bachelder RE, Wendt MA, Mercurio AM. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res. 2002;62:7203–7206. [PubMed] [Google Scholar]
- 35.Kukreja P, Abdel-Mageed AB, Mondal D, Liu K, Agrawal KC. Up-regulation of CXCR4 expression in PC-3 cells by stromal-derived factor-1alpha (CXCL12) increases endothelial adhesion and transendothelial migration: role of MEK/ERK signaling pathway-dependent NF-kappaB activation. Cancer Res. 2005;65:9891–9898. doi: 10.1158/0008-5472.CAN-05-1293. [DOI] [PubMed] [Google Scholar]
- 36.Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- 37.Helbig G, Christopherson KW, 2nd, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD, Broxmeyer HE, Nakshatri H. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003;278:21631–21638. doi: 10.1074/jbc.M300609200. [DOI] [PubMed] [Google Scholar]
- 38.Cabioglu N, Sahin A, Doucet M, Yavuz E, Igci A, OYE, Aktas E, Bilgic S, Kiran B, Deniz G, Price JE. Chemokine receptor CXCR4 expression in breast cancer as a potential predictive marker of isolated tumor cells in bone marrow. Clin Exp Metastasis. 2005;22:39–46. doi: 10.1007/s10585-005-3222-y. [DOI] [PubMed] [Google Scholar]
- 39.Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: molecular targets of Thunder God Vine. Biochem Biophys Res Commun. 2010;394:439–442. doi: 10.1016/j.bbrc.2010.03.050. [DOI] [PubMed] [Google Scholar]
- 40.Nagase M, Oto J, Sugiyama S, Yube K, Takaishi Y, Sakato N. Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci Biotechnol Biochem. 2003;67:1883–1887. doi: 10.1271/bbb.67.1883. [DOI] [PubMed] [Google Scholar]
- 41.Zhang DH, Marconi A, Xu LM, Yang CX, Sun GW, Feng XL, Ling CQ, Qin WZ, Uzan G, d’Alessio P. Tripterine inhibits the expression of adhesion molecules in activated endothelial cells. J Leukoc Biol. 2006;80:309–319. doi: 10.1189/jlb.1005611. [DOI] [PubMed] [Google Scholar]
- 42.Pang X, Yi Z, Zhang J, Lu B, Sung B, Qu W, Aggarwal BB, Liu M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010;70:1951–1959. doi: 10.1158/0008-5472.CAN-09-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]