

Abstract
Reduced risk and severity of stroke in adult females is thought to depend on normal endogenous levels of estrogen, a well-known neuroprotectant and immunomodulator. In male mice, experimental stroke induces immunosuppression of the peripheral immune system, characterized by a reduction in spleen size and cell numbers and decreased cytokine and chemokine expression. However, stroke-induced immunosuppression has not been evaluated in female mice. To test the hypothesis that estradiol (E2) deficiency exacerbates immunosuppression after focal stroke in females, we evaluated the effect of middle cerebral artery occlusion on infarct size and peripheral and CNS immune responses in ovariectomized mice with or without sustained, controlled levels of 17-β–E2 administered by s.c. implant or the putative membrane estrogen receptor agonist, G1. Both E2- and G1-replacement decreased infarct volume and partially restored splenocyte numbers. Moreover, E2-replacement increased splenocyte proliferation in response to stimulation with anti-CD3/CD28 Abs and normalized aberrant mRNA expression for cytokines, chemokines, and chemokine receptors and percentage of CD4+CD25+FoxP3+ T regulatory cells observed in E2-deficient animals. These beneficial changes in peripheral immunity after E2 replacement were accompanied by a profound reduction in expression of the chemokine, MIP-2, and a 40-fold increased expression of CCR7 in the lesioned brain hemisphere. These results demonstrate for the first time that E2 replacement in ovariectomized female mice improves stroke-induced peripheral immunosuppression.
Stroke is a sexually dimorphic disease in terms of disease risk and outcome. Women have lower risk for ischemic stroke relative to men, but this native protection diminishes after menopause. The loss of protection is thought to be related to loss of endogenous estrogen. Similarly, female animals demonstrate less tissue damage and improved functional outcome after experimental cerebral ischemia relative to their male or ovariectomized (OVX) female counterparts (for recent review, see Ref. 1). Estradiol (E2) replacement at physiological concentrations in OVX (2) and reproductively senescent female rats (3), restores the protection observed in young adult females against cerebral ischemia. Stroke not only adversely affects the brain, but also causes systemic immune dysfuntion (4). Systemic poststroke inflammatory responses have been recently described in males, including a biphasic immunopathology that consists of first stimulation, then degeneration, of the spleen and thymus (5). Activation of these lymphoid organs likely leads to immunocyte translocation into brain, exacerbating the evolving brain infarct (6). The subsequent degeneration leads to immunodepression. Humans who survive the initial brain insult, may succumb to fatal infection (4, 7, 8).
Because all previous studies have been conducted in male animals or in cells derived from male tissue, we know virtually nothing about systemic immunopathology in the female. Many members of the estrogen steroid family are well-known immunomodulators, and E2 confers benefit or enhanced pathology depending on the type of disease (9). In this study, we tested the hypothesis that peripheral immune dysfunction after focal stroke, induced by middle cerebral artery occlusion (MCAO) in the mouse, occurs in E2-deficient animals and that E2 replacement regulated to produce sustained, physiological levels restores immune function without inducing deleterious consequences for the recovering brain. We also tested whether G1, a synthetic agonist for the recently discovered G protein-coupled receptor 30 (GPR30), a putative membrane estrogen receptor, could provide similar protection afforded by E2 (10–13).
Materials and Methods
Animals
All experiments were conducted in accordance with the National Institutes of Health guidelines for the use of experimental animals. Age-matched sexually mature 8–10 wk of age, female mice C57BL/6J; Charles River Laboratories (Hollister, CA), body weight 20–25 g were used in all experiments.
Ovariectomy, E2, and G1 replacement
Intact females were studied to document effects of gonadal steroids at cyclical estrous levels. Ovariectomy was performed in female mice 1 wk before transient focal cerebral ischemia. To study sustained, controlled levels of E2, the steroid was replaced via a s.c. silastic implant containing 35 μl of 180 μg/ml E2 in sesame oil (6.3 μg total dose), as previously published, yielding physiologic levels of plasma E2 (14). G1, a synthetic specific agonist for GPR30 (15) was replaced via 30 d release s.c. pellets containing 1.8 mg G1 (Innovative Research of America, Toledo, OH) at the time of ovariectomy as previously described (16). We chose the dose based on previous studies in our laboratory (17).
MCAO in mice
Reversible focal cerebral ischemia was induced by MCAO via the intraluminal suture technique under isoflurane anesthesia for 90 min as described previously (18, 19). The animal is anesthetized initially with 4% isoflurane in oxygen-enriched air using a facemask, then maintained with 1.0–1.5%. Body temperature was maintained during the surgery by placing the animal on a heated water pad. A small laser-Doppler probe was affixed to the skull to monitor cortical perfusion and verify vascular occlusion and reperfusion. A silicone-coated 6-0 nylon monofilament was inserted into the right internal carotid artery via the external carotid artery until a drop in laser-Doppler signal was observed. After securing the filament in place, the surgical site was closed, and the animal was awakened for the 90 min of occlusion. Mice were reanesthetized, laser-Doppler probe repositioned over same site on the skull, and the occluding filament withdrawn to allow for reperfusion. Mice were then allowed to recover for 96 h.
Physiological measurements
Body and head temperatures were controlled at 36.5°C ± 1.0°C, with a warming blanket and heat lamps during surgery and ischemia. Occlusion was determined by drop in laser-Doppler signal to less than 20% of baseline. For G1, a separate, nonsurviving cohort of animals, laser-Doppler flow, mean arterial blood pressure (MABP), arterial blood gases (pH, PaO2, PaCO2), head temperature, and blood glucose values were studied just before occlusion and at 45 min during MCAO.
Infarct volume assessment
Infarction volume was measured by standard 2, 3, 5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, St. Louis, MO) histology as in previous studies (18–20). Infarct size was measured at 96 h after MCAO in 2-mm thick coronal brain sections (five total) using TTC staining and digital image analysis. Sections were incubated in 1.2% TTC in saline solution for 15 min at 37°C, and then fixed in 10% formalin for 24 h. Slices were photographed, infarcted (unstained), and uninfarcted (red color) areas were measured with MCID software (InterFocus Imaging, Linton, U.K.) and integrated across all five slices. To account for the effect of edema, the infarcted area was estimated indirectly by subtracting the uninfarcted area in the ipsilateral from the contralateral area and expressing infarct volume as a percentage of contralateral structure.
Isolation of leukocytes from spleen, thymus, and blood
Spleen and thymus from individual sham and MCAO mice were removed and a single-cell suspension was prepared by passing the tissue through a 100-μm nylon mesh (BD Falcon, Bedford, MA). The cells were washed using RPMI 1640 and the red cells lysed using 1×red cell lysis buffer (8.3 g NH4Cl in 0.01 M Tris-HCl [pH 7.4]) and incubated for 3 min (eBioscience, San Diego, CA). The cells were then washed twice with RPMI 1640, counted, and resuspended in stimulation medium (RPMI 1640, containing 10% FBS, 1% sodium pyruvate, 1% L-glutamine, 0.4% βME). Cardiac blood was collected in 3 mg/ml EDTA. Cells were pelleted and 1× red cell lysis buffer was added to the cell pellet and incubated for 10 min. Cells were washed twice with RPMI 1640, counted and resuspended in RPMI 1640.
Splenic proliferation assay
A total of 400,000 cells/well were plated on standard 96-well flat-bottom tissue-culture plates at 37°C at 7% CO2 with and without plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (1 μg/ml) (BD Falcon) Abs. Cells were incubated for 24 h and 0.5 μCi of [3H]thymidine was added 6–8 h prior to harvesting. Cells were harvested onto glass fiber filters and [3H]thymidine uptake was determined by liquid scintillation. Cells were plated in triplicate. Stimulation index (SI) is represented as an average fold change of anti-CD3/CD28 stimulated over nonstimulated counts per million.
Analysis of cell populations by FACS
All Abs were purchased from (BD Biosciences, San Jose, CA) or (eBioscience) as published (19, 20). Four-color (FITC, PE, APC, and PerCP) fluorescence flow cytometry analyses were performed to determine the phenotypes of splenocytes, thymocytes, and blood leukocytes as published (20). One million cells were washed with staining medium (PBS containing 0.1% NaN3 and 1% BSA) (Sigma-Aldrich, Chicago, IL) and incubated with the combinations of the following mAbs: CD4 (GK1.5), CD8 (53–6.7), CD3 (145–2C11), CD11b (MAC-1), CD45 (Ly-5) and CD11c (HL-3), CD19 (1D3), CD25 (PC61), CD44 (IM7), PD-1 (RMP1-30), and FoxP3 for 10 min at 4°C. One milliliter staining buffer was added to wash the cells. Propidium iodide (PI) was added to identify dead cells. The FoxP3 staining kit was used according to the manufacturer’s protocol (eBioscience). Briefly, after cells were surface stained and washed with staining buffer, the cells were fixed for 1 h and washed twice with 2 ml eBioscience permeabilization buffer. The cells were costained for 15 min with FcBlock and IgG2b PerCP (BD Pharmingen, San Jose, CA). The FoxP3 and PD-1 fluorescent-labeled Abs were directly added and incubated for 30 min. The cells were then washed twice with 2 ml per-meabilization buffer, resuspended in staining medium, and run on a BD FACSCalibur. Data were analyzed using FCS express software (De Novo Software, Los Angeles, CA).
RNA isolation and real-time PCR
Total RNA was isolated from splenocytes and brains using the RNeasy mini-kit protocol (Qiagen, Valencia, CA) and then converted to cDNA using oligo dT, random hexamers, and Superscript RT II enzyme (Invitrogen, Grand Island, NY). Real-time PCR was performed using Universal Taqman master mix (Applied Biosystems, Foster City, CA) and predesigned Taqman primers (synthesized by Applied Biosystems). Reactions were conducted on the ABI Prism 7000 Sequence Detection System (Applied Biosystems) to detect the housekeeping gene GAPDH, IFN-γ, TNF-α, IL-2, IL-10, IL-4, IL-6, IL-17, TGF-β1, IL-13, IL-1β, RANTES, MIP-2, IP-10, CCR1, CCR2, CCR3, CCR5, CCR6, CCR7, and CCR8.
Cytokine determination by Luminex Bio-Plex assay kit
Splenocytes were isolated and cultured in triplicate with plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (1 μg/ml) (BD Falcon) Abs for 24 h. Culture supernatants were assessed for cytokine levels using a Luminex Bio-Plex cytokine assay kit (Bio-Rad Laboratories, Hercules, CA), according to the manufacturer’s instructions.
Statistical analysis
Differences between infarct volumes and percentages of cellular subtypes for FACS analysis were analyzed with a Student t test. Values for infarct volumes are reported as mean ± SEM. Splenocyte counts, thymocyte counts, or SI for spleen cell proliferation assays were analyzed by either t test (for G1-replacement) or ANOVA for repeated measurements (for E2 replacement studies). Physiological parameters were analyzed by two-way ANOVA. ANOVA analyses were followed by a Neuman-Kuels post hoc test. The criterion for statistical significance was p < 0.05. Statistical analyses were carried out using SigmaStat Statistical Software (Version 3.0, SPSS, Chicago, IL).
Results
Effect of E2 on infarct size, spleen cell numbers, and thymic cell numbers
The goal of these studies was to evaluate the effects of E2 deficiency on infarct size and immune cell function and to determine whether repletion with E2, a well-known neuroprotectant and immunomodulator, could restore changes observed in the absence of E2. We thus compared infarct size and corresponding spleen and thymus cell numbers among gonadally intact, OVX and E2-repleted (OVX-E2) C57BL/6 female mice 96 h after MCAO. As expected, E2-replacement reduced infarct size in both the cortex (CTX) and striatum (STR), as well as in the total affected hemisphere (Fig. 1). The total number of spleen cells did not differ among sham-treated intact, OVX and OVX-E2 groups, but experimental stroke drastically reduced mononuclear cell numbers per spleen in all groups relative to sham-treated mice (Fig. 2A). However, sustained E2-replacement in OVX females partially restored splenic cell numbers relative to both intact, E2 cycling females and E2 deficient, OVX females (Fig. 2A). MCAO reduced thymic cell numbers in a similar fashion 96 h post reperfusion, but E2-replacement in OVX females did not restore thymic cell numbers after MCAO (Fig. 2B).
FIGURE 1.
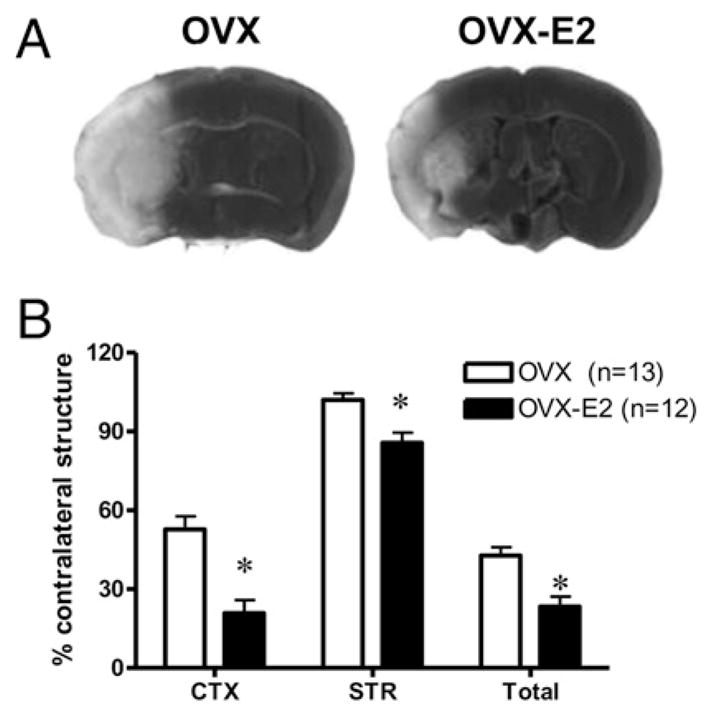
E2 conferred robust protection after MCAO. Female C57BL/ 6 mice were OVX and replaced with a s.c. E2 pellet (6.3 μg) (OVX-E2) 1 wk prior to ischemia. A, Representative TTC-stained coronal sections from OVX and OVX-E2 brains 96 h post-MCAO. B, Infarction volume (percent of contralateral structure) was assessed in the CTX, STR, and total hemisphere (Total) at 96 h after reperfusion. Data are presented as mean ±SEM.
FIGURE 2.
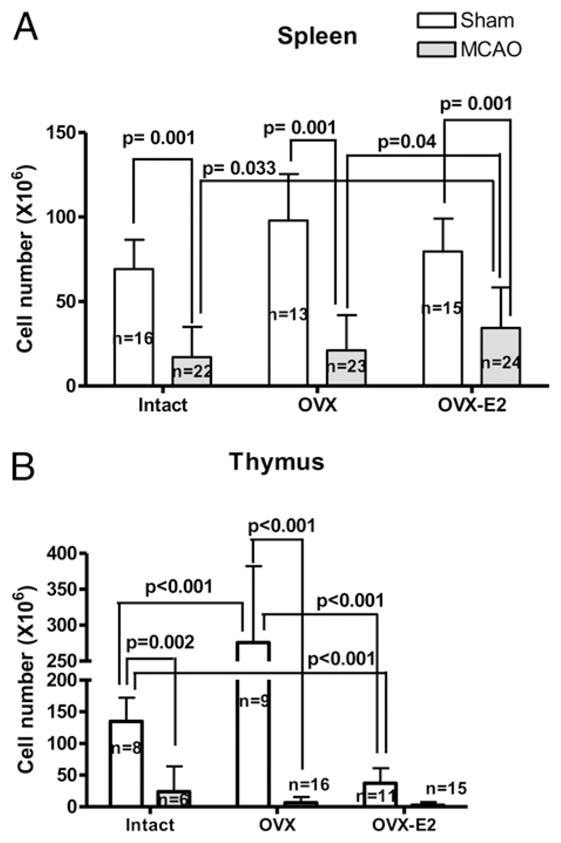
Cell counts of (A) spleen and (B) thymus from animals in Fig. 1 as compared with corresponding sham cohorts. Data are presented as mean ± SD of individual mice.
MCAO induced changes in cell populations from spleen, thymus, and blood from intact, estrogen deficient, and E2-replaced animals
As shown previously, MCAO drastically reduced spleen and thymus cell numbers (>90%) in intact and OVX females and to a lesser degree in spleen only (~60%) in E2-replaced females (Fig. 2). To determine whether E2 altered the composition of the residual peripheral leukocyte populations, spleen, thymus, and blood were isolated and evaluated for percentages of T cells (CD3+, CD4+, CD8+, CD25+, FoxP3+), B cells (CD19+), monocytes (CD11b+), and dendritic cells (DCs) (CD11c+) (Table I). In spleens of intact and OVX, but not E2-replaced females, MCAO significantly increased the percentage of CD3+, CD4+, and CD8+ T cells, but reduced CD19+ B cells versus sham-treated control mice. MCAO also increased the immunosuppressive CD4+Foxp3+ T regulatory (Treg) cell population in OVX animals and a striking observation was that the percentage of Treg cells was normalized in E2-replaced mice relative to shams and intact females. Furthermore, PD-1, an estrogen responsive factor known to contribute to the suppressive effect of Treg cells, was decreased in E2-replaced animals to levels similar to intact females. In the E2-repleted mice, CD19+ B cells remained at the level of sham-treated uninjured mice, suggesting normalized levels of B lymphocytes after stroke. MCAO did not alter the percentages of CD11b+ macrophages in the OVX and OVX-E2 groups relative to shams and intact females. Similar to intact females, the CD11c+ DC population was slightly decreased in the E2-treated group after MCAO compared with sham mice. PI positive cells, denoting dead cells, were increased after MCAO in OVX females, but did not increase in the E2-repleted animals, thus correlating with the observation that E2 repletion partially increased spleen cell numbers.
Table I.
Percentage of different cell types in spleen, thymus, and blood from intact, OVX, and OVX-E2 animals 96 h after MCAO
| Spleen | Intact Female
|
OVX
|
OVX-E2
|
|||
|---|---|---|---|---|---|---|
| Sham (n = 7) | MCAO (n = 6) | Sham (n = 12) | MCAO (n = 16) | Sham (n = 12) | MCAO (n = 19) | |
| CD3+ | 32.4 ± 5.2 | 46.8 ± 13.3a | 24.3 ± 3.4 | 37.5 ± 14.9a | 26.6 ± 5.7 | 31.1 ± 7.8b |
| CD4+ | 15.9 ± 3.3 | 22.6 ± 6.6a | 14.3 ± 2.3 | 19.6 ± 8.6a | 12.5 ± 4.1 | 14.9 ± 4.3b |
| CD8+ | 16.4 ± 3.4 | 25.2 ± 7.7a | 10.3 ± 2.6 | 17.4 ± 7.9a,b | 13.8 ± 2.4 | 15.5 ± 3.9b |
| CD19+/B220+ | 55.1 ± 2.5 | 46.5 ± 8.1a | 59.3 ± 3.4 | 45.9 ± 10.6a | 54.9 ± 4.5 | 53.7 ± 4.5b,c |
| CD11b+ | 4.2 ± 1.9 | 6.4 ± 3.0a | 5.8 ± 2.1 | 7.3 ± 5.8 | 6.2 ± 2.6 | 6.5 ± 1.9 |
| CD11c+ | 1.7 ± 0.3 | 0.9 ± 0.4a | 2.2 ± 0.5 | 1.5 ± 0.8b | 1.9 ± 0.7 | 1.2 ± 0.5a |
| CD4+ Foxp3+ | 3.4 ± 0.3 | 3.0 ± 0.9 | 2.9 ± 0.7 | 4.1 ± 0.6a,b | 3.2 ± 0.4 | 3.4 ± 1.0 |
| CD4+ PD-1+ | 0.7 ± 0.4 | 0.5 ± 0.4 | 2.2 ± 1.6 | 2.7 ± 0.5b | 1.4 ± 0.6 | 0.6 ± 0.2c |
| PI+ | 13.4 ± 2.3 | 17.0 ± 7.1 | 11.4 ± 2.4 | 18.5 ± 10.3a | 11.2 ± 4.3 | 11.3 ± 2.6b,c |
| Thymus | Sham (n = 8) | MCAO (n = 6) | Sham (n = 4) | MCAO (n = 6) | Sham (n = 3) | MCAO (n = 6) |
|
| ||||||
| CD4+ | 8.9 ± 1.9 | 42.5 ± 19.5a | 6.4 ± 1.2 | 51.5 ± 10.1a | 7.9 ± 0.6 | 49.0 ± 13.2a |
| CD8+ | 5.0 ± 0.9 | 27.5 ± 12.8a | 2.6 ± 1.1 | 22.3 ± 9.8a | 2.7 ± 0.3 | 19.5 ± 9.3a |
| CD4+CD8+ | 83.5 ± 3.2 | 22.2 ± 32.7a | 87.8 ± 1.1 | 3.3 ± 3.0a | 87.1 ± 1.5 | 16.1 ± 19.0a |
| CD4−CD8− | 2.6 ± 0.4 | 8.1 ± 2.8a | 3.2 ± 1.2 | 25.2 ± 17.5a,b | 2.4 ± 1.1 | 16.0 ± 7.8a,b |
| TCRb+ | 10.7 ± 2.5 | 56.4 ± 30.1a | 9.0 ± 3.3 | 67.6 ± 16.3a | 10.3 ± 1.0 | 62.2 ± 18.1a |
| CD44+ | 0.9 ± 0.2 | 5.1 ± 2.5a | 1.0 ± 0.2 | 7.9 ± 3.1a | 1.8 ± 0.8 | 9.8 ± 2.2a,b |
| CD25+ | 3.1 ± 0.3 | 6.7 ± 2.2a | 1.8 ± 0.6 | 4.5 ± 2.5 | 1.3 ± 0.4 | 6.1 ± 2.3a |
| Blood | Sham (n = 8) | MCAO (n = 6) | Sham (n = 3) | MCAO (n = 5) | Sham (n = 4) | MCAO (n = 5) |
|
| ||||||
| CD3+ | 26.6 ± 2.7 | 16.8 ± 2.2a | 11.7 ± 5.2 | 9.9 ± 2.8b | 9.7 ± 6 | 11.8 ± 5.3 |
| CD4+ | 11.3 ± 1.9 | 7.4 ± 1.9a | 7.9 ± 4 | 5.5 ± 1.7 | 5.5 ± 3.4 | 6.0 ± 3.4 |
| CD8+ | 16.1 ± 1.4 | 10.1 ± 1.4a | 6.2 ± 3.1 | 8.1 ± 3 | 4.7 ± 2.9 | 5.9 ± 3.6b |
| CD19+ | 55.6 ± 2.9 | 41.8 ± 19.4 | 27.5 ± 15.7 | 25.3 ± 7.9 | 17.2 ± 8.5 | 21.2 ± 11.1 |
| CD11b+ | 8.9 ± 1.4 | 32.4 ± 17.0a | 5.6 ± 2.2 | 38.4 ± 16.3a | 11.9 ± 7.3 | 20.4 ± 16.7 |
| CD11c+ | 1.0 ± 0.2 | 1.4 ± 0.6 | 0.6 ± 0.3 | 2.2 ± 1.3 | 0.6 ± 0.7 | 0.3 ± 0.3b,c |
Indicates significant difference compared with corresponding sham treated mice.
Indicates significant difference compared with intact MCAO.
Indicates significant difference compared with OVX MCAO.
CD4+CD8+ double-positive cells are the major T cell population in the thymus. MCAO strongly decreased the percentage of CD4+CD8+ double-positive cells in both OVX and OVX-E2 groups (Table I). Therefore, the profound depletion of thymocyte numbers in all groups (Fig. 2) after MCAO largely reflected the loss of this population. In contrast, the percentages of residual CD4+, CD8+, CD4−CD8−, TCRb+, CD44+, and CD25+ populations were increased after MCAO, suggesting relative resistance to the effects of stroke (Table I). In blood, MCAO significantly increased the CD11b+ cell population in both intact and OVX females relative to shams, but not in E2-replaced animals where the percentage remained similar to E2-replaced shams (Table I).
Effect of E2-replacement on splenocyte proliferation after MCAO
As shown previously, sustained E2-repletion partially preserved splenocyte numbers and normalized the distribution of mononuclear cell subpopulations after MCAO relative to sham-treated intact females, suggesting OVX-E2 animals retained greater peripheral immune potential than their OVX counterparts. As a simple indicator of immune function in splenocytes, we evaluated T cell proliferation responses induced by anti-CD3 and anti-CD28 mAb using the same number of cells per culture without regard to the large differences in the total number of recovered cells from the various treatment groups. As shown in Fig. 3, proliferation responses were easily detectable in sham-treated intact females and were significantly increased in residual T cells from intact females after MCAO, likely reflective of the increased percentage of CD3+, CD4+, and CD8+ T cells and the unaltered percentage of CD4+ FoxP3+ Treg cells in the recovered mononuclear cell population. In contrast, T cell proliferation responses were strongly reduced in both sham-treated and MCAO-treated OVX mice. In OVX-E2 mice, the proliferation response was significantly increased after MCAO compared with OVX mice, but this response was comparable to sham-treated intact mice, indicating partial restoration of peripheral immune function after E2 treatment.
FIGURE 3.
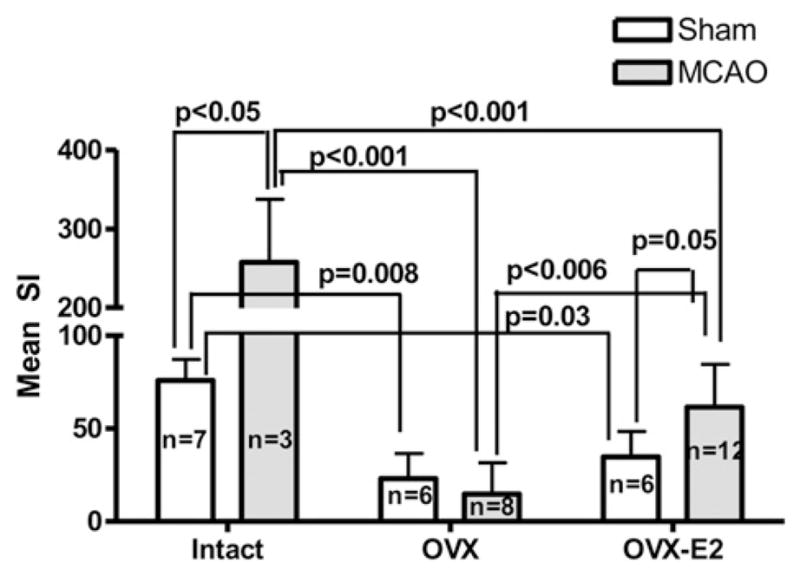
Proliferative response of splenocytes to TCR activation 96 h after MCAO or sham treatment. Splenocytes were obtained from sham-treated and MCAO-treated mice in intact, E2-deficient (OVX),or E2-replacedanimals 96 h after MCAO. Proliferation was evaluated 24 h after stimulation with anti-CD3 and anti-CD28 mAbs and is represented as mean SI. Data are presented as mean ± SD.
MCAO-induced changes in cytokines and chemokines in spleen and brain of estrogen-deficient and E2-replaced mice
We quantified changes in mRNA levels for cytokines, chemokines, and chemokine receptors (CCRs) in spleen (Fig. 4) and brain 96 h after MCAO or sham in intact, OVX, and OVX-E2 mice. In spleen, the expression of cytokines, chemokines, and CCRs was almost uniformly downregulated versus sham in estrogen-deficient animals, consistent with an immunosuppressive state. We also observed an overall decrease in protein levels at 96 h post-MCAO in OVX mice (Fig. 5), consistent with previous data demonstrating poststroke immunosuppression in male mice (20). In contrast, gene expression in spleens from E2-treated mice remained at the same level as sham, uninjured mice, with the exception of IL-17 and IL-13 that were also decreased (Fig. 4).
FIGURE 4.

Effects of MCAO on expression of (A) cytokines and (B) chemokines and chemokine receptors in spleen of intact females, and OVX female mice with or without E2 replacement measured at 96 h post-MCAO. Spleens were collected and pooled from all groups and compared with sham-operated mice for each treatment group (n = 3 for each pooled group). Relative expression of each sample was determined using the 2ΔΔCt method. The reference gene was GAPDH. Representative data are shown as fold change compared with sham-operated mice.
FIGURE 5.
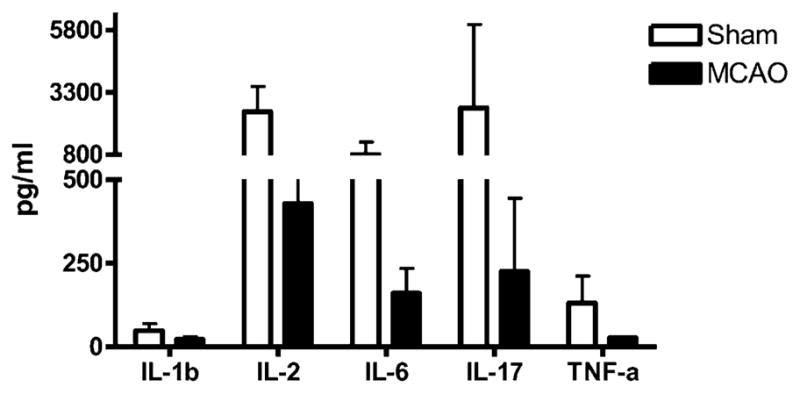
Effects of stroke on cytokines secreted from stimulated splenocytes. Spleens from OVX animals were collected 96 h after vascular occlusion and splenocytes were stimulated for 24 h with plate-bound anti-CD3/CD28 Abs. Supernatants were evaluated for levels of secreted factors (n = 2 for sham mice and n = 3 for MCAO-treated mice). Data are presented as mean ± SD.
In brain (Fig. 6), the relative levels of message for cytokines, chemokine, and CCRs were not different between OVX and E2-replaced mice, suggesting that the primary immunomodulatory effects of E2 occurred in the periphery. Two notable exceptions, however, were observed. E2 treatment profoundly decreased MIP-2 and strongly increased the chemokine receptor CCR7, compared with OVX animals after MCAO.
FIGURE 6.

Effects of MCAO on gene expression of (A) cytokines and (B) chemokines, and CCRs in brain of intact and OVX female mice with or without E2 replacement measured at 96 h post-MCAO. Ischemic (right) and nonischemic (left) brain hemispheres were collected from all groups treated with MCAO or sham-operation (n = 3 for each pooled group). Relative expression of each sample was determined using the 2ΔΔCt method. The reference gene was GAPDH. Representative data are presented as fold change as compared with sham treated mice.
Effect of G1 replacement on infarct size and splenocyte numbers
We assessed whether the novel GPR30 agonist, G1, would have similar effects as E2 in reducing infarct size and preserving spleen cell numbers after MCAO. OVX G1-replaced animals had significantly reduced infarct volume relative to placebo-replaced counterparts 96 h after MCAO in the CTX, STR, and total hemisphere (Fig 7A). Compared with E2, treatment with G1 appeared to be more effective on infarct volume in STR (44% versus 16% reduction) but less effective in CTX (23% versus 32% reduction). Like E2, G1-replaced animals retained higher spleen cell numbers relative to placebo-replaced animals, suggesting that G1 may also preserve spleen cell function after MCAO (Fig. 7B). G1 did not preserve thymocyte numbers relative to OVX (data not shown).
FIGURE 7.
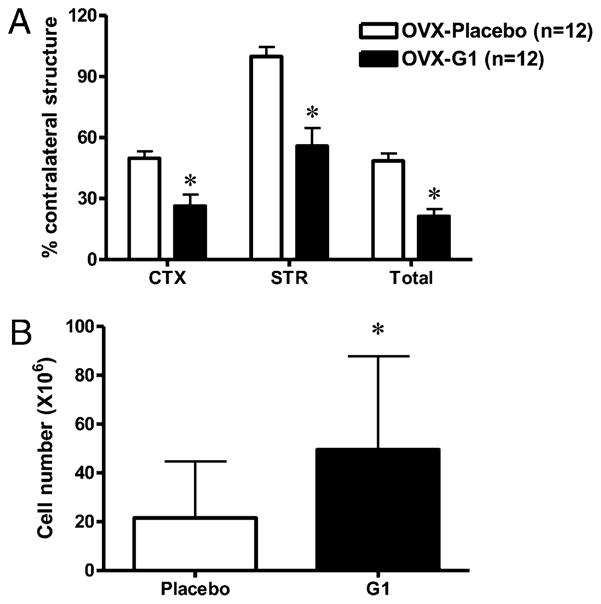
Effect of G1 on (A) infarct size and (B) spleen cell number after MCAO. Female C57BL/6 mice were OVX and replaced with 1.8 mg G1 (OVX-G1) or placebo (OVX-Placebo) pellet 1 wk prior to ischemia. Infarction volume (percent of contralateral structure) was assessed in the CTX, STR, and total hemisphere (Total) 96 h after reperfusion. Data are presented as mean ± SEM. B, Spleen and cell numbers (×106) were quantified from animals represented in Fig. 6A. Data are presented as mean ± SD.
To confirm that G1 did not alter intraischemic physiological parameters, which could influence outcome variables, we measured blood pressure, arterial oxygenation, and blood glucose at baseline and 45 min during MCAO. There were no differences between the groups for MABP, arterial blood gases, or glucose (Table II). As expected, plasma E2 levels were low in all animals: 14.87 ± 3.15 pg/ml and 15.28 ± 2.25 pg/ml for the placebo-replaced and G1-replaced animals, respectively (n = 12 for each group).
Table II.
Physiological parameters in OVX-placebo or -G1 implanted female mice before or at midpoint of vascular occlusion
| Group | pH | PaCO2 (mm Hg)a | PaO2 (mm Hg)b | Glucose (mg/dl) | MABP (mm Hg) |
|---|---|---|---|---|---|
| Placebo | |||||
| Preischemia | 7.44 ± 0.0.01 | 35 ± 2 | 198 ± 8 | 144 ± 19 | 80 ± 1 |
| MCAO | 7.39 ± 0.03 | 35 ± 3 | 190 ± 4 | 155 ± 14 | 78 ± 1 |
| G1 | |||||
| Preischemia | 7.45 ± 0.02 | 33 ± 2 | 177 ± 5 | 159 ± 10 | 80 ± 1 |
| MCAO | 7.47 ± 0.03 | 29 ± 2 | 187 ± 9 | 161 ± 22 | 79 ± 1 |
n = 4 per group.
Arterial CO2 tension.
Arterial O2 tension.
Discussion
The results presented above clearly established that stroke severity and poststroke immunosuppression were exacerbated in estrogen-deficient female mice and that E2 replacement not only reduced infarct size, but also ameliorated peripheral immunosuppression. Estrogen is the major female sex steroid in the circulation and has well-established neuroprotective effects in the brain. Our study further investigated the protective effects of E2 on the peripheral immune system after stroke, and we report three important new findings: First, MCAO in estrogen-deficient females reduced splenocyte and thymocyte cell numbers, splenocyte proliferation in response to anti-CD3 and anti-CD28 Abs and cytokine, chemokine, and CCR mRNA expression, and increased CD4+CD25+ FoxP3+ Treg cells; second, E2-replacement partially restored spleen cell numbers, normalized immune responses in spleen and brain, and reduced splenic Treg cell numbers; and third, similar to E2, replacement with the selective GPR30 agonist, G1, also reduced infarct volume and improved splenocyte numbers in estrogen-deficient animals.
Previous work has partially characterized the effects of ischemic brain injury on peripheral immune organs in male animals. However, data from females are lacking. We and others have previously demonstrated that experimental stroke leads first to stimulation and then suppression of the peripheral immune system. The events leading to stimulation of peripheral immune responses are unclear but may involve central adrenergic neural signaling (4). Recently, Ajmo et al. showed that blockade of noradrenergic receptors improved infarct severity and reduced the inflammatory response after transient focal ischemia (21). Inflammatory factors are elevated 6–22 h post-MCAO (5, 19) in spleen and are followed by a systemic decline at 96 h post-MCAO (5, 22). The subsequent immunosuppression was characterized by reduction of spleen and thymus size and increased splenocyte apoptosis (20) with corresponding loss of T and B lymphocytes (22, 23). Similarly, MCAO reduced spleen size and CD8+ T lymphocyte numbers in rats (24). These changes in systemic immune responses ultimately impact the brain. T and B cells are found in the brain after MCAO (25–27) and infarct size is improved after splenectomy (6) and in T and B cell knock-out mice (18, 27). Thus, it is thought that T and B lymphocytes are likely released from spleen, translocate to the brain, and contribute to the developing infarct.
It is increasingly well-recognized that basic immune responses differ between the sexes. The higher incidence of autoimmune diseases in women and the ability of pregnancy to reduce disease severity suggested an immunomodulatory role for sex steroids, such as E2, which has proinflammatory effects at low plasma levels (estrus levels in rodents and postmenopausal women) and anti-inflammatory effects at high physiological levels (proestrus phase in rodents, periovulatory phase of the menses, and pregnancy in women). E2 deficiency in the menopause is associated with a proinflammatory Th1 phenotype, namely, T cell expansion in bone marrow that secretes IL-1, TNF-α, and IL-6 (28). In animals, prolonged hypoestrogenicity eliminates anti-inflammatory E2 actions in postischemic brain (29). Our data affirm the role of E2 in normalizing immune responses after MCAO in the periphery and brain. E2 partially restored immune reactivity in OVX females by increasing spleen cell numbers and proliferation and cytokine responses. These effects of E2 are consistent with a role for E2 in normal spleen and thymus development in the female (9), as well as induction of anti-inflammatory cytokines in spleen after traumatic brain injury (30). In our study, E2 replacement did not restore MCAO-induced loss of thymic cell numbers, particularly CD4+CD8+ thymocytes that are known to be sensitive to deletion by E2 (9).
We measured a wide array of mRNA transcripts for cytokine, chemokines, and CCRs by quantitative real-time PCR in both spleen and brain. Although we did not directly compare estrogen-deficient females with male mice, the estrogen-deficient females showed many similar characteristics of peripheral immunosuppression previously described in males 96 h post-MCAO (20). We found that estrogen repletion maintained the levels of cytokines, chemokines, and CCRs, including TNF-α, IFN-γ, CCR3, CCR5, and their ligand, RANTES, to that of sham, suggesting prevention of immunosuppression observed in OVX females after MCAO. These effects of E2 may lead to anchoring of inflammatory cells in the spleen, blocking translocation to brain, and thereby improving infarct severity.
Proinflammatory genes are rapidly induced in brain after ischemic injury, including TNF-α (31, 32), IL-6 (33), IL-1β (32), and chemokines IP-10 (34), MIP-2, MCP-1, and RANTES, along with their respective CCRs (35). These factors promote leukocyte adhesion and infiltration into brain (35, 36). In experimental autoimmune encephalomyelitis, E2 treatment strikingly decreased recruitment of encephalitogenic T lymphocytes, TNF-α positive macrophages, and other inflammatory cells into the CNS (37, 38), and in LPS-induced brain inflammation, E2 suppressed both resident microglial activation and recruitment of peripheral T and B cells (39).
In our study, E2 replacement induced a profound decrease in MIP-2 mRNA in brain, consistent with a role for E2 in suppressing brain inflammation. Interestingly, CCR7 mRNA was markedly increased by E2-replacement relative to OVX ischemic brain. Estrogen alters expression of the CCR family of chemokine receptors (40), including CCR7 that is highly expressed on mature DCs as well as on naive and central memory T cells (41). CCR7 and its ligands, CCL19 and CCL21, are responsible for homing of T cells and DCs to lymph nodes (42), and CCR7 is also required for the in vivo function of CD4+CD25+ Treg cells (43). Although our study did not identify CNS cell types that expressed CCR7, it is possible, that estrogen increased CCR7 expression on cerebroprotective CD4+ CD25+FoxP3+ Treg cells that have been shown to exert key immunomodulatory effects in acute experimental stroke (44). E2-enhanced CCR7+ Treg cells in CNS might later be guided from the brain through the cerebral spinal fluid to deep cervical lymph nodes to influence the afferent limb of CNS immunity (45).
We previously reported that MCAO in male mice results in general loss of B and T cells and macrophage/DC in spleen, yet increases in immunosuppressive CD4+CD25+FoxP3+ Treg cells that could contribute topoststroke immunosuppression (20). In the current study, we observed that splenic CD4+ T cells were similar in OVX-E2 MCAO versus sham mice. Of importance, E2 deficiency increased, whereas E2 repletion maintained the percentage of Treg cells. In health, Tregs limit inflammation and inhibit autoimmune disease (46–48) by maintaining self-tolerance (49). However, an overabundance of Treg cells impedes immunosurveillance (50, 51) and therefore may be immunosuppressive in certain diseases, such as stroke.
The potential therapeutic benefit of G1 reported previously is a novel observation. In addition to the classical estrogen receptors, ERα and ERβ, that are known to contribute to protection in the brain after MCAO (for reviews see Refs. 52–56), an estrogen-activated GPR30 has been identified in cellular plasma membranes as well as the endoplasmic reticulum (10–13). G1, a selective agonist for GPR30, provided robust protection equivalent to E2 in other sexually dimorphic diseases, such as experimental autoimmune encephalomyelitis, and was shown to reduce inflammation, improve axonal damage and decrease the number of injured axons in spinal cord (17). However, unlike E2, G1 does not increase plasma E2 or progesterone steroid levels, or increase uterine weight, confirming a lack of interaction with nuclear estrogen receptors (16). These effects make G1 an attractive candidate for therapeutic intervention against ischemic brain injury because it lacks undesired effects on reproductive organs. However, further characterization of the efficacy of G1 and mechanisms of protecting brain and the immune system after injury is necessary.
In conclusion, replacement with either E2 or G1 improved stroke severity and partially restored cell numbers and function in the spleen. Conceptually, the dual benefit of E2 to brain and spleen argues against a prevailing hypothesis that postischemic immunosuppression is a teleological adaptation, that is, provides endogenous CNS protection from secondary injury due to influx of peripheral immunocytes, that leaves the host with a compromised defense against infection. Our data argue against this hypothesis in that E2 preserves immunocytes and restores their responsiveness to stimulation after ischemia, without causing increased brain damage. These results suggest that estrogenic compounds may have high potential for clinical application as a combined neuroprotectant and immunoprotectant after cerebral ischemia in the female.
Acknowledgments
This work was supported by National Institutes of Health Grant NS49210 and the Biomedical Laboratory Research and Development Service, Department of Veterans Affairs.
We thank Heather Hoem for assistance in submitting the manuscript.
Abbreviations used in this paper
- CTX
cortex
- DC
dendritic cell
- E2
estradiol
- GPR30
G protein-coupled receptor 30
- MABP
mean arterial blood pressure
- MCAO
middle cerebral artery occlusion
- OVX
ovariectomized
- OVX-E2
OVX E2-repleted
- PI
propidium iodide
- SI
stimulation index
- STR
striatum
- Treg
T regulatory
- TTC
2, 3, 5-triphenyltetrazolium chloride
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Herson PS, I, Koerner P, Hurn PD. Sex, sex steroids, and brain injury. Semin Reprod Med. 2009;27:229–239. doi: 10.1055/s-0029-1216276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rusa R, Alkayed NJ, Crain BJ, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD. 17beta-estradiol reduces stroke injury in estrogen-deficient female animals. Stroke. 1999;30:1665–1670. doi: 10.1161/01.str.30.8.1665. [DOI] [PubMed] [Google Scholar]
- 3.Alkayed NJ, Murphy SJ, Traystman RJ, Hurn PD, Miller VM. Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke. 2000;31:161–168. doi: 10.1161/01.str.31.1.161. [DOI] [PubMed] [Google Scholar]
- 4.Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, Prass K, Meisel A. Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007;38(2, Suppl):770–773. doi: 10.1161/01.STR.0000251441.89665.bc. [DOI] [PubMed] [Google Scholar]
- 5.Offner H, Vandenbark AA, Hurn PD. Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience. 2009;158:1098–1111. doi: 10.1016/j.neuroscience.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ajmo CT, Jr, Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res. 2008;86:2227–2234. doi: 10.1002/jnr.21661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamorro A, Urra X, Planas AM. Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke. 2007;38:1097–1103. doi: 10.1161/01.STR.0000258346.68966.9d. [DOI] [PubMed] [Google Scholar]
- 8.Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- 9.Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–574. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- 10.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346:904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 11.Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Ronnekleiv OK, Kelly MJ. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–9540. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 13.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 14.Ardelt AA, McCullough LD, Korach KS, Wang MM, Munzenmaier DH, Hurn PD. Estradiol regulates angiopoietin-1 mRNA expression through estrogen receptor-alpha in a rodent experimental stroke model. Stroke. 2005;36:337–341. doi: 10.1161/01.STR.0000153795.38388.72. [DOI] [PubMed] [Google Scholar]
- 15.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Dehghani B, Magrisso IJ, Rick EA, Bonhomme E, Cody DB, Elenich LA, Subramanian S, Murphy SJ, Kelly MJ, et al. GPR30 contributes to estrogen-induced thymic atrophy. Mol Endocrinol. 2008;22:636–648. doi: 10.1210/me.2007-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J Immunol. 2009;182:3294–3303. doi: 10.4049/jimmunol.0803205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–1805. doi: 10.1038/sj.jcbfm.9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 20.Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- 21.Ajmo CT, Jr, Collier LA, Leonardo CC, Hall AA, Green SM, Womble TA, Cuevas J, Willing AE, Pennypacker KR. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol. 2009;218:47–55. doi: 10.1016/j.expneurol.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, Hug A, Zorn M, Dalpke A, Nawroth P, Veltkamp R. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009;40:2849–2858. doi: 10.1161/STROKEAHA.109.549618. [DOI] [PubMed] [Google Scholar]
- 24.Vendrame M, Gemma C, Pennypacker KR, Bickford PC, Davis Sanberg C, Sanberg PR, Willing AE. Cord blood rescues stroke-induced changes in splenocyte phenotype and function. Exp Neurol. 2006;199:191–200. doi: 10.1016/j.expneurol.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 25.Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002;932:110–119. doi: 10.1016/s0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- 26.Subramanian S, Zhang B, Kosaka Y, Burrows GG, Grafe MR, Vandenbark AA, Hurn PD, Offner H. Recombinant T cell receptor ligand treats experimental stroke. Stroke. 2009;40:2539–2545. doi: 10.1161/STROKEAHA.108.543991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- 28.Pfeilschifter J, Köditz R, Pfohl M, Schatz H. Changes in proinflammatory cytokine activity after menopause. Endocr Rev. 2002;23:90–119. doi: 10.1210/edrv.23.1.0456. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki S, Brown CM, Dela Cruz CD, Yang E, Bridwell DA, Wise PM. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc Natl Acad Sci USA. 2007;104:6013–6018. doi: 10.1073/pnas.0610394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruce-Keller AJ, Dimayuga FO, Reed JL, Wang C, Angers R, Wilson ME, Dimayuga VM, Scheff SW. Gender and estrogen manipulation do not affect traumatic brain injury in mice. J Neurotrauma. 2007;24:203–215. doi: 10.1089/neu.2006.0163. [DOI] [PubMed] [Google Scholar]
- 31.Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concomitant cortical expression of TNF-alpha and IL-1 beta mRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol. 1994;23:103–114. doi: 10.1007/BF02815404. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Yue TL, Young PR, Barone FC, Feuerstein GZ. Expression of interleukin-6, c-fos, and zif268 mRNAs in rat ischemic cortex. J Cereb Blood Flow Metab. 1995;15:166–171. doi: 10.1038/jcbfm.1995.18. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Ellison JA, Siren AL, Lysko PG, Yue TL, Barone FC, Shatzman A, Feuerstein GZ. Prolonged expression of interferon-inducible protein-10 in ischemic cortex after permanent occlusion of the middle cerebral artery in rat. J Neurochem. 1998;71:1194–1204. doi: 10.1046/j.1471-4159.1998.71031194.x. [DOI] [PubMed] [Google Scholar]
- 35.Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 36.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Ito A, Buenafe AC, Matejuk A, Zamora A, Silverman M, Dwyer J, Vandenbark AA, Offner H. Estrogen inhibits systemic T cell expression of TNF-alpha and recruitment of TNF-alpha(+) T cells and macrophages into the CNS of mice developing experimental encephalomyelitis. Clin Immunol. 2002;102:275–282. doi: 10.1006/clim.2001.5175. [DOI] [PubMed] [Google Scholar]
- 38.Matejuk A, Adlard K, Zamora A, Silverman M, Vandenbark AA, Offner H. 17 beta-estradiol inhibits cytokine, chemokine, and chemokine receptor mRNA expression in the central nervous system of female mice with experimental autoimmune encephalomyelitis. J Neurosci Res. 2001;65:529–542. doi: 10.1002/jnr.1183. [DOI] [PubMed] [Google Scholar]
- 39.Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front Neuroendocrinol. 2008;29:507–519. doi: 10.1016/j.yfrne.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mo R, Chen J, Grolleau-Julius A, Murphy HS, Richardson BC, Yung RL. Estrogen regulates CCR gene expression and function in T lymphocytes. J Immunol. 2005;174:6023–6029. doi: 10.4049/jimmunol.174.10.6023. [DOI] [PubMed] [Google Scholar]
- 41.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 42.Ebert LM, Schaerli P, Moser B. Chemokine-mediated control of T cell traffic in lymphoid and peripheral tissues. Mol Immunol. 2005;42:799–809. doi: 10.1016/j.molimm.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 43.Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med. 2007;204:735–745. doi: 10.1084/jem.20061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 45.Kivisäkk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, Wujek J, Ravid R, Staugaitis SM, Lassmann H, Ransohoff RM. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- 46.Maloy KJ, Powrie F. Regulatory T cells in the control of immune pathology. Nat Immunol. 2001;2:816–822. doi: 10.1038/ni0901-816. [DOI] [PubMed] [Google Scholar]
- 47.Mason D, Powrie F. Control of immune pathology by regulatory T cells. Curr Opin Immunol. 1998;10:649–655. doi: 10.1016/s0952-7915(98)80084-8. [DOI] [PubMed] [Google Scholar]
- 48.Shevach EM. Regulatory T cells in autoimmmunity*. Annu Rev Immunol. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 49.Sakaguchi S, Sakaguchi N. Regulatory T cells in immunologic self-tolerance and autoimmune disease. Int Rev Immunol. 2005;24:211–226. doi: 10.1080/08830180590934976. [DOI] [PubMed] [Google Scholar]
- 50.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 51.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 52.Bryant DN, Sheldahl LC, Marriott LK, Shapiro RA, Dorsa DM. Multiple pathways transmit neuroprotective effects of gonadal steroids. Endocrine. 2006;29:199–207. doi: 10.1385/ENDO:29:2:199. [DOI] [PubMed] [Google Scholar]
- 53.Morissette M, Le Saux M, D’Astous M, Jourdain S, Al Sweidi S, Morin N, Estrada-Camarena E, Mendez P, Garcia-Segura LM, Di Paolo T. Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol. 2008;108:327–338. doi: 10.1016/j.jsbmb.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 54.Pozzi S, Benedusi V, Maggi A, Vegeto E. Estrogen action in neuroprotection and brain inflammation. Ann N Y Acad Sci. 2006;1089:302–323. doi: 10.1196/annals.1386.035. [DOI] [PubMed] [Google Scholar]
- 55.Vagnerova K, I, Koerner P, Hurn PD. Gender and the injured brain. Anesth Analg. 2008;107:201–214. doi: 10.1213/ane.0b013e31817326a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–153. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]