

Abstract
Using recently available cDNA and genomic data (Berkeley Drosophila Genome Project; http://www.fruitfly.org), we computed a large sample of 10,057 Drosophila splice sites. An information-theoretic analysis of the nucleotide sequences adjacent to these splice sites showed a strong correlation between the sizes of introns and exons and the levels of information, which is a measure of sequence conservation. The strong correlation permitted us to determine extensive consensus sequences at the donor and acceptor sites of longer introns. These sequences were further refined and extended by examining the information in regions around splice sites that only partially matched the consensus. The correlation between length and information provided the basis for determining alternative consensus arrangements associated with shorter introns, as well as general base-composition preferences that likely promote spliceosome function. We also observed a correlation between information near splice sites and the lengths of nonadjacent introns, indicating that there are long-range effects spanning multiple introns. The ordered partitioning approach used in this analysis may become increasingly useful as large genomic data sets become available.
Molecular recognition events involving DNA or RNA typically rely on specific nucleotide sequences that are selected during evolution to favor binding by the interacting molecules. Strategies to identify these “consensus” sequences normally involve the assembly of a set of sequence elements that can mediate the binding event, and then the identification and assessment by genetic, biochemical, and structural tests of the shared sequence characteristics. This approach identifies motif elements shared by a majority of the sites, but can miss degenerate parts of motifs, or parts of the motif involved in apparently dispensable molecular interactions. Genomic scale studies have opened up new possibilities for characterizing consensus sequences. Their large scale permits selection of substantial sample subsets with properties that optimize the analysis of their function.
Consensus sequences must have sufficient information to be distinguished uniquely within sequence space, and yet overspecification of sites would be wasteful and would require additional evolutionary selection away from random sequence. But these constraints present a conundrum for molecular events that sometimes involve recognition of sequence motifs that only occur at very large intervals, and have sufficiently long recognition motifs for their occurrence to be rare. When the equivalent molecular events occur on a small scale over shorter intervals, are the recognition motifs equally long, or are there compensating mechanisms to ensure that the motifs are not overspecified?
We have addressed this question in the context of RNA splicing. The lengths of introns cover an enormous range, from less than 100 to tens of thousands of nucleotides (nt), raising the possibility that different mechanisms might account for the specification of small introns compared with large introns (Lim and Burge 2001). Using the large number of sequenced cDNAs available from the Berkeley Drosophila genome project (BDGP; http://www.fruitfly.org; Spradling et al. 1995; Stapleton et al. 2002), we were able to compute a large data set, consisting of nucleotide sequences in regions around splice sites, that could be partitioned into substantial subsets, each corresponding to a relatively small range of intron lengths.
This technique allowed us to assess the information requirements for splicing over different distances. The analysis we describe below implied that there are demands on spliceosome function that increase progressively with greater distances between splice sites. These constraints permitted us to devise strategies to define extended consensus sequences near splice sites, as well as nucleotide composition preferences that likely enhance spliceosome function.
METHODS
Computation of Drosophila Splice Sites
We used conservative criteria to compute a data set of Drosophila splice sites, thereby ensuring that in most cases, the splice sites were identified correctly. To identify splice sites, we used a 5′-to-3′ scanning algorithm (Appendix 1) that matched the cDNA with corresponding genomic sequences (BDGP; http://www.fruitfly.org). Because the genomic DNA and cDNA libraries sequenced by BDGP came from genetically similar fly strains, the problem of polymorphisms within the source populations was minimized. Nevertheless, polymorphisms, in particular insertions or deletions of a few nucleotides, led to incorrect prediction of short introns or exons, or failure of the algorithm to find 3′-exons. Hence, to maintain a data set of high quality, we removed all cDNAs predicted to contain any intron or exon with <20 nt, as well as any cDNA whose scan terminated prematurely before the poly-adenylation sequence was reached.
With these constraints, the algorithm predicted the genomic DNA sequence coordinates of 10,057 introns based on 3090 cDNAs; 514 additional cDNAs were predicted to contain no intron. The predictions for 2631 of the 10,057 introns were made independently of splice-site consensus information, relying solely on the comparison of genomic and cDNA sequences. Greater than 99% of these predicted introns were bracketed at either end by GU and AG, or the secondary consensus end sequences, AU and AC (three introns; Hall and Padgett 1994; Nilsen 1998; Burge et al. 1999; Yu et al. 1999). Predicting the coordinates of the remaining 7426 introns was initially ambiguous because of short sequence duplications near the putative donor and acceptor sites, and had to be resolved by testing for the canonical GU...AG (or AU...AC) at the intron boundaries and requiring matches in at least three of four positions. Of the 10,057 introns, 99.2% had matches at all four canonical positions,0.7% had matches at three positions, and only 0.2% (19) introns had a poorer match. In eight cases, the intron matched the secondary consensus AU...AC at three or four positions.
Although splicing proceeds through formation of a lariat intermediate, defined by a branch point near the acceptor site (Nilsen 1998; Burge et al. 1999), we did not include branch points in the present analysis because, unlike the donors and acceptor sites, the branch points could not be defined independently of their consensus sequences.
The 10,057 introns in our data set had a mean length of 648 nucleotides (nt). However, the distribution of intron lengths had a very large standard deviation (2759 nt), reflecting the very large range of lengths (23-78,340 nt; Supplemental Fig. S1A available online at www.genome.org). The length distribution was skewed, with 59.5% of introns having lengths in the range 50-80 nt, and 10.6% having lengths >1000 nt. (This distribution was similar to the one reported previously by Lim and Burge [2001].) The distribution of exon lengths was similarly skewed, with a mean length of 502 nt, but the standard deviation was much smaller (514 nt), reflecting the smaller range of lengths (20-5061 nt; Supplemental Fig. S1B). It is interesting to note that 34.0% of the introns fall in a very small length range (56-64 nt) with >300 cases for each of the nine intron lengths in this interval.
Storing Splice Sites in a Relational Database
We designed a relational database using Microsoft SQL Server to store the data about exons, introns, and splice-site regions that were computed by the preceding algorithm. Each exon and intron was stored based on its transcript identity, rank in the transcript (1st exon or intron, 2nd, etc.), and nucleotide coordinates in the preprocessed transcript before splicing. Individual nucleotides at positions -32 to +32 relative to the donor and acceptor sites (Supplemental Fig. S2) were also stored. A collection of stored procedures was developed to compute the information that provided the foundation for the analyses described below. The database and stored procedures can be accessed at http://igs.wesleyan.edu.
Representing the sequence data in a relational database facilitated significantly our analysis of the sequence conservation at splice sites. We wrote stored procedures to permit efficient testing of partition contexts that enhanced consensus site information. Once contexts favoring splice function were identified (such as intron length; see below), these were introduced as parameters in procedures to test other contexts. This approach for the testing of sequence space may become increasingly useful as large genome data sets become available.
RESULTS
Splice-Site Information Increases With Genomic Distances
Previous studies have assessed whether splice sites have sufficient information for their identification given their average frequencies of occurrence in pre-mRNAs (Stephens and Schneider 1992; Burge et al. 1999). These studies measured the information at each position in aligned splice-site regions by quantifying the deviation from a random sequence using the following definition:
 |
where the quantity in square brackets is the uncertainty (or equivalently entropy; see Shannon and Weaver 1949) of the nucleotides at the given position based on the frequencies of occurrence fA,..., fU of the nucleotides A... U, at the given position, and where the correction factor γ depends on the number of splice sites that are being aligned (Stephens and Schneider 1992; Appendix 2). For sets of up to 125 splice sites, we used the exact correction discussed in Stephens and Schneider (1992); for sets of >125 splice sites, we used the approximation due to Basharin (1959). Because RNA sequences have four possible bases, the information at a particular nucleotide position of the aligned splice sites lies between 0 bits (random sequence) and 2 bits (one particular base at that nucleotide position). In the definition above, information is calculated relative to a completely random background, where each nucleotide frequency is 0.25. Information can also be calculated relative to the background nucleotide frequencies of the organism's genome (discussed in Appendix 3). However, except where noted, we have used information relative to a random background in this study.
Our measurements of information at positions -32 to 32 of the 10,057 donor and acceptor sites confirmed previous observations that most of the information (sequence conservation) was located in the introns immediately adjacent to the splice sites (Supplemental Fig. S3), as would be expected if exonic regions that contain protein open reading frames are not constrained by splicing (Stephens and Schneider 1992; Burge et al. 1999). Significant conservation of exon sequences (>0.5 bits of information) was observed only at position -1 of the donor site (D - 1). (The value of 0.5 bits corresponds, for example, to 64.5% of one nucleotide, and 11.9% for each of the other three.) The sites with at least 0.5 bits of information were donor positions D - 1 to D + 6 and acceptor positions A - 5, and A - 3 to A - 1 (Supplemental Fig. S3). These sites had the consensus sequences GGU(A/G)AGU and UNCAG, respectively (Fig. 1A, no partitioning). The nonzero information downstream from D + 5 and upstream of A - 3 was due to elevated levels of nucleotides A and U, as well as a pyrimidine (C/U)-rich region upstream of A - 3. The pyrimidine tract has been implicated in splice function (Patterson and Guthrie 1991; Guo et al. 1993; Guo and Mount 1995; Chiara et al. 1997; Yu et al. 1999), and the contributions to splicing of this tract and the elevated A and U content are discussed below.
Figure 1.

Progressive partitioning reveals splice site consensus sequences. (A) The figure summarizes the consensus sequences that emerge by using the following techniques: (i) The information profile of regions around the donor and acceptor sites of all 10,057 introns (Suppl. Fig. S3) showed >0.5 bits of information at donor positions D - 1 to D + 6 and acceptor positions A - 5 and A - 3 to A - 1. (Nucleotide positions are defined relative to the splice site junctions.) (ii) Partitioning introns by length (Figs. 2 and 3) showed that longer introns had at least 0.5 bits of information at additional positions A - 11 to A - 9 and A - 6. Underlined nucleotides indicate positions that have an increase in information of at least 0.5 bits over the lengths 64 to 32,767 nt (Fig. 3). The position D + 5 (square border) has least information for intermediate lengths, and progressively higher information for shorter and longer lengths (Fig. 3). (iii) Partitioning introns by forced mismatches (Fig. 5; Suppl. Fig. S7) showed >0.5 bits of information at the additional positions D - 2 and A - 14, and revealed a preference for A at D + 3. Segments of U1, U5, and U6 snRNAs are aligned with donor and acceptor site sequences, where they are thought to interact through base-pairing or non-Watson-Crick interactions. Pre-mRNA regions where snRNP and associated proteins are thought to bind are indicated with brackets; these include U2AF35(38), U2AF65(58), and U5 snRNP Prp8 (vertebrate homolog p220). Also illustrated are regions where forced mismatches (Fig. 5; Suppl. Figs. S5, S6, S7) led to A enrichment, and to a lesser extent, U enrichment. This bias in composition may promote the splicing reaction by favoring reduced RNA secondary structure and enhanced binding of protein or nucleic acid components of the spliceosome. Forced mismatches gave particularly strong A enrichment at D - 2 and D - 3, where U5 snRNA binds. (B) The graph shows the frequency distributions for numbers of matches to the donor consensus sequence AGGUAAGU (D - 2 through D + 6) and the acceptor consensus sequence UNNUUUNNUUNCAG (A - 14 through A - 1) for short introns (64-80 nt) and long introns (8192-32,767 nt). Assuming that the distributions are approximately normal, an approximate t-test using the means and standard deviations of the two distributions shows that in both the donor and acceptor cases, the distribution means are higher for the longer introns (p > 0.99). The distribution means and standard deviations are listed in parentheses.
One can calculate the minimum information required to define a recognition sequence of a given abundance. For example, a four-cutter restriction enzyme will cut random DNA on average every 256 (28) bases. The fact that aligned recognition sequences for this enzyme have 8 bits of information illustrates the relationship: log2(distance) = information. It has been pointed out previously that this relationship appears to apply, at least in part, to other biological contexts including splicing (Stephens and Schneider 1992; Burge et al. 1999). But because the lengths of introns (and, to a lesser extent, exons) span large size ranges, we wished to address specifically whether long and short introns (and exons) contain the same splice site information, or whether information levels depend on intron (and exon) length. The latter has been suggested previously in studies of relatively small samples of Caenorhabditis elegans and Drosophila introns (Fields 1990; Mount et al. 1992).
To assess further this question, we compared the information profiles for the set of introns with lengths between 64 and 80 nt (2156 introns) with the set of introns of length >8191 nt (152 introns; Fig. 2A). The set of longer introns had more information at several nucleotide positions, in particular, at donor position D + 4 and acceptor positions A - 6 and A - 5 (one-tailed t-test, p > 0.99; Appendix 2). Nucleotide profiles (Fig. 2B) indicated that these positions correspond to elevated levels of the nucleotides A (by 25% at D + 4) and U (65% at A - 6,53% at A - 5; see Appendix 2 for statistical analysis of nucleotide distributions). The set of longer introns also had a broader and more pronounced pyrimidine tract adjacent to the acceptor site that was particularly rich in the nucleotide U. In a more extreme case, examination of the 20 positions A - 24 to A - 5 for the 16 introns of length >32,767 nt revealed striking islands of Us with frequent runs of 4 or more and a nucleotide composition with >50% U. In addition, the set of longer introns had enhanced A-nucleotide content in the exon and intron regions surrounding the donor site (see the Discussion below). Previous analysis of 209 Drosophila introns (Mount et al. 1992) revealed similar pyrimidine enrichment at A - 21 to A - 11 in large (81-5392 nt) compared with small (51-80 nt) introns. This analysis did not reveal increased information near donor sites of the larger introns. However, analysis of 139 nematode introns (Fields 1990) showed enhanced information at D + 3 through D + 6 in introns >75 nt compared with those <75 nt, consistent with our observation in Drosophila.
Figure 2.

Donor and acceptor sites flanking long introns have more information. (A) The graphs show the information profiles for regions around the donor and acceptor sites of two sets of introns—those with lengths between 64 and 80 nt and those with lengths >8191 nt; N denotes the number of introns in each set. The set of longer introns had elevated levels of information, in particular at donor position D + 4 and acceptor positions A - 6 and A - 5 (indicated by arrows). The average standard deviation at each nucleotide position is 0.01 bits (64-80 nt) and 0.05 bits (>8191 nt; see Appendix 2 for information standard deviation calculations). The cumulative information at positions -32 to +32 for introns 64-80 is 9.57 ± 0.08 (donor) and 10.00 ± 0.08 (acceptor), and for introns >8191 is 11.80 ± 0.34(donor) and 14.27 ± 0.40 (acceptor; also see Table 3; Appendix 3). The cumulative information for donor and acceptor sites of the longer introns is significantly higher than that of the shorter introns (p > 0.99 by one-tailed t-test). (B) The graphs show the nucleotide compositions for the sets of introns used in A. These illustrate that the elevated information resulted from stronger preferences for A at D + 4 and U at A - 6 and A - 5 (indicated by arrows). The pyrimidine (C or U) tract upstream of the acceptor site was broader and more pronounced in the set of longer introns. The maximum standard deviation in the frequency at each nucleotide position is 0.011 (64-80 nt) and 0.041 (>8191 nt; see Appendix 2 for standard deviation calculations for nucleotide distributions).
We extended our analysis above by examining the information profiles for intron lengths in windows covering several contiguous size ranges (Fig. 3A). In all size ranges, longer introns had progressively more information than shorter introns. The most pronounced increases were found again at positions D + 4, A - 6, and A - 5 (see Supplemental Fig. S4 for corresponding nucleotide profiles). However, significant upward trends (>0.5 bits of increase in information) were observed at A - 11, A - 9, and A - 3, and to a lesser extent at D - 3, D - 1, D + 3, D + 6, A - 10, and some more upstream acceptor positions (Fig. 3A). Partitioning the set of introns by length (Figs. 2 and 3) revealed an extended consensus sequence with >0.5 bits of information at each nucleotide position (except those marked “N”) at the acceptor positions A - 11 to A - 1: UUUNNUUNCAG (Fig. 1A, partitioning by length).
Figure 3.


Donor and acceptor sites have increased information in the vicinity of long introns. (A) The graphs show the information profiles for regions around the donor and acceptor sites of nine sets of introns corresponding to progressively larger length ranges. They illustrate that information increases with intron length at characteristic nucleotide positions (see text). Because multiple, consecutive length ranges were analyzed, both pronounced and subtle trends are evident in the graphs. The average standard deviation in information at a nucleotide position is <0.06 bits, except for the range 16,384-32,767 (where it is ∼0.14 bits). (B) The graphs show the information profiles for regions around the donor and acceptor sites of nine sets of introns that immediately follow or precede the introns in the sets used in A. They illustrate that information increases with intron length, but the effects are more pronounced at the donor and acceptor sites closest to the intron varying in length (illustrated with a double arrow). The information is computed at the donor sites (D) and acceptor sites (A) that are shaded yellow. The number of introns in each set is listed in parentheses after each length range. The average standard deviation at a nucleotide position is <0.14 except in the lower panel, where it is ∼0.2 bits for donor and acceptor sites in the range 16,384-32,767. Cumulative information (positions -32 to +32) of donor and acceptor sites of introns preceding or following introns (64-80 nt) is significantly lower than those of introns (>8191; in all cases, p > 0.99 by one-tailed t-test). (C) Changes in nucleotide content at each position were measured for three pairwise comparisons: (i) introns >8191 compared with 64-80; (ii) 4096-8191 compared with 81-127; (iii) 2048-4095 compared with 60-63. The mean of the changes in (i), (ii), and (iii) is illustrated if all three comparisons showed a positive change, or all showed a negative change, and all three changes were significant by a two-tailed t-test (p > 0.99).
Many of the positions showing increased information correspond to sites predicted to interact with the splicing machinery (Fig. 1A). For example, the U1 snRNA interacts with D - 2 through D + 6 (Parker and Guthrie 1985; Fouser and Friesen 1986; Vijayraghavan et al. 1986; Aebi et al. 1987; Siliciano and Guthrie 1988; Rosbash and Seraphin 1991; Yu et al. 1999), and U6 snRNA interacts with D - 1 through D - 6 (Lesser and Guthrie 1993; Sontheimer and Steitz 1993; Luukkonen and Seraphin 1998). Also, U2AF35 (U2AF38 in Drosophila; Rudner et al. 1996) is thought to interact with A - 3 through A - 1, and U2AF65 (Drosophila U2AF58) interacts with the adjacent pyrimidine tract (Wu et al. 1999; Zorio and Blumenthal 1999; Reed 2000). Yeast U5 snRNP Prp8 also interacts with the pyrimidine tract and A - 3 through A - 1 (Collins and Guthrie 1999; Siatecka et al. 1999). The pronounced increases in information at A - 6 and A - 5 within the pyrimidine tract indicate that U2AF65 (U2AF58) or Prp8 might have important interactions at these positions in the pre-mRNA. In yeast and mammals, additional U5 snRNPs have also been shown to have functional interactions with the pyrimidine tract, although Drosophila homologs have not yet been characterized (Chiara et al. 1997). Our finding that longer introns have progressively more information at their donor and acceptor sites implies that spliceosome function is increasingly constrained, presumably because the molecular interactions associated with more distant splice sites need to be stronger. This point will be discussed further below.
So far, we have considered the relationship between intron length and information at adjacent donor and acceptor regions. We also considered whether the information at donor and acceptor sites is sensitive to the lengths of more distant introns. We found that increases in intron length led to increased information content at more distant splice sites. For example, there was increased information content at both the donor and acceptor sites of the intron immediately downstream (or upstream) from the intron whose length was being varied (Fig. 3B). In general, varying the lengths of introns had more pronounced influences on close-by splice sites compared with more distant sites.
We observed changes in nucleotide content (Figs. 3C and 4; Table 1) corresponding to the changes in information (Fig. 3B) observed when intron length was varied. We saw both position-specific changes at consensus positions as well as general effects. For example, the splices sites of the intron following the varied intron revealed consistent enhancement of consensus nucleotides at D - 1, D + 3 through D + 6, and A - 3 (Fig. 3C), as well as general increases in C content preceding the donor, and surrounding the acceptor (Fig. 4; Table 1). In contrast, the splice sites of the intron preceding the varied intron showed A enrichment preceding the donor and following the acceptor (Fig. 4; Table 1), and did not have pronounced effects at consensus positions (Fig. 3C) except the acceptor pyrimidine tract.
Figure 4.

General nucleotide content changes near splice sites in the neighborhood of a varied intron. Differences in the general nucleotide content in the neighborhood of introns >2047 compared with introns 60-127 (Table 1). Increasing intron length correlates with A enrichment of exon sequences flanking the varied intron, and the preceding intron, whereas C enrichment occurs for the following intron. Increasing intron length also correlates with U and C enrichment upstream of the acceptor of the varied intron (pyrimidine tract), whereas U enrichment occurs upstream of the preceding acceptor and C enrichment occurs for the following acceptor. Only significant nucleotide content changes in the various regions are illustrated based on a two-tailed t-test with p > 0.99.
Table 1.
Nucleotide Content Differences (%) for Introns >2047 nt Versus Introns 60–127 nt Long
| D − 32 to D − 1 | D+1 to D+32 | A − 32 to A − 1 | A+1 to A+32 | ||
|---|---|---|---|---|---|
| Intron before | A | 1.99 | 1.62 | −1.75 | 2.48 |
| C | −0.93 | −0.36 | −0.57 | −0.24 | |
| G | −1.59 | −0.72 | −0.59 | −2.11 | |
| U | 0.54 | −0.54 | 2.91 | −0.13 | |
| Varied intron | A | 2.32 | 0.42 | −4.31 | 1.00 |
| C | −0.57 | 0.39 | 1.26 | 0.40 | |
| G | −0.92 | 0.51 | −0.54 | −0.62 | |
| U | −0.83 | −1.33 | 3.59 | −0.78 | |
| Intron after | A | −0.33 | 1.11 | −1.61 | −0.71 |
| C | 1.31 | −0.68 | 1.75 | 1.14 | |
| G | 0.02 | 0.67 | −0.24 | 0.32 | |
| U | −1.00 | −1.11 | 0.10 | −0.76 |
Bold difference values have p > 0.99 significance by a two-tailed t-test using the observed standard deviations in nucleotide content over the 32-nt ranges. These differences were also significant by the non-parametric Mann-Whitney test, which also showed the increase in U for the varied intron (D − 32 to D − 1) to be significant (p > 0.99).
The preceding observations imply that the information around a given splice site is determined by a balance of influences from neighborhood introns. The molecular mechanisms that underlie these influences remain to be determined. One can imagine mechanisms in which more extensive and conserved splice site sequences in neighborhoods with longer introns improve binding efficiencies of spliceosome components, thereby helping to increase the local concentrations of these components. These sequences could compensate for the sites being much more distantly located, which may tend to reduce local concentrations of the spliceosome factors. Elevated information at splice sites would work alongside splicing enhancers (Hertel et al. 1997) to increase binding of spliceosome components to splice sites.
Forced Mismatches Reveal Extended Consensus Sequences
Using the criteria that each position with at least 0.5 bits of information defines a consensus nucleotide, we identified earlier the consensus sequences GGU(A/G)AGU at donor sites D - 1 through D + 6 and UNCAG at acceptor sites A - 5 through A - 1, based on the information profile of the entire data set (Fig. 1A; Supplemental Fig. S3). Partitioning the data set according to intron length (Figs. 2 and 3) extended the acceptor consensus sequence to UUUNNUUNCAG (A - 11 through A - 1; Fig. 1A).
Given the relationship between length and information, we wished to determine whether suboptimal matches to these consensus sequences were compensated for by increased information in neighboring positions. This idea is consistent with previously observed cases of splice acceptor sites lacking the consensus AG at positions A - 2 and A - 1, but containing a compensating, long pyrimidine tract (Wu et al. 1999; Reed 2000). We restricted our attention to introns with lengths in the ranges 64-255, 512-1023, and 1024-4095 nt, respectively, and examined the information profiles of sets of introns with various “suboptimal” matches to the consensus sequences. In effect, by fixing the range of lengths, we controlled the cumulative information for each set; this allowed us to assess the effect of the “degree of match” on the information profile.
Using the seven consensus donor positions D - 1 through D + 6, we compared the information profiles for the sets of introns with matches at 4-of-7,5-of-7,6-of-7, and 7-of-7 positions. The information profiles for the 5-of-7 and 7-of-7 sets are illustrated in Figure 5A, and show that the poorer match represented by the 5-of-7 set has more information outside the positions D - 1 through D + 6, when compared with the 7-of-7 set. Hence, the poorer match within the consensus range D - 1 through D + 6 led to increased information in the neighborhood, both immediately adjacent to the consensus positions, and in the vicinity, perhaps compensating for weaker spliceosome interactions at the consensus sequences.
Figure 5.

Partial-canonical splice sites reveal alternative information content. (A) The graphs show the information profiles for two sets of introns with lengths between 1024 and 4095 nt—those that match the donor consensus sequence GGU(A/G)AGU at D - 1 through D + 6 in all seven positions (7-of-7; 241 introns) compared with those that match at five positions (5-of-7; 123 introns). The 5-of-7 set had extra information outside of D - 1 through D + 6. The average standard deviation at a nucleotide position (-32 to 32) is 0.015 (7-of-7); 0.044 (5-of-7). (B) The graphs of nucleotide counts for the 5-of-7 set in A reveal a preference for A at D + 3 that would base-pair most effectively with U in U1 snRNA (Fig. 1A). The 5-of-7 set also had enriched A content at D - 3 through D - 1, which would enhance base-pairing with U5 snRNA (Fig. 1A). The standard deviation at each nucleotide position is <0.033 (7-of-7); <0.12 (5-of-7).
Examination of nucleotide content for the 5-of-7 set (Fig. 5B) showed that positions D - 3 and D - 2 were particularly enriched in the nucleotide A, consistent with the expectation that these nucleotides interact with U bases in the U5 snRNA during the latter part of the spliceosome reaction (Fig. 1A; Newman and Norman 1992; Steitz 1992; Sontheimer and Steitz 1993). Indeed, the A and U content of exons and introns in the general vicinity of the donor site was enriched in the 5-of-7 set for each restricted range of intron lengths (Supplemental Fig. S5).
The A and U enrichment may facilitate the binding of pre-mRNA to protein components of the U1-snRNP complex (perhaps by reducing secondary structure; Burkard et al. 1999). In yeast, U1-snRNP proteins have been shown to cross-link to exon and intron sequences in an extended region upstream and downstream from the donor site (Puig et al. 1999; Zhang and Rosbash 1999).
The 5-of-7 set also had a marked preference for A at D + 3, whereas the 7-of-7 set had similar preferences for A or G (Fig. 5B). An A at this position would favor interaction with U in the corresponding position in the Drosophila U1 snRNA (see Fig. 1A), and hence may facilitate spliceosome function at the donor site. Similarly, the preferred A at D - 2 (Fig. 5B) would likely base-pair with U1 (Fig. 1A). We also noticed a tendency for pronounced A enrichment at the nucleotide positions immediately flanking the predicted region of U1 or U6 binding. This was observed, for example, when we examined the nucleotide compositions for the set of introns that match AGGU(A/G)A at positions D - 2 through D + 4 but fail to match either of the consensus nucleotides at D + 5 and D + 6. This set shows pronounced A enrichment at D - 3, D + 5, and D + 6 (Supplemental Fig. S6). These observations indicate that A enrichment on either side of the region of U1 binding enhances spliceosome function.
The effects of suboptimal consensus sequences depended on the ranges of intron lengths that were considered. Suboptimal consensus sequences for smaller introns (e.g.,64-255 nt; Supplemental Fig. S5) had enrichment in both A and U content, but the levels of this combined enrichment were lower than observed with longer introns (1024-4095 nt; Supplemental Fig. S5), which were particularly enriched in A content. Analogous effects were observed when comparing different levels of mismatch with consensus sequences. For example, using introns of length 64-255 nt, comparison of 5-of-7 with 7-of-7 sets for donor positions D + 1 through D + 6 showed modest A and U enrichment, whereas the corresponding 4-of-7 set had greater enrichment, particularly in A content (Supplemental Fig. S5).
A similar analysis of suboptimal consensus sequences at acceptor sites revealed a compensating enrichment of the A and U content of intron sequences in the vicinity, again perhaps facilitating the splicing reaction (Supplemental Fig. S7A,B). The regions of elevated A content included the pyrimidine tract whose predicted interaction with U2AF65(58) or Prp8 may be enhanced by reduced secondary structure. Forced mismatches to the consensus sequence in the range A - 11 through A - 1 revealed enhanced pyrimidine content upstream of A - 11, in particular at A - 14 (Supplemental Fig. S7C).
The observation that forced mismatches are associated with elevated A content raised the possibility that these splice sites might be in genomic regions of general elevated A content that can tolerate suboptimal splice sites. However, examination of the A content of introns (512-8191 nt) and exons (512-4095 nt) flanking suboptimal donor sites (Table 2) showed that the elevation in A content was only significant in the immediate vicinity of the donor splice sites (positions -32 to +32; p > 0.99 by one-tailed approximate t-test). The A content of exons flanking suboptimal donor sites was not significantly different from that of optimal donor sites (p > 0.99), indicating that there was no A-content bias in genomic regions of suboptimal donor sites.
Table 2.
Percent A Content Near Donor Splice Sites

| ||||
|---|---|---|---|---|
| Match to donor | % A ± S.D. (n) | |||
| 7 of 7 | 25.5 ± 4.0 (517) | 26.0 ± 8.3 (517) | 27.5 ± 10.0 (478) | 29.5 ± 3.3 (478) |
| 6 of 7 | 25.8 ± 3.7 (1068) | 28.1 ± 8.4 (1068)c | 30.3 ± 10.3 (686)c | 29.9 ± 3.5 (686)c |
| 5 of 7 | 26.0 ± 3.6 (432) | 29.3 ± 9.2 (432)d | 32.1 ± 11.0 (266)d | 30.5 ± 3.6 (266) |
Exons 512−4095, introns unconstrained
Introns 512−8191, exons unconstrained
6-of-7 > 7-of-7, p > 0.99 by one-tailed t-test using observed standard deviations (S.D.) in percent A content
5-of-7 > 6-of-7, p > 0.99 by one-tailed t-test using observed standard deviations (S.D.) in percent A content
DISCUSSION
The selective partitioning of our splice-site data set based on intron lengths has revealed similar consensus sequence and general nucleotide content trends to those we observed by partitioning based on suboptimal matches to consensus sequences. Because these trends were observed in contexts that would strain the function of the spliceosome system, they likely serve to promote splicing efficiency. Indeed, many of the trends are consistent with sequence requirements indicated by previous molecular and genetic studies (Fig. 1A). Our combined results provide a view of “optimized” consensus sequences for splicing. Ordered partitioning by intron length (Fig. 3) defined consensus positions with >0.5 bits of information in longer introns at the donor site (positions -1 to 6): GGU(A/G)AGU, and at the acceptor site (positions -11 to -1): UUUNNUUNCAG (Fig. 1A). Analysis of suboptimal matches (Fig. 5; Supplemental Fig. S7C) refined and extended the donor consensus at positions -2 to 6: AGGUAAGU, and the acceptor consensus at positions -14 to -1: UNNUUUN NUUNCAG (Fig. 1A). The average numbers of matches to these consensus sequences were higher for donor and acceptor sites adjacent to longer introns (Fig. 1B). The two partitioning analyses also revealed similar general nucleotide content trends. In addition to expansion of the pyrimidine tract upstream of acceptor sites (Fig. 2), enrichment in A content, and to a lesser degree, U content in the vicinity of donor and acceptor sites, were implicated in promoting the splicing reaction (Fig. 5; Supplemental Figs. S5, S6, S7).
Our observations of local (Fig. 5B; Supplemental Fig. S5) and more general (Fig. 4; Supplemental Figs. S5, S6, S7) A enrichment indicate that spliceosome function may be facilitated when participating pre-mRNA regions are less likely to base-pair in secondary structures. This suggestion that reduced secondary RNA structure could favor splicing is supported by previous reports that cryptic acceptor sites can be activated by mutations predicted to reduce the base-pairing of A - 3 through A - 1 in stem-loops of secondary structures (Deshler and Rossi 1991). This conclusion is also supported by analysis of splicing in dicot plants in which AU-rich intron sequences have been implicated in promoting splicing (Goodall and Filipowicz 1991).
The association of enriched A and U content with enhanced splicing implies that in assessing the information requirements of donor and acceptor sites, it is appropriate to include the cumulative small contributions of information over broad regions in the vicinity of the splice sites. For example, summing the information at positions -32 to 32 for all donor and acceptor sites (see Supplemental Fig. S3) revealed information estimates of 9.64 ± 0.04 (donor) and 10.15 ± 0.04 (acceptor) bits. (After subtracting an adjustment for Drosophila genome background nucleotide frequencies, these cumulative measures are 9.23 [donor] or 9.76 [acceptor]; see Appendix 3.) Interestingly, these values exceed the average log2(distance) between donor sites (9.07) and acceptor sites (8.86) in our data set, indicating that these information estimates are sufficient for the observed spacing between donor sites and acceptor sites, although the latter may also require information at the splice branch points (Chua and Reed 2001; Lim and Burge 2001). As expected from our analysis at individual nucleotide positions, the cumulative information at positions -32 to +32 increases with intron length (Supplemental Fig. S8). Moreover, the cumulative information for donor and acceptor sites of small introns (64-80 nt) is significantly lower than those of longer introns (>8191 nt; Fig. 2A; p > 0.99 by one-tailed approximate t-test).
Our analysis has allowed us to define quality measures at different nucleotide positions relative to the donor and acceptor sites. We have confirmed that the canonical GU...AG bookends of introns are extremely highly conserved for all lengths of introns (Fig. 3A). We have also shown that certain positions have strikingly more information with progressively longer introns (e.g., D + 4, A - 6, A - 5). The position D + 5 stands out in having a very high incidence of G (>83%) for all intron length ranges (Supplemental Fig. S4). We suggest that because the three hydrogen bonds of G would contribute more to the interaction with U1 or U6 snRNA than would neighboring As and Us, this may represent an optimal choice providing greater interaction strength with minimal increase in information. Interestingly, D + 5 was also found to be invariant in a genome-wide analysis of 228 yeast introns (Spingola et al. 1999).
Our observation that information and nucleotide content at splice sites is affected by the lengths of nonadjacent introns implies the existence of long-range effects spanning multiple introns. Although identification of molecular processes mediating these long-range effects awaits future analyses, it is striking that the effects are both sequence-specific (e.g., enhanced consensus sequences in the donor site following the varied intron; Fig. 3C), as well as more general (e.g., C enrichment near splice sites following, and A enrichment near splice sites preceding the varied intron; Table 1; Fig. 4). One can speculate that these observations are consistent with there being a molecular complex involving multiple splice sites. It is particularly striking that the levels of C and U enrichment in the pyrimidine tracts of acceptor sites correlate with the lengths of nonadjacent introns−C enrichment reflects a longer upstream intron, whereas U enrichment reflects a longer downstream intron−consistent with the possibility that C and U enrichment may facilitate molecular interactions with upstream and downstream splice sites, respectively.
The large size of our data set (10,057 introns) permitted its partitioning into subsets of substantial size. The stored procedures in our database permitted us to select ordered subsets corresponding to progressive changes in selected parameters (in our case, intron length or extent of mismatch to consensus). In contrast to random sampling, this approach allowed us to uncover rather subtle trends. For example, although the information increases at positions A - 18 through A - 13 (Fig. 3A) are quite subtle, they are highly suggestive because they derive from ordered partitioning according to intron length.
In the future, as larger data sets become available, it will become possible to extract progressively more information with the analytical approach used in this paper. We have shown that partitioning of splice-site data based on intron lengths, and conformity to consensus sequences, provides an enhanced view of splice-site sequence requirements that was not possible with previous smaller data sets. We expect that similar studies of selected subsets of large data sets reflecting special contexts that strain the functioning of molecular machines, for example, involving protein-DNA interactions, will provide useful insights into their functions.
Acknowledgments
We thank Laurel Appel, Anne Baranger, Deborah Eastman, and Robert Lane for critical reading and suggestions for the paper. We also thank one of the reviewers for suggestions. We thank Matt Eaton for implementing the scanning algorithm for splice site detection, and Will Gladstone for providing programming support. This work was supported in part by funds from the Howard Hughes Medical Institute to support undergraduate initiatives in the life sciences. We thank especially the Berkeley Drosophila Genome Project—this work would not have been possible without the crucial contributions of members of BDGP.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
APPENDIX 1
Scanning Algorithm to Identify Splice Sites
To determine the splice sites for a given cDNA transcript, we used the following algorithm with the transcript and the corresponding genomic DNA.
Step 1: Starting initially at the 5′-end of the cDNA, find the first exact match between the next 20 bases in the cDNA and the genomic DNA.
Step 2: Continue scanning the cDNA and genomic DNA one base at a time until the first mismatch is found; this determines a maximal matching region on the genomic DNA. Starting at the first mismatch in the cDNA, repeat step 1 to determine a second downstream matching region on the genomic DNA. The intervening region is the predicted intron.
Step 3: If the first base upstream of the intron and the last base in the intron do not match, use the intron to define the donor and acceptor sites and go to step 4.
Otherwise, while the first base upstream of the intron and the last base in the intron match, perform the following steps:
If a GT...AG or AT...AC consensus is found at the ends of the intron, use it to define the donor and acceptor sites and go to step 4.
Move the start and finish positions of the intron one base upstream.
If no weak form of consensus (3 out of 4 match) is found, terminate the processing of the transcript.
Step 4: Repeat step 2 using the second matching region of the genomic DNA and the corresponding cDNA matching region.
An extended version of the algorithm also partially handles SNPs in step 2 by using the following rule: The maximal matching region is extended as long as at most 1 mismatch is encountered in every 10 bases.
APPENDIX 2
As discussed in the Appendix to Schneider et al. (1986), a bias is introduced by using the standard formula for computing the uncertainty at a given position p based on the frequencies of nucleotide occurrences in a sample size N. The correct estimate for the uncertainty is obtained by assuming a multinomial distribution of the nucleotides at a position p and computing the expected value of the random variable Xp that assumes the uncertainty value
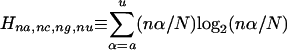 |
with the probability
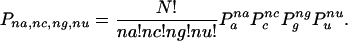 |
where Pa, Pc, Pg, Pu represent fixed estimates for the frequencies of nucleotide occurrences and na, ng, nc, and nu represent the number of occurrences of A, C, G, and U, respectively, at position p.
The expected value of Xp is defined by
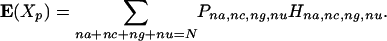 |
Numerical estimates show that the expected value E(Xp) converges to the standard uncertainty value Hp = - {Pα log2(Pα): α = a, c, g, u} as the sample size N increases. These estimates also indicate that the correction factor γp = Hp - E(Xp) has approximately the same value for each distribution {Pα}. Therefore, for N ≤ 125, we calculate γp using Pa = Pc = Pg = Pu = 0.25. For N > 125, we estimate the correction factor using the following approximation due to Basharin (1959): γ ≈ p 1.5/(ln(2) × N).
By definition, the variance of the random variable Xp is
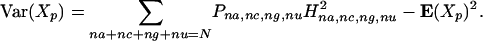 |
For N ≤ 125, we use this expression to compute the standard deviation
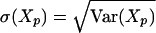 |
of the sampling error at position p. Numerical estimates indicate that as the sample size N increases the variance Var(Xp) converges to the following expression due to Basharin (1959):
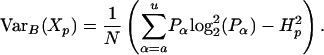 |
Therefore, for N > 125, we estimate the standard deviation σ(Xp) using 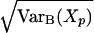 .
.
Assuming that the random variables Xpi at the contiguous distinct positions p = p1, p2,..., pr are independent, it follows that the variance on the site p1p2... pr is given by the expression
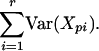 |
The expected values and standard deviations of Xp and 〈Xp1, Xp2,..., Xpr〉, were used in approximate t-tests (Zar 1999, section 8.1) to compare information estimates at positions and regions near donor and acceptor sites.
Other methods based on techniques from large-deviation statistics have also been used to estimate P values of information content scores (Hertz and Stormo 1999).
Assuming a sample size N, we will assume a multinomial distribution for the nucleotides at a position p, where Pa, Pc, Pg, and Pu represent the standard frequency estimates for the occurrences of the respective nucleotides. In this case, the probability that na, ng, nc, and nu represent the number of occurrences of A, C, G, and U, respectively, is given by the expression
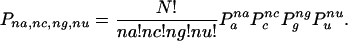 |
Based on this assumption, it is well known that the random variable Yα representing the nucleotide count of a particular nucleotide α (α = A, C, G, or U) at position p has a binomial distribution. Therefore, the expected value of Yα is E(Yα) = NPα and the variance of Yα is Var(Yα) = NPα(1 - Pα).
Assuming that the random variables Yiα at the contiguous distinct positions p = p1, p2,..., pr are independent, it follows that the variance on the site p1p2... pr is given by the expression
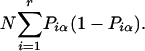 |
In particular, because the maximum value of NPiα(1 - Piα) is N/4 when Piα = 0.5, the maximum value of the variance for a site r bases wide is Nr/4. The expected values and standard deviations of Yα and 〈Y1α, Y2α,..., Yrα〉 were used in approximate t-tests (Zar 1999, section 8.1) to compare nucleotide counts and nucleotide percentages at positions and regions near donor and acceptor sites.
APPENDIX 3
In this paper, we calculate information relative to random background where each nucleotide frequency is 0.25. However, information can also be calculated relative to background nucleotide frequencies of the Drosophila genome: 28.8% A, T; 21.2% G, C (BDGP releases 2 and 3). One can calculate information relative to these genomic frequencies using different methods, either by subtracting background information, or by calculating relative entropy (Lim and Burge 2001). Because the average genomic background information in Drosophila is 0.017 bits/nucleotide position, the values relative to a random background may be inflated by as much as 0.017 bits. Hence, information can be corrected by subtracting this amount (0.017 bits; Supplemental Fig. S3B). These corrections (background subtraction or use of relative entropy) lead to very small adjustments at any given position, but to modest changes in cumulative information (see, e.g., Table 3). We note that the statistical corrections described in this study do not necessarily apply to either type of genomic frequency adjustment.
Table 3.
Cumulative Information for Positions -32 to +32
| Intron length | Site | Information (random)a | Information (genomic)b | Relative entropyc |
|---|---|---|---|---|
| 64−80 | D | 9.57 | 9.14 | 9.24 |
| 64−80 | A | 10.00 | 9.58 | 9.15 |
| All | D | 9.64 | 9.23 | 9.10 |
| All | A | 10.15 | 9.76 | 9.23 |
| >8191 | D | 11.80 | 11.00 | 11.61 |
| >8191 | A | 14.27 | 13.48 | 14.22 |
Information relative to random nucleotide frequencies: 25% A, C, G, T
Absolute differences between information relative to random nucleotide frequencies and background information computed from Drosophila genomic nucleotide frequencies (28.8% A, T; 21.2% G, C). The sampling error (See Appendix 2) is also subtracted, but the final value is not allowed to be negative
Relative entropy based on the Drosophila genomic nucleotide frequencies with no correction for sampling error
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1715204.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Aebi, M., Hornig, H., and Weissmann, C. 1987. 5′ cleavage site in eukaryotic pre-mRNA splicing is determined by the overall 5′ splice region, not by the conserved 5′ GU. Cell 50: 237-246. [DOI] [PubMed] [Google Scholar]
- Basharin, G.P. 1959. On a statistical estimate for the entropy of a sequence of independent random variables. Theor. Prob. Appl. 4: 333-336. [Google Scholar]
- Burge, C.B., Tuschl, T., and Sharp, P.A. 1999. Splicing of precursors to mRNAs by the spliceosomes. In The RNA world (eds. R.F. Gesteland et al.), pp. 525-560. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Burkard, M.E., Turner, D.H., and Tinoco, I. 1999. The interactions that shape RNA structure. In The RNA world (eds. R.F. Gesteland et al.), pp. 233-264. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Chiara, M.D., Palandjian, L., Feld Kramer, R., and Reed, R. 1997. Evidence that U5 snRNP recognizes the 3′ splice site for catalytic step II in mammals. EMBO J. 16: 4746-4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, K. and Reed, R. 2001. An upstream AG determines whether a downstream AG is selected during catalytic step II of splicing. Mol. Cell. Biol. 21: 1509-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, C.A. and Guthrie, C. 1999. Allele-specific genetic interactions between Prp8 and RNA active site residues suggest a function for Prp8 at the catalytic core of the spliceosome. Genes & Dev. 13: 1970-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshler, J.O. and Rossi, J.J. 1991. Unexpected point mutations activate cryptic 3′ splice sites by perturbing a natural secondary structure within a yeast intron. Genes & Dev. 5: 1252-1263. [DOI] [PubMed] [Google Scholar]
- Fields, C. 1990. Information content of Caenorhabditis elegans splice site sequences varies with intron length. Nucleic Acids Res. 18: 1509-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouser, L.A. and Friesen, J.D. 1986. Mutations in a yeast intron demonstrate the importance of specific conserved nucleotides for the two stages of nuclear mRNA splicing. Cell 45: 81-93. [DOI] [PubMed] [Google Scholar]
- Goodall, G.J. and Filipowicz, W. 1991. Different effects of intron nucleotide composition and secondary structure on pre-mRNA splicing in monocot and dicot plants. EMBO J. 10: 2635-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. and Mount, S.M. 1995. Localization of sequences required for size-specific splicing of a small Drosophila intron in vitro. J. Mol. Biol. 253: 426-437. [DOI] [PubMed] [Google Scholar]
- Guo, M., Lo, P.C., and Mount, S.M. 1993. Species-specific signals for the splicing of a short Drosophila intron in vitro. Mol. Cell. Biol. 13: 1104-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, S.L. and Padgett, R.A. 1994. Conserved sequences in a class of rare eukaryotic nuclear introns with non-consensus splice sites. J. Mol. Biol. 239: 357-365. [DOI] [PubMed] [Google Scholar]
- Hertel, K.J., Lynch, K.W., and Maniatis, T. 1997. Common themes in the function of transcription and splicing enhancers. Curr. Opin. Cell Biol. 9: 350-357. [DOI] [PubMed] [Google Scholar]
- Hertz, G.Z. and Stormo, G.D. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563-577. [DOI] [PubMed] [Google Scholar]
- Lesser, C.F. and Guthrie, C. 1993. Mutations in U6 snRNA that alter splice site specificity: Implications for the active site. Science 262: 1982-1988. [DOI] [PubMed] [Google Scholar]
- Lim, L.P. and Burge, C.B. 2001. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. 98: 11193-11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen, B.G. and Seraphin, B. 1998. Genetic interaction between U6 snRNA and the first intron nucleotide in Saccharomyces cerevisiae. RNA 4: 167-180. [PMC free article] [PubMed] [Google Scholar]
- Mount, S.M., Burks, C., Hertz, G., Stormo, G.D., White, O., and Fields, C. 1992. Splicing signals in Drosophila: Intron size, information content, and consensus sequences. Nucleic Acids Res. 20: 4255-4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, A.J. and Norman, C. 1992. U5 snRNA interacts with exon sequences at 5′ and 3′ splice sites. Cell 68: 743-754. [DOI] [PubMed] [Google Scholar]
- Nilsen, T.W. 1998. RNA-RNA interactions in nuclear pre-mRNA splicing. In RNA structure and function (eds. R.W. Simons and M. Grunberg-Manago), pp. 279-307. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Parker, R. and Guthrie, C. 1985. A point mutation in the conserved hexanucleotide at a yeast 5′ splice junction uncouples recognition, cleavage, and ligation. Cell 41: 107-118. [DOI] [PubMed] [Google Scholar]
- Patterson, B. and Guthrie, C. 1991. A U-rich tract enhances usage of an alternative 3′ splice site in yeast. Cell 64: 181-187. [DOI] [PubMed] [Google Scholar]
- Puig, O., Gottschalk, A., Fabrizio, P., and Seraphin, B. 1999. Interaction of the U1 snRNP with nonconserved intronic sequences affects 5′ splice site selection. Genes & Dev. 13: 569-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, R. 2000. Mechanisms of fidelity in pre-mRNA splicing. Curr. Opin. Cell Biol. 12: 340-345. [DOI] [PubMed] [Google Scholar]
- Rosbash, M. and Seraphin, B. 1991. Who's on first? The U1 snRNP-5′ splice site interaction and splicing. Trends Biochem. Sci. 16: 187-190. [DOI] [PubMed] [Google Scholar]
- Rudner, D.Z., Kanaar, R., Breger, K.S., and Rio, D.C. 1996. Mutations in the small subunit of the Drosophila U2AF splicing factor cause lethality and developmental defects. Proc. Natl. Acad. Sci. 93: 10333-10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.D., Stormo, G.D., Gold, L., and Ehrenfeucht, A. 1986. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 188: 415-431. [Schneider, T.D., Haemer, J.S., and Stormo, G.D. Appendix: Calculation of sampling uncertainty and variance.] [DOI] [PubMed] [Google Scholar]
- Shannon, C.E. and Weaver, W. 1949. The mathematical theory of communication. University of Illinois Press, Urbana, IL.
- Siatecka, M., Reyes, J.L., and Konarska, M.M. 1999. Functional interactions of Prp8 with both splice sites at the spliceosomal catalytic center. Genes & Dev. 13: 1983-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano, P.G. and Guthrie, C. 1988. 5′ splice site selection in yeast: Genetic alterations in base-pairing with U1 reveal additional requirements. Genes & Dev. 2: 1258-1267. [DOI] [PubMed] [Google Scholar]
- Sontheimer, E.J. and Steitz, J.A. 1993. The U5 and U6 small nuclear RNAs as active site components of the spliceosome. Science 262: 1989-1996. [DOI] [PubMed] [Google Scholar]
- Spingola, M., Grate, L., Haussler, D., and Ares, M. 1999. Genome-wide bioinformatic and molecular analysis of yeast introns. RNA 5: 221-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling, A.C., Stern, D.M., Kiss, I., Roote, J., Laverty, T., and Rubin, G.M. 1995. Gene disruptions using P transposable elements: An integral component of the Drosophila genome project. Proc. Natl. Acad. Sci. 92: 10824-10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton, M., Liao, G., Brokstein, P., Hong, L., Carninci, P., Shiraki, T., Hayashizaki, Y., Champe, M., Pacleb, J., Wan, K., et al. 2002. The Drosophila gene collection: Identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 12: 1294-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz, J.A. 1992. Splicing takes a holliday. Science 257: 888-889. [DOI] [PubMed] [Google Scholar]
- Stephens, R.M. and Schneider, T.D. 1992. Features of spliceosome evolution and function inferred from an analysis of the information at human splice sites. J. Mol. Biol. 228: 1124-1136. [DOI] [PubMed] [Google Scholar]
- Vijayraghavan, U., Parker, R., Tamm, J., Iimura, Y., Rossi, J., Abelson, J., and Guthrie, C. 1986. Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J. 5: 1683-1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S., Romfo, C.M., Nilsen, T.W., and Green, M.R. 1999. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature 402: 832-835. [DOI] [PubMed] [Google Scholar]
- Yu, Y.-T., Scharl, E.C., Smith, C.M., and Steitz, J.A. 1999. The growing world of small nuclear ribonucleoproteins. In The RNA world (eds. R.F. Gesteland et al.), pp. 487-524. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Zar, J.H. 1999. Biostatistical analysis, 4th ed. Prentice Hall, Upper Saddle River, NJ.
- Zhang, D. and Rosbash, M. 1999. Identification of eight proteins that cross-link to pre-mRNA in the yeast commitment complex. Genes & Dev. 13: 581-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio, D.A. and Blumenthal, T. 1999. Both subunits of U2AF recognize the 3′ splice site in Caenorhabditis elegans. Nature 402: 835-838. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://igs.wesleyan.edu; Integrative Genomic Sciences at Wesleyan University.
- http://www.fruitfly.org; Berkeley Drosophila Genome Project (BDGP).