

Abstract
Purpose
Hepatitis C virus (HCV) infection can promote the development of hepatocellular carcinoma (HCC). Published data implicate the HCV core gene in oncogenesis. We tested the hypothesis that core gene sequences from HCC patients differ from those of patients without cirrhosis/HCC.
Experimental Design
Full-length HCV sequences from HCC patients and controls were obtained from the investigators and GenBank and compared to each other. A logistic regression model was developed to predict the HCC risk of individual point mutations and other sequence features. Mutations in partial sequences (bases 36–288) from HCC patients and controls were also analyzed. The first base of the AUG start codon was designated position 1.
Results
A logistic regression model developed through analysis of full-length core gene sequences identified seven polymorphisms significantly associated with increased HCC risk (36G/C, 209A, 271U/C, 309A/C, 435A/C, 481A, 546A/C) and an interaction term (for 209A-271U/C) that had an odds ratio < 1.0. Three of these polymorphisms could be analyzed in the partial sequences. Two of them, 36G/C and 209A, were again associated with increased HCC risk, but 271U/C, was not. The odds ratio of 209A-271U/C was not significant.
Conclusions
HCV core genes from patients with and without HCC differ at several positions. Of interest, 209A has been associated with interferon resistance and HCC in previous studies. Our findings suggest that HCV core gene sequence data might provide useful information about HCC risk. Prospective investigation is needed to establish the temporal relationship between the appearance of the viral mutations and development of HCC.
Keywords: HCV, HCC, core, RNA, diversity, complexity
Introduction
Viruses are among the most important human carcinogens. Several viruses, including the human papilloma virus (HPV), encode oncogenic proteins that promote cellular transformation(1). HPV strains differ in their oncogenic potential(2), showing that specific mutations can modulate viral transforming activities. Chronic infection with the hepatitis C virus (HCV) increases the risk of hepatocellular carcinoma (HCC) in patients who progress to liver cirrhosis. The role of HCV proteins in the development of HCC is unclear. Based on the HPV model, if HCV has direct oncogenic effects, it is likely that the sequences of HCV in patients with HCC are different from the viral sequences in patients with early-stage liver disease.
Of the several HCV proteins reported to alter cellular growth(3–6), the core protein is the one most strongly associated with cellular transformation. Expression of the core protein enhances cell proliferation, DNA synthesis, cell cycle progression, cellular transformation and liver cancer in experimental systems(7–15). Transgenic mice containing the HCV core gene develop steatosis and liver cancer (16–18).
The HCV core gene is a complex genetic region of the viral genome. It contains two open reading frames (19, 20), three confirmed RNA structures (21, 22) and several additional putative RNA signals/structures(23). Mutations in the core gene therefore have the potential to alter the proteins encoded by the two open reading frames, the RNA secondary structures, and/or the RNA signals.
HCV sequences differ in patients with early versus late stage liver disease (24–29). It is not known whether the sequence differences are present because viruses with certain polymorphisms have enhanced potential to cause liver damage and consequently viruses that carry these mutations are more likely to be found in patients with liver cancer. Alternatively, the sequence differences arise because liver disease progression changes the cellular environment in which HCV replicates and selects for variants carrying polymorphisms that enhance survival in the damaged liver. HCV core genes differ in tumor versus non-tumor samples of the same liver (30–35). Interestingly, analyses of full-length core gene sequences from serum indicate that there may be specific substitutions in the circulating HCV RNA of patients with HCC; however, these investigations are not definitive because they were performed on small sets of sequences from a single geographic region [n=15(36) and n=13(37)].
Regardless of the events that give rise to HCV disease-specific polymorphisms, knowledge of these polymorphisms is potentially useful. If HCV mutations can be associated with liver disease status, then HCV sequencing can become a non-invasive and economical tool for obtaining information about the condition of a patient’s liver and HCC risk. In addition, understanding the causal relationship between viral mutations and liver disease may suggest new targets for therapeutic intervention. The first step in harnessing HCV sequence data for these applications is to rigorously test the hypothesis that specific viral polymorphisms are, in fact, associated with clinically-distinct stages of liver disease.
The investigation reported here is a systematic analysis of an international collection of HCV genotype 1b core gene sequences isolated from human serum. Our findings provide a framework for designing prospective studies to determine the temporal relationship between the appearance of the viral mutations, the development of advanced liver disease, and the progression to liver cancer.
Materials and Methods
Assembly of sequences for analysis
Full-length core gene sequences (bases 1–573) were downloaded from Genbank using the HCV database interface at http://hcv.lanl.gov/components/sequence/HCV/combined_search/searchi.html in November 2007. Genotype 1b sequences were included in further analysis if they were isolated from human serum and represented a unique patient who was treatment-naïve, had not undergone liver transplantation, was not actively infected with either human immunodeficiency virus (HIV) or hepatitis B virus (HBV), whose liver disease could be identified as either non-cirrhotic, cirrhotic without HCC, or HCC; did not have additional noted co-morbidities (e.g., hemophilia) and contained the complete HCV core gene, without deletions or insertions. Publications linked to the sequences or the GenBank entries were reviewed to identify sequences that satisfied the inclusion criteria. Sequences were excluded if insufficient information was available. Sequences meeting the inclusion criteria were separated into two groups based on the disease state of the patients: [1]acute and chronic HCV infection without cirrhosis; [2]chronic infection with HCC, recognizing that most, if not all, HCV patients with HCC also have cirrhosis. To provide a second set of sequences to use in limited confirmatory testing, partial core gene sequences were downloaded from GenBank using the HCV database interface in November 2007. Partial sequences were retained in the study if they met the inclusion criteria applied to the full-length sequences, contained bases 36–288, but did not contain the complete core gene sequence.
Identification of polymorphisms and interactions of interest
Identifying polymorphisms associated with the country of origin
We first analyzed the full-length and partial sets of control sequences to identify any polymorphisms associated with either Japanese or Spanish origin. This was done because many of the full-length HCC sequences came from Japan and all of the partial HCC sequences came from Spain. To seek region-associated polymorphisms, we first constructed a consensus sequences derived from the multiple sequence alignment of the full-length non-HCC (control) HCV sequences. The majority nucleotide at each position was designated the consensus nucleotide. The first nucleotide of the AUG start codon was numbered nucleotide 1. At each position of the full-length sequences, the chi-squared test or Fisher’s exact test (R package), as appropriate, was used to compare the proportion of the non-HCC (control) sequences from Japan that had the consensus nucleotide to the proportion of control sequences from other locations that had the consensus nucleotide. Polymorphisms with a p-value<0.05 were identified and analyzed further using forward, backward, and stepwise multivariable logistic regression. Polymorphisms that remained significantly associated with Japanese origin were identified (p<0.05) and all subsequent analyses of full-length sequences adjusted for these Japan-associated polymorphisms and for Japanese origin. A similar process was used to seek Spain-associated polymorphisms among the partial sequences, but none were found and no adjustment was made for Spanish origin in analyses of the partial sequences.
Building a logistic regression model of the HCC risk associated with polymorphisms in the HCV core gene
We used a multi-step process to identify mutations that were more prevalent among the HCV sequences from HCC patients than among the HCV sequences from patients without HCC. The proportion of the consensus nucleotide at each position in the full-length HCC HCV sequences was first compared to the proportion of the consensus nucleotide at each position in the full length non-HCC HCV sequences using a multivariable logistic regression model which included the following covariates: the Japan-associated mutations, Japanese origin, and the position under analysis. Polymorphisms that were more common in the HCC HCV set (p<0.05) were investigated further, using multivariable logistic regression to identify mutations that remained significantly associated with HCC (p<0.05). We next created interaction covariates using all possible two-ways combinations of the point mutations found to be associated with HCC in the previous analysis and analyzed a series of multivariable logistic regression models to find the best fit. All tested models included the covariates for Japan-associated mutations and Japanese origin. We used forward, backward, and stepwise logistic regression to identify the polymorphisms and interactions significantly associated with HCC (p<0.05). The best fit was determined by the Wald statistic.
Analysis of partial core gene sequences
To look for additional evidence that the polymorphisms identified by multivariable logistic regression were associated with HCC, the prevalence of these mutations in partial core gene sequences (bases 36–288) of HCC and control patients were evaluated using chi-square or Fishers exact test, as appropriate, to the extent permitted by the length constraints. Sequence features with p-values <0.05 were considered significant. Calculations were performed using SAS Version 9.0 (Cary NC) and Epi Info Version 3.4.3.
Analysis of potential RNA structures
RNA secondary structural models were generated using M-fold (38, 39) (http://frontend.bioinfo.rpi.edu/applications/mfold/cgi-bin/rna-form1.cgi) with default parameters. The inputs were portions of a consensus sequence of all the full-length genotype 1b core sequences analyzed in this study. Structures of interest were evaluated using R-fold analyzer, which is freely available at http://tandem.bu.edu/foldsupport/foldsupport.html and by request, in PERL format.
Analysis of sequence diversity and complexity
Genetic distances at the nucleotide level were calculated using the Kimura-two-parameter metric and these distances were used to construct phylogenetic trees using the neighbor joining algorithm (Phylip). Consensus trees were constructed from 1000 bootstrap repetitions. Protein distances were calculated using the Dayhoff PAM model in Protidist (Phylip). The number of nucleotide and amino acid substitutions in the sequences of the HCC set and the control set were tallied relative to Con 1 (Genbank AJ238799), which served as a genotype 1b reference sequence, using custom Perl scripts and compared using the Students T-test.
The genetic complexity of the sequences from patients with HCC was compared to that of sequences from the control group by calculating the Shannon entropy at each position of the gene using the Los Alamos National Lab Entropy-Two tool from http://hcv.lanl.gov/content/sequence/ENTROPY/entropy.html. Global Shannon entropies were compared by the Wilcoxon rank sum test (R package version 2.6.0).
Results
Sequence collection, selection, and characterization
A total of 1439 full-length sequences were downloaded from Genbank. Eight hundred and ninety-eight were eliminated for the following reasons: 532 lacked sufficient information for inclusion; 208 were isolated from liver tissue; 104 were from later time points of patients whose earlier time point was already included; 19 were from chimpanzees; 10 were from pooled serum samples; eight were cell culture-adapted; four were from patients with HIV and/or HBV co-infection; four were non-genotype 1b recombinants; three were post-interferon treatment; three were post-liver transplantation; two contained insertions or deletions; and one was isolated from ascites fluid. In total, 541 satisfied the inclusion criteria. Two hundred and sixty sequences were products of direct sequencing. The remaining 281 were quasispeices clones from 18 patients. When only two clones were available, one was chosen at random, when more than two clones were available, the consensus sequence of the clones was constructed and used.
In total, 278 of the full-length sequences in this study were obtained from Genbank; 214 from patients with either acute or chronic HCV and without cirrhosis or HCC (the control group); 58 from patients with HCC; and six from cirrhotic patients without HCC. Seven additional sequences from patients with HCC were produced by the investigators, yielding a total of 65 sequences from patients with HCC (the HCC group; Figure S1A, Table S1A Supplement). The sequences from the control group included 97 (45%) from Japan and the sequences from the HCC group included 58 (89%) from Japan. We assume that most of the patients with HCC were cirrhotic, as HCC in HCV patients almost always arises in the background of cirrhosis. The number of available sequences from patients with cirrhosis but without HCC was too small to allow detailed analysis.
A total of 1824 partial core sequences were downloaded from Genbank. We verified that all these sequences were from patients not represented in the full-length set. Of these, 1132 were eliminated for the following reasons: 744 lacked sufficient information for inclusion; 153 were from patients with HIV and/or HBV infection; 68 were isolated from liver tissue; 59 were from pooled serum samples; 39 were isolated from non-serum extrahepatic compartments; 38 were post-liver transplantation; 18 were from later time points of patients whose earlier time point was already included; 11 were non-genotype 1b recombinants; one contained deletions; one was isolated from a patient with hemophilia. In total, 692 sequences satisfied the inclusion criteria. Of these, 71 were quasispecies clones from a total of 7 patients. A consensus of the clones from each subject was determined and used. The remaining 621 sequences were either directly sequenced or represented one of two available clones. In total, 628 sequences were obtained; 543 from patients with acute or chronic HCV without cirrhosis or HCC (the control group); 55 from patients with HCC (the HCC group); and 30 from cirrhotic patients without HCC (Figure S1B, Table S1B Supplement). Since the region spanning nucleotides 36–288 had the highest representation in the partial sequences, the analysis was carried out on this region. Sequences from 49 from patients with HCC and 309 from control patients were analyzed.
Identification of HCV core gene polymorphisms associated with HCC in full-length sequences and construction of a logistic regression model
Univariable and then multivariable analysis found five Japan-associated polymorphisms: 9A, 264C, 273A, 330G, and 534C. All subsequent analyses controlled for both Japanese origin and for the effects of the Japan-associated polymorphisms (see methods).
The multivariable logistic regression models built to evaluate individual positions identified ten HCV core gene polymorphisms significantly associated with increased HCC risk, 36G/C, 209A, 271C/U, 309A/C, 384U, 408U, 435A/C, 465U, 481A, 546A/C, and one significantly associated with decreased HCC risk, 78U (Table 1). Each of these polymorphisms was examined to determine its impact on the amino acid sequence of the core protein and/or ARFPs. Of the 11, four alter the amino acid sequence of the core protein: 36G/C (K12Silent/N); 209A (Q70R); 271U/C (M91L); 481A (G161S). Six are predicted to change the amino acid sequence of the ARFPs: 36G/C (N11S/T), 78U (A25V), 209A (G69S), 309C/A (L102P), 384U (A127V), 408U (T135I). In addition, six are predicted to modify RNA structural elements. Specifically, 36G/C alters the sequence of the portion of the internal ribosome entry site that overlaps with the beginning of the core protein coding sequence; 78U alters SLV (a required translation element); 271U/C and 309A/C alter SL_248; 465U and 481A alter the terminal stem loops element (TSLE). In addition, 271U/C changes the codon at a site of internal protein synthesis initiation (40). (Table 2, Figure 1).
Table 1.
Gene sequences of patients with hepatitis C infection and hepatocellular carcinoma compared to gene sequences of subjects with hepatitis C infection and without hepatocellular carcinoma, GenBank, NCBI, (N=279). Odds ratios and p-values were calculated using logistic regression while controlling for Japanese origin.
| HCC Sequence Set (N=65) (%) | Control Sequence Set (N=214)(%) | Individual Analysis | Multivariable Analysis | |||
|---|---|---|---|---|---|---|
| OR (95%CI) | p-value | OR (95% CI) | p-value | |||
| Gene Position | ||||||
| 36C/G | 3(5) | 3(2) | 8.38 (1.28, 54.62) | 0.026 | 14.79 (2.01,108.35) | 0.008 |
| 36A | 62(95) | 211(98) | ||||
| 78U | 2(3) | 43(20) | 0.16 (0.03, 0.71) | 0.016 | ||
| 78C | 63(96) | 171(80) | ||||
| 209A | 37(56) | 74(35) | 2.93 (1.57, 5.42) | <0.001 | 13.50 (4.48, 40.68) | <0.001 |
| 209G | 28(43) | 140(65) | ||||
| 271U/C | 42(65) | 61(29) | 3.84 (1.91, 7.70) | <0.001 | 10.57 (3.42, 32.66) | <0.001 |
| 271A | 23(35) | 153(71) | ||||
| 309C/A | 29(45) | 55(26) | 2.04 (1.05, 3.98) | 0.034 | 2.66 (1.17, 6.05) | 0.018 |
| 309U | 36(55) | 159(74) | ||||
| 384U | 3(5) | 1(1) | 12.70 (1.07,149.80) | 0.043 | ||
| 384C | 62(95) | 213(99) | ||||
| 408U | 4(6) | 2(1) | 9.98 (1.38, 72.12) | 0.023 | ||
| 408C | 61(94) | 212(99) | ||||
| 435A/C | 23(35) | 27(13) | 2.12 (1.05, 4.26) | 0.035 | 4.09 (1.73, 9.68) | 0.001 |
| 435G | 42(65) | 187(87) | ||||
| 465U | 5(8) | 4(2) | 4.93 (1.07, 22.64) | 0.040 | ||
| 465C | 60(92) | 210(98) | ||||
| 481A | 13(20) | 8(4) | 3.47 (1.31, 9.13) | 0.012 | 3.825 (1.2, 11.89) | 0.02 |
| 481G | 52(80) | 206(96) | ||||
| 546A/C | 8(12) | 9(4) | 3.85 (1.16, 12.80) | 0.027 | 10.04 (2.17, 46.37) | 0.003 |
| 546G | 57(88) | 205(96) | ||||
| 209A/271U/C | 0.16 (0.03, 0.72) | 0.01 | ||||
Table 2.
Effects of significant substitutions on potential secondary structures and coding function in the core gene. Consensus nucleotide is shown in bold.
| Position | Control Type | HCC Type | RNA effect | Core effect | ARFP effect | ||
|---|---|---|---|---|---|---|---|
| Control | HCC | Control | HCC | ||||
| 36 | A | G/C | None | Lys | Silent/Asn | Asn | Ser/Thr |
| Weakens SLV | Silent (Gly) | Ala | Val | ||||
| 78 | U | C | GCGU | ||||
| 209 | G | A | None | Gln | Arg | Gly | Ser |
| Strengthens SL_248 | |||||||
| 271 | A | U/C | Introduces GU or GC pair Weakens SL_248 | Met | Leu | Silent (Ala) | |
| 309 | U | C/A | Disrupts AU base pair | Silent (Ser) | Leu | Pro | |
| 384 | C | U | None | Silent (Cys) | Ala | Val | |
| 408 | C | U | None | Silent (Tyr) | Thr | Ile | |
| 435 | G | A/C | None | Silent (Gly) | |||
| 465 | C | U | Weakens TSLE (GCGU) | Silent (Val) | |||
| 481 | G | A | Strengthens TSLE, UGUA | Gly | Ser | ||
| 546 | G | A/C | None | Silent (Leu) | |||
Figure 1. Characteristic substitutions in the core gene may impact putative RNA secondary structures and alter core and ARFP function.

The position of the nucleotide mutations on the RNA secondary structures of the core gene region are shown in white boxes. ARFP mutations are shown in grey shaded boxes, and core mutations are shown in black shaded boxes. The domains of the core protein are shown below the secondary structure map of the core region.
Multivariable logistic regression analysis of the 11 polymorphisms identified in the individual analysis showed that seven of these mutations were significantly associated with increased risk: 36G/C, 209A, 271C/U, 309A/C, 435A/C, 481A, 546 A/C. These seven mutations and their 21 pair-wise combinations were evaluated in a final logistic regression analysis. All seven polymorphisms and the 209A-271C/U pair were significantly associated with HCC in the best fit model (p<0.05). The odds ratio (OR) of the 209A-271C/U interaction term was 0.16, indicating that the presence of both mutations in the same sequence conferred less risk than the product of their individual ORs.
Analysis of a second set of partial sequences
The OR of four of the sequence features in the best fit model could be examined in a second set of partial sequences, which spanned bases 36–288. The ORs for 36G/C, 209A, 271C/U and the 209A-271C/U pair are presented in Table 3. The significant increased risk of HCC associated with 36G/C and 209A were also found in this analysis (p=0.003 and p<0.001, respectively), both at the usual level of significance (p<0.05) and when a Bonferroni correction is applied (p<0.0125). Contrary to the first analysis, 271U/C was not significantly associated with HCC as an individual term or as part of the 209A-271C/U pair.
Table 3.
Risk associated with HCC in the partial sequences.
| HCC HCV N=49 (%) |
Control HCV N=309 (%) |
OR (95%CI) | p-value | |
|---|---|---|---|---|
| Gene Position | ||||
| 36C/G | 5 (10) | 4 (1) | 8.64 (1.92, 40.14) | 0.003 |
| 36A | 44 (90) | 304 (99) | ||
| 209A | 38 (77) | 116 (37) | 5.72 (2.69, 2.40) | <0.001 |
| 209G | 11 (23) | 192 (63) | ||
| 271U/C | 11 (23) | 87 (28) | 0.74 (0.34,1.57) | 0.39 |
| 271A | 38 (77) | 221 (72) | ||
| 209A/271UC | 8 (16) | 26 (8) | 2.12 (0.82, 5.34) | 0.08 |
Further analysis of the full-length sequences
Analysis of positions in and around the TSLE
The TSLE is a putative RNA secondary structure composed of bases 438–516 (41). Mutations in the TSLE region of HCV RNA are associated with HCC (42). Multivariable logistic regression showed that 435A/C and 481A individually increased HCC risk, and individual analysis showed that 465U was associated with increased HCC risk. We explored the possibility that the TLSE might have a 5′ extension involving base 435. M-fold identified a 10 base pair helix in which bases 427 and 435 are predicted to pair; this pair cannot form in sequences with the HCC 435 A/C polymorphism (Figure 2). R-fold Analyzer (see Methods), an automated bioinformatics tool that reports the percentage of the sequences in an alignment capable of forming predicted interactions, showed that the new helix has phylogenetic support in all three sets of full-length sequences that we analyzed: HCV HCC (n=65); HCV non-HCC (n=214); consensus sequences of genotypes 1a, 1b, 2, 3, 4, 5, and 6 (n=8). The impacts of the 435 A/C, 465U and 481A HCC polymorphisms on helical elements in the TSLE are depicted in Figure 2.
Figure 2. Analysis of two potential stem loop structures in the C-terminal region of the core gene.
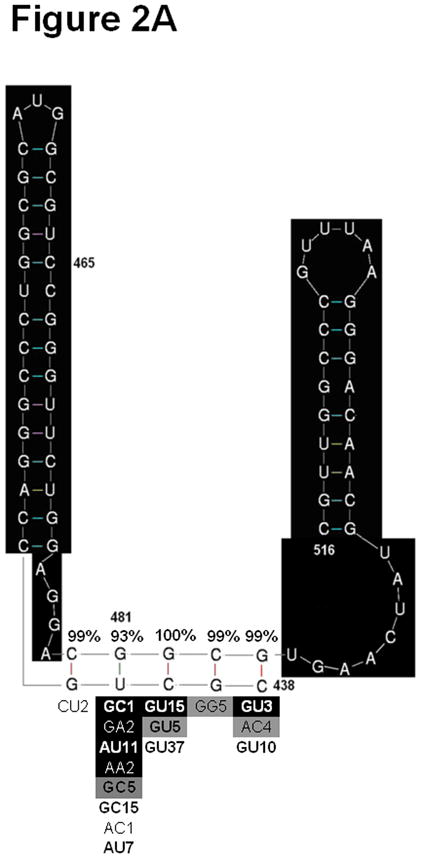
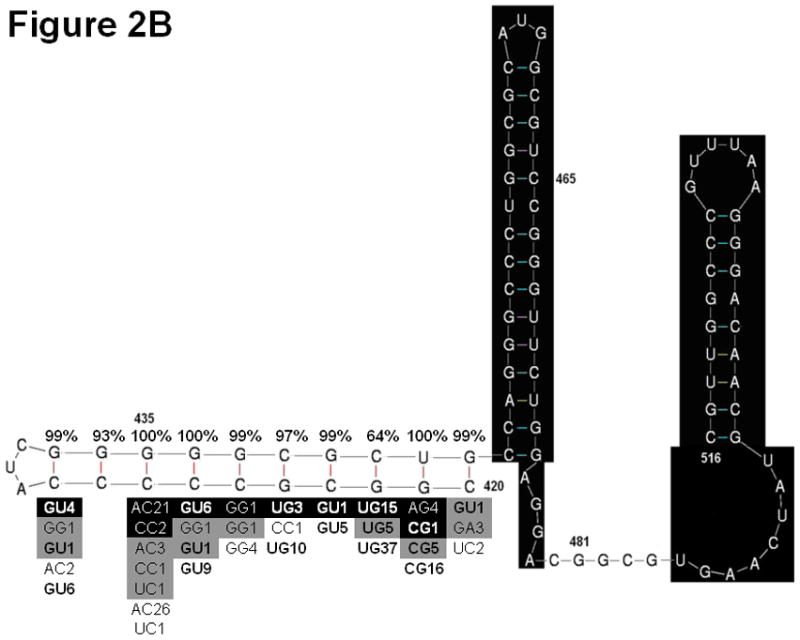
A) Original model of the TSLE. B) Modified model of the TSLE to include position 435. Sections highlighted in black are common between the two structures. Phylogenetic analysis of the original TSLE (A) and modified TSLE (B) as assessed by R-fold Analyzer are shown surrounding the helices of interest. The percentage of sequences in an alignment of HCC sequences supporting base pair formation is given above each base pair in the heix. Below the helix, alternative base-pair combinations found in an alignment of HCC sequences (black shaded), an alignment of HCC genotype consensus sequences (grey shaded) and an alignment of control sequences (unshaded) are shown. The base pairs are presented with the 3′ base on the left and the 5′ base on the right. The bold font indicates combinations of nucleotides that support Watson-Crick or wobble base pairing. The number following the base pair indicates the number of sequences observed in the alignment with the given combination of nucleotides.
Analysis of sequence diversity and complexity
We compared full-length HCV HCC and control HCV sequences to identify regions where one group of sequences was more variable than the other. The core gene, core protein, and ARFPs were individually investigated. In the first analysis, we identified nucleotides in the RNA and amino acids in the core protein and ARFP that differed from an external reference sequence, Con1 (Genbank Accesion number AJ238799), and then compared the number of substitutions per sequence between groups. The average number of nucleotide substitutions in full-length core gene sequences of patients with HCC was 22.5 ± 4.8 (mean ± S.D) and the average number of nucleotide substitutions in full-length core gene sequences of patients without HCC was 20.6 ± 5.4 (p=0.01, Student’s t-test). The average number of amino acid substitutions in ARFPs of HCC patients was 18.3 ± 3.9 versus 16.8 ± 4.3 in ARFPs of control patients(p=0.01). The average number of amino acid substitutions in the core protein did not differ between HCC and non-HCC patients. Within the RNA structural element SLV, the sequences from patients with HCC had fewer substitutions than the control sequences (0.44 ± 0.66 versus 0.72 ± 0.78; p=0.010), suggesting that the sequences from patients with HCC were under a greater selection pressure to preserve the wildtype function of this translation control element than the sequences from control patient, although the high degree of conservation of SLV makes it difficult to draw firm conclusions. The sequences from patients with HCC did not differ from sequences from control patients in the density of mutations in SLVI and SL_248, but had more mutations in the TSLE (3.50 ± 2.2 versus 2.76 ± 1.7; p=0.004). The number of sequences containing at least one mutation in the TSLE was 34/65 (52%) in the sequences from patients with HCC versus 35/214 (16%) of the sequences from the control patients (p<0.001).
We next examined the Shannon entropy and mean genetic distance. The total entropy of the HCC set was slightly higher (42.6 versus 41.0; p<0.001, Wilcoxon rank sum test). The Shannon entropy at each nucleotide position of the core gene was compared between the two groups. A Monte-Carlo randomization strategy was used to identify positions with a significant difference in entropy. In the HCC set compared to the control set, twelve positions, 28, 270, 271, 378, 384, 408, 435, 465, 472, 481, 517, 546, were more variable; and one position, 78, was less variable (Figure 3). In the sequences from patients with HCC, the mean pair-wise genetic distance for the core protein was 17% greater than in the sequences from control patients (0.027 versus 0.023; p<0.001) and for the ARFP reading frame it was 11% greater (0.063 versus 0.056; p<0.001). Taken together, these analyses show that there is a small but statistically significant increase in the variability and diversity of the sequences from patients with HCC. Countering this overall trend, SLV is more conserved and less diverse in the HCV HCC set. Of note, three of the seven HCV polymorphisms associated with HCC, 36G/C, 209A, and 309A/C, do not involve positions that have greater entropy in the HCC set than in the control set. Thus, it is unlikely that they reflect random sequence change or pressure to escape from anti-core antibodies, as both of these processes typically increase entropy.
Figure 3. Difference in Entropy between control and HCC sequences.

The difference in Shannon entropy was calculated for each position of the core gene. A monte-carlo randomization strategy was used to identify sites that differed significantly (shown in grey and labeled). A positive difference in entropy indicates more conservation (less variability) in the HCC set. A negative difference in entropy indicates more conservation in the control set.
Lack of clustering by disease status
A phylogenetic tree was generated from an alignment of all full-length HCC and control sequences. The sequences from patients with HCC did not form a separate cluster (data not shown).
Discussion
In this study, we examined HCV core gene sequences to identify features associated with HCC. All available genotype 1b core sequences were examined and those with sufficient information about the clinical status of the patient were included. The most striking result of this study is the finding that although the HCC core genes do not represent a separate clade or strain they do have characteristic mutations and other distinctive sequence features. HCC core gene sequences differed from controls at seven positions that remained associated with increased risk of HCC after controlling for country of origin. Although sequences from patients with HCC were significantly more diverse than sequences from the control patients, the actual difference in the number of mutations relative to an external consensus was small, about 8%, suggesting that the mutations in the sequences from patients with HCC do not reflect non-specific sequence drift but rather are the result of positive selection pressure. Longitudinal studies are needed to determine whether the HCC-associated polymorphisms are associated with the risk of developing HCC or with the presence of HCC.
The HCV core gene is a complex genetic region with overlapping functions. Many of the nucleotides have additional functions above coding for the amino acids of core protein including encoding the ARFPs and maintaining required RNA signals and structures. Other research will be needed to determine how the polymorphisms identified in this study contribute to the oncogenesis observed.
Seven polymorphisms were identified as having significant associations with HCC: 36G/C, 209A, 271U/C, 309A/C, 435A/C, 481A, and 546A/C. Further investigation is needed to determine whether the four polymorphisms that did not remain significantly associated with HCC in the final analysis (78U, 384U, 408U, 465U) are in fact associated with HCC. Positions of particular interest and their potential impact on HCV gene expression are discussed below.
36G/C may be involved in translation of the HCV polyprotein
The 36C/G polymorphism had an OR of 14.79 (95% CI, 2.01, 108.35) and a p-value of 0.008 in multivariable linear regression analysis of full-length sequences and an OR of 8.64 (95% CI, 1.92, 40.14) and p-value of 0.003 in the analysis of the partial sequences. Because 36C/G was associated with HCC in both sets of sequences, the association between mutations at position 36 and HCC is likely to be biologically significant. The prevalence of the A36G/C mutation was low in both the full length (5%) and partial (10%) sequences from patients with HCC, in keeping with the high level of conservation, over 95% across HCV genotypes, at this position in particular, and in the region from codons 4 to 15. Although this polymorphism may not be epidemiologically significant, the multiple roles of this position, in coding for both the core protein, (K12), ARFP (N11) and as part of the IRES (43, 44) is noteworthy, as mutations at this position may have a global impact on HCV protein production and replication.
G209A, a mutation previously linked to interferon resistance and HCC
The G209A substitution replaces the basic amino acid, arginine, with the neutral amino acid glutamine. The 209A polymorphism was present in 56% of the full-length sequences from patients with HCC sequences and in only 35% of the control sequences (p<0.001); 209A was present in 74% of the partial sequences from patients with HCC and in only 40% of the control partial sequences (p<0.001). The 209A mutation remained significantly associated with HCC in multivariate analysis of the full-length core sequences. The increased risk associated with this polymorphism was also observed in the set of partial sequences. In addition, this mutation was previously reported in 4/5 liver sequences from patients with HCC (45). These results indicate that there is a strong association between the G209A substitution and the presence of HCC.
In studies of Akuta and colleagues, the G209A polymorphism has been linked to both interferon treatment failure and HCC (46). Genotype 1b sequences obtained from the study of Viral Resistance to Antiviral Therapy of Chronic Hepatitis C (Virahep-C) (47), a prospective study of pre-treatment sequences from serum designed to assess rates of response of to peg-interferon and ribavirin therapy, were also analyzed at position 209. The 209A polymorphism was present in 4/16 (25%) of the subjects with a marked response to interferon treatment and in 12/16 of subjects with a poor response (75%) (p=0.01). Thus, the significance of 209A is heightened by its association with both interferon resistance and HCC risk. The interferon pathway’s anti-proliferative effects help protect cells from neoplastic transformation. It is important to determine if the 209A polymorphism confers interferon resistance and the pathway of this activity. This critical pathway may not only lead to treatment failure, but may also override cellular control mechanisms and contribute to cellular transformation and the development of HCC.
The significance of position 271
Further investigations are needed to determine the significance of mutations at position 271. Base 271 is part of codon 91, which encodes either leucine (271 U/C) or methionine (271A) in the genotype 1b core protein. Analysis of position 271 in over 300 sequences from Japanese patients with high viral load genotype 1b HCV showed associations between methionine-91 (271A) and interferon resistance and HCC(48). In contrast, smaller direct sequencing studies from Japan involving 28 HCC and 15 control sequences showed an association between leucine-91 (271U/C) and HCC (49, 50). In our investigation, 271U/C was associated with increased HCC risk in the full-length sequences, but not in the partial sequences. The relationship between 271U/C and HCC risk may clearer when the molecular changes that accompany mutations at position 271 are understood. Recent data indicate that the 271U/C polymorphism decreases the strength of a signal for the internal initiation of HCV protein synthesis (51). This modulation of a translation signal may be the key functional change that occurs as a result of mutations at position 271. The associated amino acid change (methionine to leucine) is highly conservative and not predicted to cause significant changes in the structure of the core protein (52).
The TSLE and positions 435, 465, and 481
The C-terminal region of the core gene contains a putative RNA structure called the TSLE originally proposed to comprise nucleotides 438–516. Three of the 11 positions identified in our individual analysis are in this region. Positions 465 and 481 alter the proposed structure of the TSLE. Our analysis of the region upstream of the TSLE, suggested that the structural model of the TSLE is improved by the addition of a third helix. In this new helix, base 435 is predicted to be base paired in 187/214 (87%) control sequences, but is only paired in 43/65 (64%) of sequences from patients with HCC due to the 435A/C polymorphism. In our set of full-length sequences, mutations that altered the TSLE were present in 34 (52%) of the sequences from patients with HCC versus 35 (16%) of the control sequences (p<0.001). In addition to the individually significant point mutations, we found that sequences from patients with HCC have a significantly, but only slightly higher, diversity in this region. This characteristic was also observed by Ogata et al., who reported a high density of mutations in this region in HCV sequences from patients with HCC (53).
Strengths and limitations of this study
In this study, we characterized all available sequences in Genbank. We examined every position in the core gene and combined multiple approaches to uncover a number of distinguishing features of sequences from patients with HCC. Since the majority of sequences in the full-length set were from Japan, we adjusted for Japanese origin in our multivariable analyses. While the high percentage of Japanese sequences may have biased our findings, two of the three mutations associated with HCC in the full-length set of sequences were also associated with HCC in the partial set of sequences, which were from Spain. This result suggests that our results have global relevance. Finally, the positions identified in this study provide an important foundation for future experiments to understand how the mutations change the biochemical properties of the core gene and confer a selective advantage. In the future, it will be important to learn whether these mutations enhance cellular proliferation and interferon resistance.
Limitations of our study include its cross-sectional design. It is possible and likely that a number of the control sequences were from patients who went on to develop HCC or who had undiagnosed HCC at the time of specimen collection. This may have lead to an underestimate of the risk caused by some sequence features and/or an underestimate of the mutations associated with HCC. Now that polymorphisms associated with HCC have been identified, it is important to determine when during the course of disease progression these mutations arise. Finally, our data set did not contain information about certain possible confounders such as age, race, sex, duration of infection, and long-range interactions between nucleotides in the core gene and in other portions of the HCV genome.
Conclusions
We identified seven polymorphisms in the HCV core gene associated with increased HCC risk in a multivariable linear regression model. Two of them, 209A and 36G/C, were also associated with increased HCC risk in an independent set of partial sequences. Many of the seven polymorphisms alter predicted RNA structures, which may indicate that these structures are under selection pressure and that they regulate processes that contribute to oncogenesis.
While many host and viral factors contribute to the development of HCC, this study identified polymorphisms in the HCV core gene that may be indicative of the presence or forthcoming development of HCC. It remains unclear whether these mutations are inherently oncogenic, or if they are benign adaptations to the altered environment that exists in damaged livers. In either case, they are associated with HCC, and thus may serve as clinical markers of HCC. Patients harboring HCV strains with these mutations may benefit from closer surveillance. Further studies are warranted to assess the diagnostic value of HCV sequencing in cirrhosis/HCC identification and to identify the molecular pathways underlying the association between certain HCV mutations and advanced liver disease and HCC.
Supplementary Material
Statement of Translational Relevance.
Our study shows that sequences of the hepatitis C virus (HCV) in patients with hepatocellular carcinoma differ from those of patients with early stage liver disease. We map sequence differences in the viral core gene, the HCV gene most strongly implicated in cellular transformation and the development of liver cancer. One polymorphism was particularly strongly associated with liver cancer. Specifically, 209A was present in 56% of the full-length sequences from patients with liver cancer, but only 35% of patients with early stage disease. Our findings contribute to information about the evolution of advanced liver disease and liver cancer in patients with chronic HCV infection. Future studies are planned to determine the prognostic significance of the sequence features we detected and to determine their impact on HCV gene expression and core protein function.
Acknowledgments
This work was supported in part by NIH grants DA016156 and DK066939 (to ADB)
Footnotes
The authors have no conflicts of interest to report
Reference List
- 1.Huibregtse JM, Beaudenon SL. Mechanism of HPV E6 proteins in cellular transformation. Semin Cancer Biol. 1996;7:317–326. doi: 10.1006/scbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 2.Franco EL, Villa LL, Ruiz A, Costa MC. Transmission of cervical human papillomavirus infection by sexual activity: differences between low and high oncogenic risk types. J Infect Dis. 1995;172:756–763. doi: 10.1093/infdis/172.3.756. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh AK, Steele R, Meyer K, Ray R, Ray RB. Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J Gen Virol. 1999;80 ( Pt 5):1179–1183. doi: 10.1099/0022-1317-80-5-1179. [DOI] [PubMed] [Google Scholar]
- 4.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 5.Sakamuro D, Furukawa T, Takegami T. Hepatitis C virus nonstructural protein NS3 transforms NIH 3T3 cells. J Virol. 1995;69:3893–3896. doi: 10.1128/jvi.69.6.3893-3896.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park JS, Yang JM, Min MK. Hepatitis C virus nonstructural protein NS4B transforms NIH3T3 cells in cooperation with the Ha-ras oncogene. Biochem Biophys Res Commun. 2000;267:581–587. doi: 10.1006/bbrc.1999.1999. [DOI] [PubMed] [Google Scholar]
- 7.Sato Y, Kato J, Takimoto R, et al. Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut. 2006;55:1801–1808. doi: 10.1136/gut.2005.070417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho JW, Baek WK, Suh SI, et al. Hepatitis C virus core protein promotes cell proliferation through the upregulation of cyclin E expression levels. Liver. 2001;21:137–142. doi: 10.1034/j.1600-0676.2001.021002137.x. [DOI] [PubMed] [Google Scholar]
- 9.Erhardt A, Hassan M, Heintges T, Haussinger D. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology. 2002;292:272–284. doi: 10.1006/viro.2001.1227. [DOI] [PubMed] [Google Scholar]
- 10.Marusawa H, Hijikata M, Chiba T, Shimotohno K. Hepatitis C virus core protein inhibits Fas- and tumor necrosis factor alpha-mediated apoptosis via NF-kappaB activation. J Virol. 1999;73:4713–4720. doi: 10.1128/jvi.73.6.4713-4720.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruggieri A, Harada T, Matsuura Y, Miyamura T. Sensitization to Fas-mediated apoptosis by hepatitis C virus core protein. Virology. 1997;229:68–76. doi: 10.1006/viro.1996.8420. [DOI] [PubMed] [Google Scholar]
- 12.Ray RB, Meyer K, Steele R, et al. Inhibition of tumor necrosis factor (TNF-alpha)-mediated apoptosis by hepatitis C virus core protein. J Biol Chem. 1998;273:2256–2259. doi: 10.1074/jbc.273.4.2256. [DOI] [PubMed] [Google Scholar]
- 13.Fukutomi T, Zhou Y, Kawai S, et al. Hepatitis C virus core protein stimulates hepatocyte growth: correlation with upregulation of wnt-1 expression. Hepatology. 2005;41:1096–1105. doi: 10.1002/hep.20668. [DOI] [PubMed] [Google Scholar]
- 14.Honda M, Kaneko S, Shimazaki T, et al. Hepatitis C virus core protein induces apoptosis and impairs cell-cycle regulation in stably transformed Chinese hamster ovary cells. Hepatology. 2000;31:1351–1359. doi: 10.1053/jhep.2000.7985. [DOI] [PubMed] [Google Scholar]
- 15.Ray RB, Lagging LM, Meyer K, Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol. 1996;70:4438–4443. doi: 10.1128/jvi.70.7.4438-4443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122:352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 17.Lemon SM, Lerat H, Weinman SA, Honda M. A transgenic mouse model of steatosis and hepatocellular carcinoma associated with chronic hepatitis C virus infection in humans. Trans Am Clin Climatol Assoc. 2000;111:146–156. [PMC free article] [PubMed] [Google Scholar]
- 18.Moriya K, Fujie H, Shintani Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 19.Branch AD, Stump DD, Gutierrez JA, Eng F, Walewski JL. The hepatitis C virus alternate reading frame (ARF) and its family of novel products: the alternate reading frame protein/F-protein, the double-frameshift protein, and others. Semin Liver Dis. 2005;25:105–117. doi: 10.1055/s-2005-864786. [DOI] [PubMed] [Google Scholar]
- 20.Smith DB, Simmonds P. Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J Mol Evol. 1997;45:238–246. doi: 10.1007/pl00006226. [DOI] [PubMed] [Google Scholar]
- 21.Tuplin A, Evans DJ, Simmonds P. Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J Gen Virol. 2004;85:3037–3047. doi: 10.1099/vir.0.80141-0. [DOI] [PubMed] [Google Scholar]
- 22.Smith DB, Simmonds P. Characteristics of nucleotide substitution in the hepatitis C virus genome: constraints on sequence change in coding regions at both ends of the genome. J Mol Evol. 1997;45:238–246. doi: 10.1007/pl00006226. [DOI] [PubMed] [Google Scholar]
- 23.Tuplin A, Evans DJ, Simmonds P. Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J Gen Virol. 2004;85:3037–3047. doi: 10.1099/vir.0.80141-0. [DOI] [PubMed] [Google Scholar]
- 24.Qin H, Shire NJ, Keenan ED, et al. HCV quasispecies evolution: association with progression to end-stage liver disease in hemophiliacs infected with HCV or HCV/HIV. Blood. 2005;105:533–541. doi: 10.1182/blood-2004-04-1452. [DOI] [PubMed] [Google Scholar]
- 25.Gimenez-Barcons M, Franco S, Suarez Y, et al. High amino acid variability within the NS5A of hepatitis C virus (HCV) is associated with hepatocellular carcinoma in patients with HCV-1b-related cirrhosis. Hepatology. 2001;34:158–167. doi: 10.1053/jhep.2001.25512. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi K, Iwata K, Matsumoto M, et al. Hepatitis C virus (HCV) genotype 1b sequences from fifteen patients with hepatocellular carcinoma: the ‘progression score’ revisited. Hepatol Res. 2001;20:161–171. doi: 10.1016/s1386-6346(00)00141-8. [DOI] [PubMed] [Google Scholar]
- 27.Nagayama K, Kurosaki M, Enomoto N, et al. Time-related changes in full-length hepatitis C virus sequences and hepatitis activity. Virology. 1999;263:244–253. doi: 10.1006/viro.1999.9924. [DOI] [PubMed] [Google Scholar]
- 28.Nagayama K, Kurosaki M, Enomoto N, et al. Characteristics of hepatitis C viral genome associated with disease progression. Hepatology. 2000;31:745–750. doi: 10.1002/hep.510310327. [DOI] [PubMed] [Google Scholar]
- 29.Ogata S, Nagano-Fujii M, Ku Y, Yoon S, Hotta H. Comparative sequence analysis of the core protein and its frameshift product, the F protein, of hepatitis C virus subtype 1b strains obtained from patients with and without hepatocellular carcinoma. J Clin Microbiol. 2002;40:3625–3630. doi: 10.1128/JCM.40.10.3625-3630.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogata S, Nagano-Fujii M, Ku Y, Yoon S, Hotta H. Comparative sequence analysis of the core protein and its frameshift product, the F protein, of hepatitis C virus subtype 1b strains obtained from patients with and without hepatocellular carcinoma. J Clin Microbiol. 2002;40:3625–3630. doi: 10.1128/JCM.40.10.3625-3630.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruster B, Zeuzem S, Krump-Konvalinkova V, et al. Comparative sequence analysis of the core- and NS5-region of hepatitis C virus from tumor and adjacent non-tumor tissue. J Med Virol. 2001;63:128–134. [PubMed] [Google Scholar]
- 32.De Mitri MS, Mele L, Chen CH, et al. Comparison of serum and liver hepatitis C virus quasispecies in HCV-related hepatocellular carcinoma. J Hepatol. 1998;29:887–892. doi: 10.1016/s0168-8278(98)80115-3. [DOI] [PubMed] [Google Scholar]
- 33.Delhem N, Sabile A, Gajardo R, et al. Activation of the interferon-inducible protein kinase PKR by hepatocellular carcinoma derived-hepatitis C virus core protein. Oncogene. 2001;20:5836–5845. doi: 10.1038/sj.onc.1204744. [DOI] [PubMed] [Google Scholar]
- 34.Horie C, Iwahana H, Horie T, et al. Detection of different quasispecies of hepatitis C virus core region in cancerous and noncancerous lesions. Biochem Biophys Res Commun. 1996;218:674–681. doi: 10.1006/bbrc.1996.0121. [DOI] [PubMed] [Google Scholar]
- 35.Alam SS, Nakamura T, Naganuma A, et al. Hepatitis C virus quasispecies in cancerous and noncancerous hepatic lesions: the core protein-encoding region. Acta Med Okayama. 2002;56:141–147. doi: 10.18926/AMO/31716. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi K, Iwata K, Matsumoto M, et al. Hepatitis C virus (HCV) genotype 1b sequences from fifteen patients with hepatocellular carcinoma: the ‘progression score’ revisited. Hepatol Res. 2001;20:161–171. doi: 10.1016/s1386-6346(00)00141-8. [DOI] [PubMed] [Google Scholar]
- 37.Nagayama K, Kurosaki M, Enomoto N, et al. Characteristics of hepatitis C viral genome associated with disease progression. Hepatology. 2000;31:745–750. doi: 10.1002/hep.510310327. [DOI] [PubMed] [Google Scholar]
- 38.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 40.Eng F, McMullan LK, Klepper A, et al. A Novel IRES in the Core-encoding Region Stimulates Production of Mini-Core, a Small Protein Comprised of the C-terminal Portion of the Core Protein. 8th International Conference on HCV and Related Viruses; Glasgow, Scotland. 2007. [Google Scholar]
- 41.Walewski JL, Gutierrez JA, Branch-Elliman W, et al. Mutation Master: profiles of substitutions in hepatitis C virus RNA of the core, alternate reading frame, and NS2 coding regions. RNA. 2002;8:557–571. doi: 10.1017/s1355838202029023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogata S, Nagano-Fujii M, Ku Y, Yoon S, Hotta H. Comparative sequence analysis of the core protein and its frameshift product, the F protein, of hepatitis C virus subtype 1b strains obtained from patients with and without hepatocellular carcinoma. J Clin Microbiol. 2002;40:3625–3630. doi: 10.1128/JCM.40.10.3625-3630.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reynolds JE, Kaminski A, Kettinen HJ, et al. Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 1995;14:6010–6020. doi: 10.1002/j.1460-2075.1995.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Honda M, Ping LH, Rijnbrand RC, et al. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology. 1996;222:31–42. doi: 10.1006/viro.1996.0395. [DOI] [PubMed] [Google Scholar]
- 45.Honda M, Kaneko S, Unoura M, Kobayashi K, Murakami S. Sequence analysis of putative structural regions of hepatitis C virus isolated from 5 Japanese patients with hepatocellular carcinoma. Arch Virol. 1993;128:163–169. doi: 10.1007/BF01309797. [DOI] [PubMed] [Google Scholar]
- 46.Akuta N, Suzuki F, Kawamura Y, et al. Amino acid substitutions in the hepatitis C virus core region are the important predictor of hepatocarcinogenesis. Hepatology. 2007;46:1357–1364. doi: 10.1002/hep.21836. [DOI] [PubMed] [Google Scholar]
- 47.Donlin MJ, Cannon NA, Yao E, et al. Pretreatment sequence diversity differences in the full-length hepatitis C virus open reading frame correlate with early response to therapy. J Virol. 2007;81:8211–8224. doi: 10.1128/JVI.00487-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akuta N, Suzuki F, Kawamura Y, et al. Amino acid substitutions in the hepatitis C virus core region are the important predictor of hepatocarcinogenesis. Hepatology. 2007;46:1357–1364. doi: 10.1002/hep.21836. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi K, Iwata K, Matsumoto M, et al. Hepatitis C virus (HCV) genotype 1b sequences from fifteen patients with hepatocellular carcinoma: the ‘progression score’ revisited. Hepatol Res. 2001;20:161–171. doi: 10.1016/s1386-6346(00)00141-8. [DOI] [PubMed] [Google Scholar]
- 50.Nagayama K, Kurosaki M, Enomoto N, et al. Characteristics of hepatitis C viral genome associated with disease progression. Hepatology. 2000;31:745–750. doi: 10.1002/hep.510310327. [DOI] [PubMed] [Google Scholar]
- 51.Eng F, Klepper A, Walewski JL, et al. Infectious HCV produces a family of mini-core proteins. Hepatology. 2008;48:754A–754A. [Google Scholar]
- 52.Boulant S, Montserret R, Hope RG, et al. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J Biol Chem. 2006;281:22236–22247. doi: 10.1074/jbc.M601031200. [DOI] [PubMed] [Google Scholar]
- 53.Ogata S, Nagano-Fujii M, Ku Y, Yoon S, Hotta H. Comparative sequence analysis of the core protein and its frameshift product, the F protein, of hepatitis C virus subtype 1b strains obtained from patients with and without hepatocellular carcinoma. J Clin Microbiol. 2002;40:3625–3630. doi: 10.1128/JCM.40.10.3625-3630.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.