

Abstract
We present a computational method for de novo identification of gene function using only cross-organismal distribution of phenotypic traits. Our approach assumes that proteins necessary for a set of phenotypic traits are preferentially conserved among organisms that share those traits. This method combines organism-to-phenotype associations,along with phylogenetic profiles,to identify proteins that have high propensities for the query phenotype; it does not require the use of any functional annotations for any proteins. We first present the statistical foundations of this approach and then apply it to a range of phenotypes to assess how its performance depends on the frequency and specificity of the phenotype. Our analysis shows that statistically significant associations are possible as long as the phenotype is neither extremely rare nor extremely common; results on the flagella,pili, thermophily,and respiratory tract tropism phenotypes suggest that reliable associations can be inferred when the phenotype does not arise from many alternate mechanisms.
The increasing number of fully sequenced genomes has made it possible to infer protein function using comparative genome techniques. Most current computational methods assign function to proteins by matching them to other proteins with known function (for review, see Bork et al. 1998); this matching has traditionally relied on sequence homology (Altschul et al. 1990), but nonhomology-based methods have also been introduced recently. The Clusters of Orthologous Groups (COGs) database (http://www.ncbi.nlm.nih.gov/COG/) is a homology-based method that establishes COGs as groups of homologs that are found in at least three major phylogenetic lineages, and enables transfer of functional information from one ortholog to the entire set of proteins within a COG (Tatusov et al. 1997). Phylogenetic profiles (Gaasterland and Ragan 1998; Pellegrini et al. 1999), gene clusters (Overbeek et al. 1999), and gene fusion analysis (Enright et al. 1999; Marcotte et al. 1999; Snel et al. 2000) are methods that can group together proteins that do not necessarily share sequence homology. Phylogenetic profiles describe the presence or absence of proteins in different genomes, and proteins with similar phylogenetic profiles are thought to share similar functions (Pellegrini et al. 1999). Gene cluster analysis (Overbeek et al. 1999; Tamames et al. 2001) infers functional relationships between genes from conservation of chromosomal proximity. Gene fusion analysis (Enright et al. 1999; Marcotte et al. 1999; Snel et al. 2000) identifies proteins that either belong to a protein complex or catalyze consecutive steps in a pathway by looking for corresponding genes that are separate in one organism, but are fused into one sequence in another. For a comparison of these nonhomology techniques see Huynen et al. (2000).
This study introduces an alternative method that infers protein function without requiring any prior functional annotation on any proteins. Instead, the method uses organism-level phenotype annotations and phylogenetic profiles to identify proteins with high propensities for a given phenotype. The method has broad applicability, as there are many well-characterized phenotypes, and phylogenetic profiles can be directly computed from sequenced genomes. A recent work (Levesque et al. 2003) described a related but different approach for predicting protein function on the basis of phenotypic traits, and applied them to identify flagellar proteins; that approach uses various settheoretic algorithms and phylogenetic information in the form of orthologous gene sets obtained from the COGs database. Another recent work (Martin et al. 2003) used clusters of phylogenetic profiles to identify proteins that differentiate Gram-positive from Gram-negative bacterial genomes.
We first present the statistical foundations of our approach. Then, we apply our method to the flagella phenotype to show that our method works better than earlier approaches that use phylogenetic profiles (Pellegrini et al. 1999; Levesque et al. 2003). In addition, we apply our approach on three new phenotypes that have not been tried previously (pili, thermophily, and respiratory tract tropism), and make novel predictions. Our analyses show that reliable associations can be inferred when the phenotype is unlikely to arise from many alternate mechanisms. As opposed to previous approaches, our method has the advantage that it can eliminate annotations that are not statistically significant; additionally, our theoretical analysis shows that phenotypes that are either extremely rare or extremely common do not permit annotations of gene function. These features are critical for general application of the approach to a wide range of phenotypes.
METHODS
Each protein in a reference organism with the phenotype of interest is analyzed by identifying whether it is preferentially conserved among organisms exhibiting the phenotype. For each protein, a BLAST search (Altschul et al. 1990) against the nonredundant (nr) database (http://www.ncbi.nih.gov/BLAST/blast_databases.html) reveals possible homologs, and a genome is considered to contain a homolog when one of its proteins has an alignment to the query protein sequence with e-value below 1.0 e-10, and when the length of the alignment is at least 2/3 the length of the query sequence (the latter requirement is useful for screening out good alignments from shorter motifs).
Once homologs in all genomes are identified, proteins are matched to the phenotype of interest as follows. The extent to which a protein i is associated with a given phenotype f is quantified by a propensity score Φf(i):
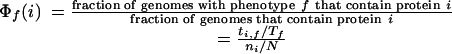 |
1 |
in which Tf is the number of genomes that exhibit phenotype f, N is the total number of genomes, ti,f is the number of genomes that both exhibit phenotype f and contain homologs to gene i, and ni is the total number of genomes that contain homologs to gene i.
The hypergeometric distribution is then used to screen out statistically insignificant protein-phenotype associations. For a given gene i, if its homologs are found in a total of ni genomes, then
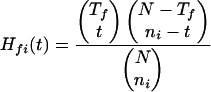 |
2 |
gives the probability that by random chance alone the gene is found in t genomes exhibiting phenotype f. The probability that a gene is found in at least ti,f genomes with phenotype f by random chance alone is 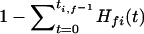 . Finally, using the conservative Bonferroni correction (Miller Jr. 1991) to account for multiple testing, the probability that some gene i is found in at least ti,f genomes with phenotype f among a set of X genes is given by
. Finally, using the conservative Bonferroni correction (Miller Jr. 1991) to account for multiple testing, the probability that some gene i is found in at least ti,f genomes with phenotype f among a set of X genes is given by
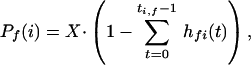 |
3 |
in which X is the number of genes in the organism whose genes we are annotating. These Pf(i) values are used for eliminating protein-phenotype associations that are not statistically significant.
Theoretical Limitations
The number of organisms exhibiting a phenotype limits the maximum propensity score. Φf(i) is maximized when gene i is found only in the target genomes (i.e., when ti,f = ni). Therefore, the maximum propensity 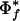 for phenotype f is:
for phenotype f is:
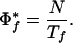 |
4 |
The number of organisms exhibiting a given phenotype also limits the statistical significance of the results. In particular, Pf(i) is minimized (most significant) when ti,f = ni = Tf, and the minimum 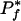 on the propensity scores for a phenotype f is:
on the propensity scores for a phenotype f is:
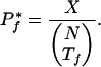 |
5 |
Equations 4 and 5 describe a trade-off between statistical significance and propensity when choosing the query phenotype. For a given number of sequenced genomes N, a smaller Tf (i.e., a more rare phenotype) will allow for higher propensity scores but at lower statistical significance limits. Intuitively, a large 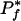 indicates that phenotype f is too rare or too common, and a small
indicates that phenotype f is too rare or too common, and a small 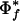 indicates that the phenotype is too common. With 86 genomes and 4000 genes,
indicates that the phenotype is too common. With 86 genomes and 4000 genes, 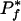 is <4.0 e-07 when 7<Tf <79(see Fig. 1).
is <4.0 e-07 when 7<Tf <79(see Fig. 1).
Figure 1.

Relationship between maximum propensity Φ and minimum estimated 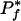 as a function of the number of organisms exhibiting phenotype f, given that there are N = 86 total genomes and that we are testing X = 4000 genes.
as a function of the number of organisms exhibiting phenotype f, given that there are N = 86 total genomes and that we are testing X = 4000 genes.
RESULTS
We apply our method to identify proteins associated with flagella, pili, thermophily, and respiratory tropism phenotypes using 86 sequenced genomes (13 archaeabacteria and 73 eubacteria) annotated for these phenotypes (see online Supplemental Material available at www.genome.org for the list of organisms and their phenotype annotations). The phenotype annotations were obtained by reading through all matching PubMed (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db-PubMed) abstracts supplemented by an exhaustive search of relevant online research Web pages; however, it is possible that a few phenotype annotations are missing in our data set. The frequency of each of these phenotypes does not preclude statistically significant associations (see Table 1), and thus, our approach is applicable.
Table 1.
Maximum Propensities and Minimum Pf Values for Flagella, Pili, Thermophily, and Respiratory Tract Tropism Phenotypes
| Phenotype f | Tf | Max Propensity Φf* | X | Min Estimated p-Value Pf* |
|---|---|---|---|---|
| Flagella | 40 | 2.15 | 4289 | 7.93e-22 |
| Pili | 14 | 6.14 | 5565 | 1.22e-12 |
| Thermophily | 15 | 5.73 | 2588 | 1.19e-13 |
| Respiratory Tract Tropism | 14 | 6.14 | 2240 | 4.93e-13 |
The values are computed with N = 86 genomes. X is the number of ORFs in the organism that we are annotating, and is used to apply the Bonferroni correction to the minimum Pf values.
Flagellar Proteins
The many annotated flagellar proteins in Escherichia coli allow us to assess the performance of our method. Additionally, previous work on this phenotype allows us to benchmark and compare our method with other approaches. Table 2 shows the 60 most statistically significant Escherichia coli genes with flagellar propensity scores >1.9(90% of the maximum flagellar propensity 2.15). This list includes 24 known flagellar genes, one putative motility gene (mbhA, b0230), and five nonflagellar genes known to be involved in chemotaxis.
Table 2.
The 60Most Statistically Significant Escherichia coli Genes With Flagellar Propensity Scores Greater Than 1.9 (90% of Max Propensity)
| Locus | Gene | Propensity | p value | t/n | Identification |
|---|---|---|---|---|---|
| b1077 | flgF | 2.15 | 2.950E-11 | 30/30 | +flagellar biosynthesis cell-proximal portion of basal-body rod |
| b1078 | flgG | 2.15 | 2.950E-11 | 30/30 | +flagellar biosynthesis cell-distal portion of basal-body rod |
| b1939 | fliG | 2.15 | 2.950E-11 | 30/30 | +flagellar biosynthesis, component of motor switch and energizing |
| b0229 | fhiA | 1.97 | 5.730E-10 | 33/36 | +polar flagellar assembly protein |
| b1879 | flhA | 1.95 | 8.642E-08 | 30/33 | +flagellar biosynthesis; possible export of flagellar proteins |
| b1880 | flhB | 1.97 | 5.730E-10 | 33/36 | +putative part of export apparatus for flagellar proteins |
| b1948 | fliP | 1.97 | 5.730E-10 | 33/36 | +flagellar biosynthesis protein flip |
| b1938 | fliF | 2.15 | 7.081E-10 | 28/28 | +flagellar biosynthesis; basal-body membrane ring and collar protein |
| b1074 | flgC | 2.08 | 7.231E-10 | 30/31 | +flagellar biosynthesis cell-proximal portion of basal-body rod |
| b1941 | fliI | 1.92 | 4.597E-09 | 33/37 | +flagellum-specific ATPase |
| b1076 | flgE | 2.15 | 1.378E-08 | 26/26 | +flagellar biosynthesis hook protein |
| b1950 | fliR | 2.15 | 1.378E-08 | 26/26 | +flagellar biosynthesis |
| b1884 | cheR | 2.01 | 4.494E-08 | 29/31 | response regulator for chemotaxis; protein glutamate methyltransferase |
| b1887 | cheW | 2.15 | 5.606E-08 | 25/25 | positive regulator of CheA protein activity |
| b1945 | fliM | 2.15 | 5.605E-08 | 25/25 | +flagellar biosynthesis component of motor switch |
| b1080 | flgI | 2.15 | 2.172E-07 | 24/24 | +homolog of Salmonella P-ring of flagella basal body |
| b1883 | cheB | 2.00 | 8.063E-07 | 27/29 | response regulator for chemotaxis (cheA sensor); protein methylesterase |
| b1888 | cheA | 2.07 | 1.113E-06 | 25/26 | sensory transducer kinase between chemo- signal receptors and CheB and CheY |
| b1079 | flgH | 2.15 | 2.863E-06 | 22/22 | +flagellar biosynthesis basal-body outer-membrane L ring protein |
| b1082 | flgK | 2.15 | 9.791E-06 | 21/21 | +flagellar biosynthesis hook-filament junction protein 1 |
| b1890 | motA | 2.15 | 3.187E-04 | 18/18 | +proton conductor component of motor; no effect on switching |
| b0230 | mbhA | 2.15 | 9.560E-04 | 17/17 | * putative motility protein |
| b1889 | motB | 2.15 | 9.560E-04 | 17/17 | +enables flagellar motor rotation, linking torque machinery to cell wall |
| b1924 | fliD | 2.15 | 9.560E-04 | 17/17 | +flagellar biosynthesis; filament capping protein; enables filament assembly |
| b0316 | yahB | 2.15 | 2.789E-03 | 16/16 | putative transcriptional regulator LYSR-type |
| b1659 | ydhB | 2.15 | 2.789E-03 | 16/16 | putative transcriptional regulator LYSR-type |
| b1339 | ydaK | 1.95 | 1.062E-02 | 19/21 | putative transcriptional regulator LYSR-type |
| b0504 | ybbS | 2.03 | 1.179E-02 | 17/18 | putative transcriptional regulator LYSR-type |
| b3105 | yhaJ | 2.02 | 3.211E-02 | 16/17 | putative transcriptional regulator LYSR-type |
| b1925 | fliS | 2.15 | 2.789E-03 | 16/16 | +flagellar biosynthesis; repressor of class 3a and 3b operons (RflA activity) |
| b1946 | fliN | 2.15 | 2.789E-03 | 16/16 | +flagellar biosynthesis, component of motor switch and energizing |
| b0494 | tesA | 2.04 | 4.199E-03 | 18/19 | acyl-CoA thioesterase I; also functions as protease I |
| b0387 | yaiI | 2.15 | 7.921E-03 | 15/15 | orf, hypothetical protein |
| b2213 | ada | 2.15 | 7.921E-03 | 15/15 | O6-methylguanine-DNA methyltransferase; transcription activator/repressor |
| b2681 | 2.15 | 7.921E-03 | 15/15 | putative transport protein | |
| b3686 | glnG | 2.15 | 7.921E-03 | 15/15 | response regulator for gln |
| b3687 | ibpA | 2.15 | 7.921E-03 | 15/15 | heat shock protein |
| b3081 | ygjL | 1.95 | 1.062E-02 | 19/21 | putative NADPH dehydrogenase |
| b0898 | ycaD | 2.03 | 1.179E-02 | 17/18 | putative transport |
| b4119 | melA | 2.03 | 1.179E-02 | 17/18 | alpha-galactosidase |
| b1542 | ydfI | 1.94 | 2.896E-02 | 18/20 | putative oxidoreductase |
| b2172 | yeiQ | 1.94 | 2.896E-02 | 18/20 | putative oxidoreductase |
| b4323 | uxuB | 1.94 | 2.896E-02 | 18/20 | D-mannonate oxidoreductase |
| b1521 | uxaB | 2.02 | 3.211E-02 | 16/17 | altronate oxidoreductase |
| b3356 | yhfA | 1.94 | 2.896E-02 | 18/20 | orf, hypothetical protein |
| b3924 | fpr | 1.94 | 2.896E-02 | 18/20 | ferredoxin-NADP reductase |
| b1256 | yciD | 2.02 | 3.211E-02 | 16/17 | putative outer membrane protein |
| b1813 | yeaB | 2.02 | 3.211E-02 | 16/17 | orf, hypothetical protein |
| b2069 | yegD | 2.02 | 3.211E-02 | 16/17 | putative heat shock protein |
| b3775 | ppiC | 2.02 | 3.211E-02 | 16/17 | peptidyl-prolyl cis-trans isomerase C (rotamase C) |
| b0610 | rnk | 2.15 | 5.931E-02 | 13/13 | regulator of nucleoside diphosphate kinase |
| b1075 | flgD | 2.15 | 5.931E-02 | 13/13 | +flagellar biosynthesis, initiation of hook assembly |
| b1083 | flgL | 2.15 | 5.931E-02 | 13/13 | +flagellar biosynthesis; hook-filament junction protein |
| b1688 | 2.15 | 5.931E-02 | 13/13 | orf, hypothetical protein | |
| b2922 | yggE | 2.15 | 5.931E-02 | 13/13 | putative actin |
| b3010 | yqhC | 2.15 | 5.931E-02 | 13/13 | putative ARAC-type regulatory protein |
| b3328 | hofG | 2.15 | 5.931E-02 | 13/13 | putative general protein secretion protein |
| b4355 | tsr | 2.15 | 5.931E-02 | 13/13 | methyl-accepting chemotaxis protein I, serine sensor receptor |
| b1073 | flgB | 2.02 | 8.488E-02 | 15/16 | +flagellar biosynthesis, cell-proximal portion of basal-body rod |
The genes marked with + in the identification column are know flagellar genes. t is the number of flagellar bacteria that contain homologs to the gene, and n is the total number of genomes that contain homologs to the gene. Genes in adjacent shaded rows are paralogs.
The list in Table 2 contains 12 additional known flagellar genes that were not identified using the original phylogenetic profile approach described in Pellegrini et al. (1999). This list also includes all of the genes already identified in that approach except for fliQ, which has a propensity score of 2.15, but is not included in Table 2 because its Pflagella value is not significant. Note that the original phylogenetic profile approach does not use phenotype information and instead transfers functional annotations between proteins with similar profiles.
We also compared our method to the Similarity Measure (Levesque et al. 2003) method. To make direct comparisons, we applied this method to the 86 genomes considered here. At a similarity threshold of 0.65, the least restrictive cutoff used in Levesque et al. (2003), their method identified 12 known flagellar genes; all of them are a subset of the 24 identified by our approach.
Both the Similarity Measure and our method have similar performance when applied to the COGs data with the 21 genomes considered in Levesque et al. (2003); each identifies 29 known flagellar genes, among 34 top scoring genes for the Similarity Measure algorithm and 31 top scoring genes for our method.
More rigorously, the Receiver Operating Characteristic (ROC) curves in Figure 2 compare the sensitivity versus specificity tradeoffs when all three approaches are applied to the 86 genomes considered here. These curves show that our approach consistently produces fewer false positives at each level of sensitivity. It is important to note that the false-positive rates in Figure 2 are upper bounds, because we cannot assume that all flagellar proteins have been annotated (i.e., some of the putative false positives may be flagellar proteins). Figure 2 also shows that propensity scores can be used to improve performance independently of the estimated P values. At high specificity, the ROC curves improve (move closer to the upper, left corner) as we increase the propensity cutoffs from 1 to 1.8. Larger propensity cutoffs increase the number of false negatives, and eventually at cutoffs ≥2.0, the flagella ROC curves begin to worsen.
Figure 2.

Receiver Operating Characteristic (ROC) curves comparing our approach with the approaches of Levesque et al. (2003) and Pellegrini et al. (1999) on the same flagella data set. Each ROC curve for our approach is obtained by keeping all genes with propensity scores greater than a fixed cutoff and varying the P-value cutoffs. The ROC curve for the Levesque et al. (2003) approach is obtained by varying the similarity threshold cutoff. The ROC curve for the Pellegrini et al. (1999) approach is obtained by comparing phylogenetic profiles against the FlgL gene (used in their study) and varying the Manhattan distance cutoff.
Proteins Associated With Pili
Pili are another structural feature of some bacteria for which some of the component proteins are known. Table 3 shows the 40 most statistically significant Pseudomonas aeruginosa proteins with propensity scores >4.5 for organisms that have pili (Sauer et al. 2000). Five of the seven known proteins in this list are known fimbrial biogenesis proteins (pilA, pilN, pilO, pilP, and pilQ); their corresponding Bonferroni corrected Pf values are <0.109, with three of these five having Pf values <0.05.
Table 3.
The 40Most Statistically Significant Pseudomonas aeruginosa Genes With Pili Propensity Scores Greater Than 4.5.
| Locus | Gene | Propensity | p value | t/n | Identification |
|---|---|---|---|---|---|
| PA0454 | 5.27 | 2.87E-07 | 12/14 | conserved hypothetical protein | |
| P3651 | cdsA | 5.20 | 6.01E-06 | 11/13 | phosphatidate cytidylyltransferase |
| PA4525 | pilA | 6.14 | 2.42E-05 | 9/9 | type 4 fimbrial precursor PilA |
| PA0618 | 6.14 | 2.42E-05 | 9/9 | probable bacteriophage protein | |
| PA0619 | 6.14 | 2.42E-05 | 9/9 | probable bacteriophage protein | |
| PA3020 | 4.83 | 2.69E-05 | 11/14 | probable soluble lytic transglycosylase | |
| PA0936 | 6.14 | 3.15E-04 | 8/8 | hypothetical protein | |
| PA4512 | 4.91 | 1.23E-02 | 8/10 | hypothetical protein | |
| PA4115 | 5.03 | 1.18E-03 | 9/11 | conserved hypothetical protein | |
| PA3235 | 5.46 | 2.64E-03 | 8/9 | conserved hypothetical protein | |
| PA0209 | 6.14 | 3.56E-03 | 7/7 | conserved hypothetical protein | |
| PA0616 | 6.14 | 3.56E-03 | 7/7 | hypothetical protein | |
| PA0617 | 6.14 | 3.56E-03 | 7/7 | probable bacteriophage protein | |
| PA0622 | 6.14 | 3.56E-03 | 7/7 | probable bacteriophage protein | |
| PA0623 | 6.14 | 3.56E-03 | 7/7 | probable bacteriophage protein | |
| PA1376 | aceK | 4.91 | 1.23E-02 | 8/10 | isocitrate dehydrogenase kinase/phosphatase |
| PA5043 | pilN | 4.91 | 1.23E-02 | 8/10 | type 4 fimbrial biogenesis protein PilN |
| PA5040 | pilQ | 4.91 | 1.23E-02 | 8/10 | type 4 fimbrial biogenesis protein PilQ |
| PA0289 | 4.91 | 1.23E-02 | 8/10 | probable transcriptional regulator | |
| PA1009 | 4.91 | 1.23E-02 | 8/10 | hypothetical protein | |
| PA1661 | 4.91 | 1.23E-02 | 8/10 | hypothetical protein | |
| PA4476 | 4.91 | 1.23E-02 | 8/10 | hypothetical protein | |
| PA4970 | 4.91 | 1.23E-02 | 8/10 | conserved hypothetical protein | |
| PA5225 | 4.91 | 1.23E-02 | 8/10 | hypothetical protein | |
| PA0461 | 5.38 | 2.62E-02 | 7/8 | conserved hypothetical protein | |
| PA0834 | 5.38 | 2.62E-02 | 7/8 | conserved hypothetical protein | |
| PA0612 | 5.38 | 2.62E-02 | 7/8 | hypothetical protein | |
| PA0628 | 5.38 | 2.62E-02 | 7/8 | conserved hypothetical protein | |
| PA4879 | 5.38 | 2.62E-02 | 7/8 | conserved hypothetical protein | |
| PA2017 | 6.14 | 3.56E-02 | 6/6 | hypothetical protein | |
| PA3209 | 6.14 | 3.56E-02 | 6/6 | conserved hypothetical protein | |
| PA5536 | 6.14 | 3.56E-02 | 6/6 | conserved hypothetical protein | |
| PA0080 | 4.78 | 1.09E-01 | 7/9 | hypothetical protein | |
| PA0502 | 4.78 | 1.09E-01 | 7/9 | probable biotin biosynthesis protein bioH | |
| PA1727 | 4.78 | 1.09E-01 | 7/9 | conserved hypothetical protein | |
| PA3726 | 4.78 | 1.09E-01 | 7/9 | conserved hypothetical protein | |
| PA4605 | 4.78 | 1.09E-01 | 7/9 | conserved hypothetical protein | |
| PA4777 | 4.78 | 1.09E-01 | 7/9 | probable two-component sensor | |
| PA5041 | pilP | 4.78 | 1.09E-01 | 7/9 | type 4 fimbrial biogenesis protein PilP |
| PA5042 | pilO | 4.78 | 1.09E-01 | 7/9 | type 4 fimbrial biogenesis protein PilO |
t is the number of organisms with pili that contain homologs to the gene, and n is the total number of genomes that contain homologs to the gene. Genes in adjacent shaded rows are paralogs.
Proteins Associated With Thermophily
Thermoanaerobacter tengcongensis is an anaerobic thermophilic eubacterium whose genome was sequenced recently (Bao et al. 2002). How thermophiles have adapted to survive at high temperatures is not fully understood. Radiation sensitivity studies indicate that thermophiles repair DNA efficiently, but sequencing results suggest that many of their DNA repair genes are still unrecognized because they are too different from those of well-studied organisms (Grogan 1998). Here, we use our method to uncover the 40 most statistically significant T. tengcongensis genes with thermophily propensity scores >3.0 (Table 4). This list includes three DNA repair genes, one of which is reverse gyrase. Reverse gyrase is the only known topoisomerase that induces positive supercoiling in DNA, and hence, improves DNA stability at high temperatures (Forterre et al. 2000). This list also includes nine components of ferredoxin oxidoreductase. Anaerobic metabolism involving ferredoxin oxidoreductase appears to be unique to hyperthermophiles (Kelly and Adams 1994), and oxidoreductases related to hydrogen evolution have been shown recently to be crucial in the central metabolism of hyperthermophiles, replacing dehydrogenases in many key steps of metabolism (Borges et al. 1996). In addition, the recent isolation of a strain of microorganisms from hydrothermal vents that use Fe(III) as the electron acceptor and can grow at 121°, suggests that Fe(III) reduction may be an important process for growing in hydrothermal environments (Kashefi and Lovely 2003). Altogether, Table 4 identifies at least 21 genes that may be associated with thermophily: three DNA repair genes, nine ferredoxin oxidoreductase genes, and nine additional hypothetical genes that currently have unknown function.
Table 4.
The 40Most Statistically Significant T. tengcongensis Proteins With Thermophily Propensity Scores Greater Than 3.0
| Locus | Gene | Propensity | p value | t/n | Identification |
|---|---|---|---|---|---|
| TTE2470 | MesJ4 | 5.38 | 2.40E-12 | 15/16 | predicted ATPase of the PP-loop superfamily implicated in cell cycle control |
| TTE0073 | MesJ | 5.06 | 1.65E-11 | 15/17 | predicted ATPase of the PP-loop superfamily implicated in cell cycle control |
| TTE0285 | 5.73 | 9.29E-12 | 14/14 | conserved hypothetical protein | |
| TTE1955 | PflA2 | 4.78 | 9.78E-11 | 15/18 | Pyruvate-formate lysase-activating enzyme (protein modification & repair) |
| TTE0474 | Gcd14 | 4.30 | 1.84E-09 | 15/20 | predicted SAM-dependent methyltransferase involved in tRNA-Met maturation |
| TTE1745 | rgy | 5.73 | 7.71E-09 | 12/12 | Reverse gyrase (DNA replication, recombination, and repair) |
| TTE1895 | SmtA4 | 5.29 | 9.63E-08 | 12/13 | SAM-dependent methyltransferases |
| TTE2198 | PorA6 | 3.58 | 1.55E-07 | 15/24 | ferredoxin oxidoreductase, alpha subunit (anaerobic metabolism) |
| TTE1209 | PorA3 | 3.34 | 1.46E-05 | 14/24 | ferredoxin oxidoreductase, alpha subunit (anaerobic metabolism) |
| TTE1340 | PorA4 | 3.34 | 1.46E-05 | 14/24 | ferredoxin oxidoreductase, alpha subunit (anaerobic metabolism) |
| TTE0961 | PorA2 | 3.39 | 1.22E-04 | 13/22 | 2-oxoacid ferredoxin oxidoreductase, alpha subunit (fermentation) |
| TTE1354 | SpeD | 4.01 | 3.05E-07 | 14/20 | S-adenosylmethionine decarboxylase (polyamine biosynthesis) |
| TTE1210 | PorB2 | 3.44 | 3.88E-07 | 15/25 | ferredoxin oxidoreductase, beta subunit (anaerobic metabolism) |
| TTE1341 | PorB3 | 3.44 | 3.88E-07 | 15/25 | ferredoxin oxidoreductase, beta subunit (anaerobic metabolism) |
| TTE1276 | Nfo | 4.91 | 6.50E-07 | 12/14 | Endonuclease IV (DNA degradation) |
| TTE1779 | PflX | 4.85 | 1.02E-05 | 11/13 | pyruvate formate lyase activating enzyme (protein modification and repair) |
| TTE0960 | PorB | 3.34 | 1.46E-05 | 14/24 | 2-oxoacid ferredoxin oxidoreductase, beta subunit (fermentation) |
| TTE1571 | 3.73 | 2.01E-05 | 13/20 | conserved hypothetical protein | |
| TTE2189 | 3.24 | 2.72E-04 | 13/23 | conserved hypothetical protein | |
| TTE2193 | PorG3 | 4.50 | 4.50E-05 | 11/14 | indolepyruvate ferredoxin oxidoreductase, beta subunit |
| TTE1537 | HypE3 | 3.09 | 6.99E-05 | 14/26 | Hydrogenase maturation factor (electron transport) |
| TTE2532 | 3.82 | 1.13E-04 | 12/18 | predicted Zn-dependent hydrolases of beta-lactamase fold | |
| TTE0444 | 5.73 | 3.13E-04 | 8/8 | conserved hypothetical protein | |
| TTE1518 | LigT | 5.73 | 3.13E-04 | 8/8 | 3-5 RNA Ligase (cell envelope: synthesis of murein sacculus & peptidoglycan) |
| TTE0818 | GltB | 4.69 | 1.34E-03 | 9/11 | Glutamate synthase domain 3 (methanogenesis) |
| TTE1866 | 3.28 | 1.58E-03 | 12/21 | conserved hypothetical protein | |
| TTE0714 | 5.10 | 2.59E-03 | 8/9 | putative integrase-resolvase (DNA replication, recombination, repair) | |
| TTE2659 | 5.10 | 2.59E-03 | 8/9 | putative RecB family exonuclease | |
| TTE0820 | GltB3 | 5.73 | 3.11E-03 | 7/7 | amidophophoribosyltransferase (purine ribonucleotide synthesis) |
| TTE1891 | 5.73 | 3.11E-03 | 7/7 | MinD P-loop ATPase containing an inserted ferredoxin domain (electron transport) | |
| TTE1892 | 5.73 | 3.11E-03 | 7/7 | MinD P-loop ATPase containing an inserted ferredoxin domain (electron transport) | |
| TTE1893 | 5.73 | 3.11E-03 | 7/7 | conserved hypothetical protein | |
| TTE1898 | 5.73 | 3.11E-03 | 7/7 | predicted methyltransferases | |
| TTE2657 | 5.73 | 3.11E-03 | 7/7 | conserved hypothetical protein | |
| TTE0705 | 4.59 | 1.20E-02 | 8/10 | putative integrase-resolvase (DNA replication, recombination, repair) | |
| TTE0715 | 4.59 | 1.20E-02 | 8/10 | predicted transposase | |
| TTE1551 | Dap2 | 3.97 | 1.50E-02 | 9/13 | putative acyl-peptide hydrolase |
| TTE2658 | 3.97 | 1.50E-02 | 9/13 | conserved hypothetical protein | |
| TTE2633 | 5.02 | 2.26E-02 | 7/8 | conserved hypothetical protein | |
| TTE2194 | PorA5 | 3.37 | 2.72E-02 | 10/17 | indolepyruvate ferredoxin oxidoreductase, alpha subunit |
t is the number of thermophiles that contain homologs to the protein, and n is the total number of genomes that contain homologs to the protein. Genes in adjacent shaded rows are paralogs.
Proteins Associated With Respiratory Tract Tropism
We identified 14 bacteria with respiratory tract tropism, and used this list to compute respiratory tract tropism propensity scores for Streptococcus pneumoniae genes. In this case, there are no genes with statistically significant propensities for the respiratory tract tropism phenotype – none of the top propensity scores have Pf values <2. Perhaps this is because the phenotype description is too general, as bacterial tropism is known to involve a wide variety of mechanisms that include immune evasion, metabolic adaptation, and physical attachment and invasion. The lack of statistically significant associations indicates that respiratory tropism is difficult to study as a single phenotype, at least using our method.
DISCUSSION
We have described an approach that combines organism-to-phenotype associations along with phylogenetic profiles to identify proteins with high propensities for a given phenotype; such an approach can be used to annotate proteins with phenotype information. We validated this approach by demonstrating its ability to identify known flagellar and pili proteins, and then applied it to the identification of proteins associated with thermophily.
Phenotype annotations are usually more general than traditional protein functional annotations; typically, several proteins spanning multiple functional complexes and pathways contribute to a given phenotype, and the same phenotype can be accomplished in more than one way. Correspondingly, we have found that it is insufficient to simply search for proteins that are conserved in a majority of the organisms exhibiting the query phenotype. For example, none of the identified flagellar proteins are conserved in all 40 flagellar genomes, and most of them are conserved in 20 or fewer flagellar genomes. By using propensity scores, our approach is able to match proteins to phenotype without requiring that the proteins be conserved in a majority of the organisms with that phenotype.
Proteins with the same propensity scores can have very different phylogenetic profiles, and therefore, it is unlikely that a single representative protein can be used to match and identify the set of proteins responsible for a phenotype. Figure 3 shows the average Hamming distance between phylogenetic profiles of E. Coli proteins at each flagellar propensity level. The average Hamming distance between the phylogenetic profiles of the proteins with highest flagellar propensity scores is 4.0, whereas proteins with lower propensity scores can have Hamming distances >30. In addition, Figure 4 depicts the hierarchical clustering of the top proteins associated with flagella5 and thermophily (as given in Tables 2 and 4), and shows that the phylogenetic profiles of the top proteins can vary considerably. Hence, even if it is possible to identify a representative protein for a given phenotype (e.g., as in Pellegrini et al. 1999), it is not possible to find all relevant proteins by simply searching for other proteins with similar phylogenetic profiles. Our approach is robust against these large distances between phylogenetic profiles, because it uses propensity scores as opposed to raw phylogenetic profiles.
Figure 3.

Average phylogenetic distances between E. Coli proteins at each flagellar propensity level.
Figure 4.

Hierarchical clustering (average-linkage) of the top proteins associated with flagella and thermophily (see Tables 2 and 4), on the basis of their phylogenetic profiles. Genomes are on the x-axis, and genes are on the y-axis. Gray coloring indicates the presence of a gene in a genome.
An artifact of previous phylogenetic comparison approaches is that distances between phylogenetic profiles are sensitive to the size of the set of background genomes. For example, arbitrarily expanding the set of background genomes usually increases the distances between phylogenetic profiles. In our approach, this scaling relationship is automatically captured by propensity scores, and expanding the set of background genomes will, in general, increase the statistical significance (i.e., lower Pf values) of the top proteins. Follow-up work along these lines should address evolutionary distances between species; it is not obvious how to handle statistical significance in an analytical way, and nonparametric approaches may be more promising in this regard.
These initial results are encouraging, and provide a statistical framework for the general application of the approach to a large class of well-characterized phenotypes. This process might begin by looping through organism phenotype annotations and computing their 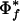 and
and 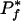 scores in order to filter out phenotypes that are too common or too rare, and then match the remaining phenotypes to individual proteins by checking each protein's propensity for that phenotype. With the rapidly increasing pace of whole-genome sequencing, and the commensurate accumulation of novel genes, approaches such as ours can efficiently generate high-yield hypotheses for experimental validation of gene function. In this regard, whole-organism characterization of phenotypic traits may become a central activity in the post-genomic approach to understanding biological networks.
scores in order to filter out phenotypes that are too common or too rare, and then match the remaining phenotypes to individual proteins by checking each protein's propensity for that phenotype. With the rapidly increasing pace of whole-genome sequencing, and the commensurate accumulation of novel genes, approaches such as ours can efficiently generate high-yield hypotheses for experimental validation of gene function. In this regard, whole-organism characterization of phenotypic traits may become a central activity in the post-genomic approach to understanding biological networks.
Acknowledgments
We thank the anonymous referees for many helpful suggestions. M.S. is supported in part by NSF PECASE award MCB-0093399 and DARPA grant MDA972-00-1-0031. S.T. is supported in part by NSF CAREER award MCB-0133750 and DARPA grant N66001-02-1-8929.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1586704.
Footnotes
References
- Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman, D. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Bao, Q., Tian, Y., Li, W., Xu, Z., Xuan, Z., Hu, S., Dong, W., Yang, J., Chen, Y., Xue, Y., et al. 2002. A complete sequence of the T. tengcongensis genome. Genome Res. 12: 689-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges, K.M., Brummet, S.R., Bogert, A., Davis, M.C., Hujer, K.M., Domke, S.T., Szasz, J., Ravell, J., DiRuggiero, J., Fuller, C., et al. 1996. A survey of the genome of the hyperthermophilic archaeon, pyrococcus furiosus. Genome Sci. Technol. 1: 37-46. [Google Scholar]
- Bork, P., Dandekar, T., Diaz-Lazcoz, Y., Eisenhaber, F., Huynen, M., and Yuan, Y. 1998. Predicting function: From genes to genomes and back. J. Mol. Biol. 283: 707-725. [DOI] [PubMed] [Google Scholar]
- Enright, A.J., Iliopoulos, I., and Kyrpides, N.C. 1999. Protein interaction maps for complete genomes based on gene fusion events. Nature 402: 86-90. [DOI] [PubMed] [Google Scholar]
- Forterre, P., de la Tour, C.B., Philippe, H., and Duguet, M. 2000. Reverse gyrase from hyperthermophiles: Probable transfer of a thermoadaptation trait from archaea to bacteria. Trends Genet. 16: 152-154. [DOI] [PubMed] [Google Scholar]
- Gaasterland, T. and Ragan, M. 1998. Constructing multigenome views of whole microbial genomes. Microbiol. Comp. Genomics 3: 177-192. [DOI] [PubMed] [Google Scholar]
- Grogan, D.W. 1998. Hyperthermophiles and the problem of DNA instability. Mol. Microbiol. 28: 1043-1050. [DOI] [PubMed] [Google Scholar]
- Huynen, M., Snel III, B., Lathe, W., and Bork, P. 2000. Predicting protein function by genomic context: Quantitative evaluation and qualitative inferences. Genome Res. 10: 1024-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashefi, K. and Lovley, D.R. 2003. Extending the upper temperature limit for life. Science 301: 934. [DOI] [PubMed] [Google Scholar]
- Kelly, R.M. and Adams, M.W.W. 1994. Metabolism in hyperthermophilic microorganisms. Antonie van Leeuvenhook 66: 247-270. [DOI] [PubMed] [Google Scholar]
- Levesque, M., Shasha, D., Kim, W., Surette, M.G., and Benfey, P.N. 2003. Trait-to-gene: A computational method for predicting the function of uncharacterized genes. Curr. Biol. 13: 129-133. [DOI] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Ng, H.-L., Rice, D.W., Yeates, T.O., and Eisenberg, D. 1999. Detecting protein function and protein–protein interactions from genome sequences. Science 285: 751-753. [DOI] [PubMed] [Google Scholar]
- Martín, M.J., Herrero, J., Mateos, A., and Dopazo, J. 2003. Comparing bacterial genomes through conservation profiles. Genome Res. 13: 991-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Jr., R.G. 1991. Simultaneous statistical inference. In Springer series in statistics Springer-Verlag, New York.
- Overbeek, R., Fonstein, M., D'Souza, M., Pusch, G., and Maltsev, N. 1999. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. 96: 2896-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini, M., Marcotte, E.M., Thompson, M.J., Eisenberg, D., and Yeates, T.O. 1999. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. 96: 4285-4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer, F., Barnhart, M., Choudhury, D., Knight, S., Waksman, G., and Hultgren, S. 2000. Chaperone-assisted pilus assembly and bacterial attachment. Curr. Opin. Struc. Biol. 10: 548-556. [DOI] [PubMed] [Google Scholar]
- Snel, B., Bork, P., and Huynen, M. 2000. Genome evolution: Gene fusion versus gene fission. Trends Genet. 16: 9-11. [DOI] [PubMed] [Google Scholar]
- Tamames, J., González-Moreno, M., Mingorance, J., Valencia, A., and Vicente, M. 2001. Bringing gene order into bacterial shape. Trends Genet. 17: 124-126. [DOI] [PubMed] [Google Scholar]
- Tatusov, R.L., Koonin, E.V., and Lipman, D.J. 1997. A genomic perspective on protein families. Science 278: 631-637. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.ncbi.nlm.nih.gov/COG/; COGs database.
- http://www.ncbi.nih.gov/BLAST/blast_databases.html; NCBI non-redundant peptide sequence database.
- http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=PubMed; PubMed.