

Abstract
A highly efficient enantioselective C–H insertion of azavinyl carbenes into unactivated alkanes has been developed. These transition metal carbenes are directly generated from readily available and stable 1-sulfonyl-1,2,3-triazoles in the presence of chiral Rh(II) carboxylates and are used for C–H functionalization of alkanes to access a variety of β-chiral sulfonamides.
Direct and chemo– and stereoselective C–H functionalization of alkanes remains a formidable challenge for synthetic organic chemists due to the inherently poor reactivity of C–H bonds. The consequent use of highly reactive species imposes inherent limitations on both chemo- and stereoselectivity of the reactions. Intermolecular insertions of highly reactive metal-bound carbenes into sp3 C–H bonds of unactivated alkanes have emerged as promising means for functionalization of these seemingly inert compounds, and excellent results have been obtained with rhodium(II)-stabilized donor/acceptor carbenes.1 However, this strategy has been mainly limited to rhodium(II) carbenes i derived from diazoesters 1 (eq 1).2
We have recently demonstrated that readily available 1-sulfonyl triazoles 3 can serve as direct precursors to related azavinyl carbenes ii (eq 2).3 The latter share many reactivity features of the well-studied carbenes i, and can be efficiently used to introduce an aldehyde or amino group into organic molecules.3b,c Herein we report the first Rh(II)-catalyzed enantioselective intermolecular C–H insertion of azavinyl carbenes ii, derived from 1-sulfonyl-1,2,3-triazoles 3, to access a variety of chiral N-sulfonylamines 5 in good yield and high enantioselectivity (eq 2).
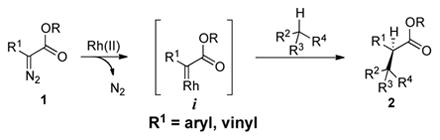 |
(1) |
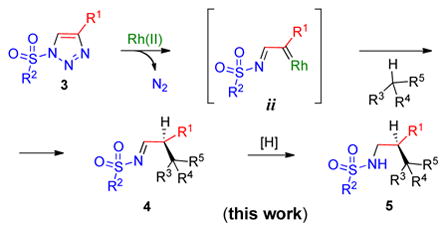 |
(2) |
Our recent success in the development of the rhodium-catalyzed asymmetric cyclopropanation reaction3b largely capitalized on utilizing 1-(alkylsulfonyl) triazoles in combination with the highly efficient Rh2(S-NTTL)44 complex 6 (Figure 1). Accordingly, we attempted enantioselective functionalization of the secondary C–H bonds of cyclohexane using 1-(methanesulfonyl)-substituted triazole 3a in the presence of 0.5 mol % of said catalyst (eq 3). We were pleased to find that the corresponding C–H insertion product, chiral imine 4a, was formed in excellent yield at ambient temperature using a 1:1 (v/v) mixture of the alkane and chloroform (eq 3).5
Figure 1.

Chiral Rh(II) carboxylates used in this study.
The attempted hydrolysis of 4a using K2CO3 in wet methanol delivered the corresponding carbaldehyde 8a as a racemic mixture, presumably due to high acidity of the hydrogen at the stereocenter. In contrast, one-pot treatment of the intermediate imine with sodium borohydride or lithium aluminum hydride resulted in smooth and quantitative reduction of 4a to afford mesyl-protected amine 5a with 96% ee (eq 3).6 Naturally, this strategy for C–H insertion with subsequent in situ reduction of the imine was adopted for further studies.
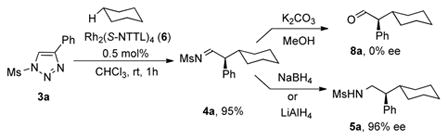 |
(3) |
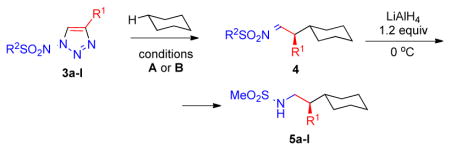
Next, we examined the scope of this rhodium-catalyzed C–H insertion into cyclohexane with respect to the substitution pattern of the triazole. First, various aryl substituents at C-4 of the triazole were examined (Table 1). It was found that 1-mesyl-substituted triazoles with both electron-withdrawing and electron-donating aryl groups at C-4 underwent C–H insertion to afford amines 5a–f with excellent enantioselectivity. Triazole with p-methoxyphenyl electron-donating group reacted noticeably faster than the parent phenyl-substituted compound. However, the yield of the corresponding product 5b was somewhat lower, and formation of carbene dimers was observed. In contrast, the methoxy group was tolerated at the meta-position of the aryl substituent in the triazole to produce the corresponding C–H insertion product 5f in excellent yield. Similarly, triazoles with electron-withdrawing groups at C-4 reacted smoothly to furnish products 5d and 5e in good yields and with excellent enantioselectivity.
Table 1.
Asymmetric C–H Functionalization of Cyclohexane with 1-Sulfonyl Triazolesa
 | ||
|---|---|---|
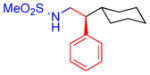 5a, 95%, 96% ee (A) |
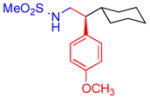 5b, 61%, 97% ee (A) |
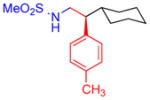 5c, 72%, 94% ee (A) |
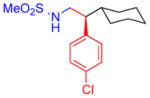 5d, 75%, 97% ee (A) |
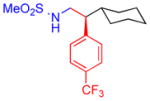 5e, 92%, 95% ee (A) |
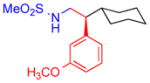 5f, 98%, 95% ee (A) |
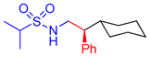 5g, 82%, 94% ee (A) |
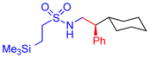 5h, 93%, 86% ee (A) |
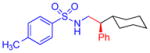 5i, 71%, 88% ee (B) |
|
5j, 72%, 88% ee (B) |
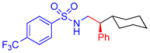 5k, 86%, 93% ee (B)b |
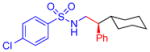 5l, 85%, 92% ee (B)b |
All reactions were carried out on a 1.0 mmol scale in a mixture of 2.5 mL of cyclohexane and 2.5 mL of chloroform. Conditions A: Rh2(S-NTTL) 4 (6, 0.5 mol%), room temperature, 1–24 hrs. Conditions B: Rh2(S-PTAD)4 (7, 0.5 mol%), 40 °C, 2–7 hrs.
Performed at room temperature.
Following the initial studies, the scope of this transformation with respect to the sulfonyl moiety of the triazole was examined. As illustrated by the results summarized in Table 1, isopropylsulfonyl and 2-(trimethylsilyl)-ethylsulfonyl7 derivatives 5g and 5h could be obtained using this method in good yield and high enantioselectivity. Arylsulfonyl-substituted triazoles, however, underwent C–H insertion in the presence of Rh2(S-NTTL)4 sluggishly. In contrast, the use of the less sterically demanding, commercially available Rh2(S-PTAD)48 complex 7 (Figure 1) resulted in smooth formation of the corresponding arylsulfonyl-protected amines 5i–l at 40 °C or at room temperature. Notably, the electronic nature of the aryl groups at the sulfonyl substituents had little or no effect on overall reaction outcome.
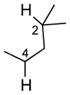
We further explored this novel protocol for C–H functionalization by examining its scope with respect to the alkane component (Table 2). Not surprisingly, secondary C–H bonds of cyclopentane and cyclooctane were efficiently functionalized using mesyl-substituted triazole 3a as the azavinyl carbene source, producing the corresponding amines 5m and 5n in 75 and 86% yield, respectively, and 93% ee for both compounds (Table 2, entries 1–2). In accord with earlier reports,2 the insertion occurred at the tertiary site of adamantane, furnishing product 5o in 95% yield and 94% ee (entry 3). Gratifyingly, C–H functionalization of 2-methylbutane proceeded selectively at the tertiary site (C-2), leaving the sterically encumbered secondary C–H bonds at C-3 intact,9 and delivering product 5p in 87% yield and 92% ee (entry 4). Moreover, the reaction of the homologous 2-methylpentane, which has accessible methylene C–H bonds, produced a mixture of products with remarkably high (5:1)10 selectivity towards insertion into tertiary C–H bonds (C-2 vs. C-4, entry 5) tertiary C–H bonds (C-2 vs. C-4, entry 5) forming sulfonamide 5q in 63% yield and 91% ee. The last two entries demostrate considerably higher levels of enantiocontrol in tertiary C–H insertions compared to previously reported protocols.11
Table 2.
Rh(II)-Catalyzed C–H Insertion with 1-Mesyl-Substituted Triazole 1a: Scope of Alkanesa
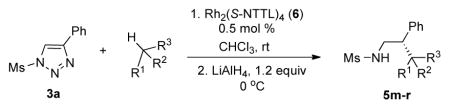
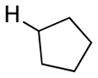
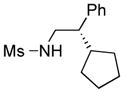 5m
5m
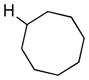
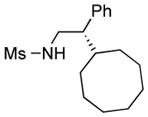 5n
5n
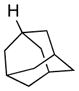
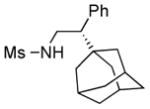 5o
5o
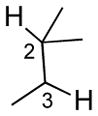
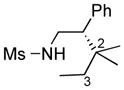 5p
5p
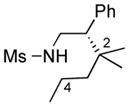 5q
5q
Conditions: triazole 3a (1.0 mmol), Rh2(S-NTTL) 4 (6, 0.5 mol%), alkane (2.5 mL) and chloroform (2.5 mL), 2–3 hrs at room temperature. One-pot subsequent reduction of imine was carried out using 0.5 mL of 2.5M solution of LiAlH4 in THF at 0 °C.
Isolated yield.
Determined by chiral HPLC.
12% of the C-4 insertion product was also observed.
The significantly higher chemoselectivity toward the insertion into tertiary over secondary C–H bonds observed for azavinyl carbenes, as compared to the carboxylic ester congeners (vide supra), prompted us to further evaluate reactivity of the former using kinetic isotope effect studies.
To this end, we subjected triazole 3a to the C–H insertion reaction conditions using a 1:1 mixture of cyclohexane and its fully deuterated analog (eq 4). The observed value of the deuterium KIE, calculated from product distribution, was very close to previously reported kH/kD for the C–H insertion of carboxylic ester-substituted carbenes (KIE = 2.0 for phenyl diazoacetate2b). While these preliminary studies should not be used to draw direct parallels between the two classes of carbenes, they point to electronic similarities of carboxylic ester and imine–substituted rhodium(II) carbenes.
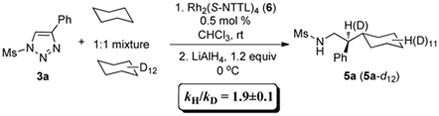 |
(4) |
We attribute the dramatic increase in the selectivity towards insertion into tertiary C–H bonds observed for azavinyl carbenes to the lower steric demand of the aldimine group compared to the carboxylic ester.
Similar to other methods that use convenient and easily available 1-sulfonyl 1,2,3-triazoles12 as direct precursors for rhodium(II) carbene complexes,3a,b the C–H insertion protocol described here is practical and experimentally simple. The inherent ability of diazoimines to exist in equilibrium that favors the closed 1,2,3-triazole form13 obviates any special slow addition techniques that are often employed to ensure a low concentration of the highly reactive carbenes and parent diazo-compounds in the reaction mixture. Since the triazole form is the predominant tautomer, concentration of the diazo species remains very low at any given time. From the synthetic viewpoint, this method provides direct access to β-chiral amine derivatives,14 expanding the range of useful products that can be directly accessed from 1,2,3-triazoles.
As a corollary of their straightforward synthesis, the electronic and steric properties of azavinyl carbenes can be easily tuned by the variation of both the imine (N1 of the parent triazole) and the carbene carbon (C4 of the parent triazole) functional groups. These variations, especially the introduction of the imine functionality in place of the traditional carboxylic ester group in diazo carbonyl compounds, appear to have a profound influence on the reactivity of the carbene and allow further derivatization of the products.
In summary, this work demonstrates that azavinyl carbenes derived from 1,2,3-triazoles can be efficiently used for highly enantioselective C–H functionalization of simple unactivated alkanes. Insertions into secondary and tertiary C–H bonds afford the corresponding chiral sulfonamide products in very good yields and with high enantiomeric excess. Further studies of this C–H insertion reaction and its applications are currently underway in our laboratories.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of General Medical Sciences, National Institutes of Health (GM-087620) and National Science Foundation (CHE-0848982).
Footnotes
This paper is dedicated to Prof. K. Barry Sharpless on the occasion of his 70th birthday.
Supporting Information Available: Experimental details, characterization data, NMR spectra and crystallographic data. This material is available free of charge via the internet at http://pubs.acs.org/
References
- 1.For reviews, see: Davies HML, Morton D. Chem Soc Rev. 2011;40:1857. doi: 10.1039/c0cs00217h.Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704. doi: 10.1021/cr900239n.Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485.Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861. doi: 10.1021/cr0200217.Lebel H, Marcoux JF, Molinaro C, Charette AB. Chem Rev. 2003;103:977. doi: 10.1021/cr010007e.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. Wiley; NY: 1998.
- 2.(a) Davies HML, Hansen T. J Am Chem Soc. 1997;119:9075. [Google Scholar]; (b) Davies HML, Hansen T, Churchill MR. J Am Chem Soc. 2000;122:3063. [Google Scholar]
- 3.Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J Am Chem Soc. 2008;130:14972. doi: 10.1021/ja805079v.Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. J Am Chem Soc. 2009;131:18034. doi: 10.1021/ja908075u.Grimster N, Zhang L, Fokin VV. J Am Chem Soc. 2010;132:2510. doi: 10.1021/ja910187s.For related 2-pyrydyl analogues, see: Davies HML, Townsend RJ. J Org Chem. 2001;66:6595. doi: 10.1021/jo015617t.Chuprakov S, Gevorgyan V. Org Lett. 2007;9:4463. doi: 10.1021/ol702084f.Chuprakov S, Hwang FW, Gevorgyan V. Angew Chem, Int Ed. 2007;46:4757. doi: 10.1002/anie.200700804.
- 4.For recent applications, see: Müller P, Allenbach YF, Robert E. Tetrahedron: Asymmetry. 2003;14:779.Müller P, Bernardinelli G, Allenbach YF, Ferry M, Flack HD. Org Lett. 2004;6:1725. doi: 10.1021/ol049554n.Marcoux D, Charette AB. Angew Chem, Int Ed. 2008;47:10155. doi: 10.1002/anie.200804586.Marcoux D, Azzi S, Charette AB. J Am Chem Soc. 2009;131:6970. doi: 10.1021/ja902205f.DeAngelis A, Shurtleff VW, Dmitrenko O, Fox JM. J Am Chem Soc. 2011;133:1650. doi: 10.1021/ja1093309.See also ref. 3b–c.
- 5.Similarly to the previously reported Rh-catalyzed C–H insertion with diazoesters, dry and degassed solvents were used to ensure efficiency of the reaction. See Supporting Information for details. See also ref. 2b.
- 6.Absolute configuration of 5a was unambiguously assigned by single crystal X-ray crystallographic analysis (CCDC 813723), and homogeneity of the crystalline materials was confirmed by chiral HPLC. See Supporting Information for details.
- 7.Generally, removal of an alkylsulfonyl protecting group to liberate amine requires harsh conditions. In contrast, 2-(trimethylsilyl)ethanesulfonyl (SES) group can be easily removed by treatment with fluoride ion. See: Weinreb SM, Demko DM, Lessen TA, Demers JP. Tetrahedron Lett. 1986;27:2099.
- 8.(a) Reddy RP, Lee GH, Davies HML. Org Lett. 2006;8:3437. doi: 10.1021/ol060893l. [DOI] [PubMed] [Google Scholar]; (b) Denton JR, Sukumaran D, Davies HML. Org Lett. 2007;9:2625. doi: 10.1021/ol070714f. [DOI] [PubMed] [Google Scholar]; (c) Denton JR, Davies HML. Org Lett. 2009;11:787. doi: 10.1021/ol802614j. [DOI] [PubMed] [Google Scholar]
- 9.Similar chemoselectivity was reported for carbenes derived from aryl- and vinyl diazoacetates. See ref. 2b.
- 10.Approximately 1:1 ratio of regioisomers was reported for the related reaction of phenyldiazoacetate and 2-methylpentane. See ref. 2b.
- 11.For the Rh2(S-DOSP)4-catalyzed reaction, 68 and 26% ee were reported for the insertion into tertiary sites of 2-methylbutane and 2-methylpentane, respectively.
- 12.Raushel J, Fokin VV. Org Lett. 2010;12:4952. doi: 10.1021/ol102087r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Dimroth O. Ann. 1909;364:183. [Google Scholar]; (b) Gilchrist TL, Gymer GE. Adv Heterocycl Chem. 1974;16:33. [Google Scholar]
- 14.β-Chiral amines can also be accessed indirectly by asymmetric 1,4-addition of C-nucleophiles to nitroolefins followed by reduction of nitro group. For recent reviews, see: Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713. doi: 10.1021/cr068373r.Berner OM, Tedeschi L, Enders D. Eur J Org Chem. 2002;1877
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.