

Abstract
To discover novel RA risk loci, we systematically examined 370 SNPs from 179 independent loci with p<0.001 in a published meta-analysis of RA GWAS of 3,393 cases and 12,462 controls1. We used GRAIL2, a computational method that applies statistical text mining to PubMed abstracts, to score these 179 loci for functional relationships to genes in 16 established RA disease loci1,3-11. We identified 22 loci with a significant degree of functional connectivity. We genotyped 22 representative SNPs in an independent set of 7,957 cases and 11,958 matched controls. Three validate convincingly: CD2/CD58 (rs11586238, p=1×10−6 replication, p=1×10−9 overall), and CD28 (rs1980422, p=5×10−6 replication, p=1×10−9 overall), PRDM1 (rs548234, p=1×10−5 replication, p=2×10−8 overall). An additional four replicate (p<0.0023): TAGAP (rs394581, p=0.0002 replication, p=4×10−7 overall), PTPRC (rs10919563, p=0.0003 replication, p=7×10−7 overall), TRAF6/RAG1 (rs540386, p=0.0008 replication, p=4×10−6 overall), and FCGR2A (rs12746613, p=0.0022 replication, p=2×10−5 overall). Many of these loci are also associated to other immunologic diseases.
Rheumatoid arthritis is a chronic autoimmune disease characterized by an inflammatory polyarthritis12. Genetic studies have now identified multiple risk alleles for autoantibody positive RA within the MHC region, a PTPN22 missense allele, and risk alleles in 14 other loci (see Table 1)1,3-11. Most RA risk loci contain multiple genes, and currently the causal genes are unknown. However, most contain at least one plausible biological candidate gene involved in immune regulation, and these genes suggest an important set of processes involved in RA pathogenesis. For example, risk alleles highlight genes involved in T-cell activation by antigen presenting cells (class II MHC region, PTPN22, STAT4, and CTLA4), the NF-κB signaling pathway (CD40, TRAF1, TNFSF14, and TNFAIP3, and the recent report of REL13), citrullination (PADI4), natural killer cells (CD244), and chemotaxis (CCL21).
Table 1. Validated RA loci used in functional analyses.
We list each of the 16 established RA loci (column 1), and representative SNPs (column 2). Also we list all of the genes in LD with the SNP (column 3); for each SNP the gene in bolded font is the one that GRAIL selected as the most functionally connected gene when that locus was scored against the 15 other validated risk loci.
| Validated RA Locus |
Representative Allele (SNPs) |
Genes within Associated Regions |
|---|---|---|
| † 1p13.2 | rs2476601 | PTPN22 AP4B1 RSBN1 BCL2L15 DCLRE1B MAGI3 PHTF1 |
| † 1p36.13 | rs2240340 | PADI3 PADI4 |
| 1p36.32 | rs3890745 | PANK4 MMEL1 PLCH2 HES5 TNFRSF14 |
| 1q23.3 | rs6682654 | LY9 CD244 |
| † 2q33.2 | rs3087243 | ICOS CTLA4 |
| 2q32.3 | rs7574865 | STAT1 GLS STAT4 |
| 4q27 | rs6822844 | IL2 IL21 ADAD1 KIAA1109 |
| 6q23.3 | rs10499194, rs6920220 |
OLIG3 TNFAIP3 |
|
† 6p21.32 (MHC class II) |
rs6457620, DRB1*0401, *0101 |
HLA-DRA HLA-DQB1 BTNL2 HLA-DQA1 HLA-DRB5 HLA- DRB1 |
| 7q21.2 | rs42041 | PEX1 FAM133B GATAD1 CDK6 |
| 9q33.2 | rs3761847 | PHF19 CEP110 TRAF1 RAB14 C5 |
| 9p13.3 | rs2812378 | CCL21 |
| 10p15.1 | rs4750316 | RBM17 PFKFB3 PRKCQ |
| 12q13.3 | rs1678542 |
DTX3 METTL1 AVIL DDIT3 XRCC6BP1 MBD6 GLI1
CYP27B1 KIF5A GEFT CTDSP2 MARS CDK4 AGAP DCTN2 TSPAN31 FAM119B MARCH9 TSFM B4GALNT1 OS9 PIP4K2C ARHGAP9 SLC26A10 |
| 20q13.12 | rs4810485 | SLC12A5 NCOA5 CD40 |
| 22q12.3 | rs3218253 | IL2RB |
Loci discovered prior to December 2006.
Based on these observations, we hypothesized that as yet undiscovered autoantibody positive RA risk loci might also contain genes with functions similar to those of genes in known risk loci. Therefore, known RA risk loci can be used to prioritize SNPs for replication from GWAS, especially SNPs with modest statistical support, in independent samples (Figure 1).
Figure 1. Using Gene Relationships Across Implicated Loci (GRAIL) to prioritize candidate RA SNPs.

We select a set of candidate SNPs to pursue in an independent genotyping experiment by starting with all SNPs that obtain p<0.001 in an independent GWAS meta-analysis. Then for each candidate SNP, GRAIL identifies the genomic region in LD, and identifies overlapping genes. It then checks to see how many other loci, already known to be associated with disease, contain functionally related genes. SNPs representing those candidate loci with significantly related genes are forwarded for genotyping in large numbers of independent case-control samples.
To objectively quantify the degree of functional similarity between genes within candidate loci (identified from GWAS) and genes within validated RA risk loci, we used a published functional genomics method, GRAIL (Gene Relationships Across Implicated Loci)2. GRAIL quantifies functional similarity between genes by applying established statistical text mining methods14 to text from a reference database of 250,000 published scientific abstracts about human and model organism genes. For each locus, GRAIL identifies the gene with the greatest number of observed relationships. GRAIL estimates the statistical significance of the number of observed relationships with a null model where relationships between genes near SNPs occur by random chance. This significance score, ptext, represents the output GRAIL score. GRAIL is already able to effectively identify functional inter-connectivity between genes within the previously known RA loci (Figure 2); it might also be able to establish connections between these 16 loci and as yet undiscovered RA risk loci.
Figure 2. GRAIL identifies inter-connectivity among genes in RA loci.
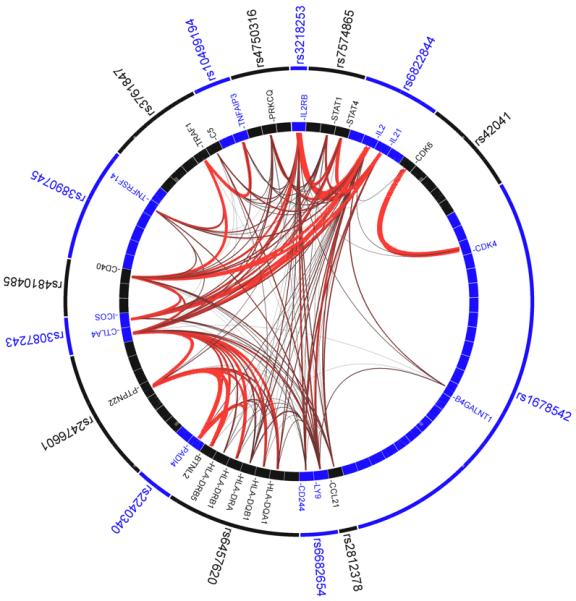
We place the known RA associated SNP along the outer ring; the internal ring represents the genes near each SNP (as listed in Table 1) with a box. We illustrate the literature-based functional connectivity between these genes with lines drawn between them - the redder and thicker the lines are the stronger the connectivity between the genes is. RA SNPs implicate a small number of highly connected genes – those genes are indicated by labeled boxes.
Since GRAIL might demonstrate variable performance across different phenotypes, we wanted to carefully quantify its predictive ability in RA before using it to prioritize SNPs for replication. To estimate GRAIL’s ability to distinguish true RA loci from spurious associations, we examined 12 RA risk loci discovered since 2006 (see Table 1, Supplementary Table 1 online). The current GRAIL implementation is based on PubMed abstracts published prior to December 2006. As these 12 risk loci were subsequently discovered, they constitute a representative set to evaluate GRAIL’s performance. In a leave-one-out analysis, we used GRAIL to score each of these loci for functional relationships to the other 15 validated RA risk loci. A total of 10 of the 12 loci obtain GRAIL scores of ptext<0.01. This analysis suggests that at that ptext threshold, GRAIL has an ~83% true positive rate (or sensitivity). To assess the false positive rate of this same ptext threshold, we modeled spurious loci by sampling 10,000 random SNPs from the Affymetrix 500K array; we scored these SNPs against all 16 RA loci. Of the random SNPs, 5.4% scored ptext<0.01; this corresponds to a specificity of ~95%. Assessment of true and false positive rates at different cutoffs revealed an area under the curve (or C statistic) of 0.97 (see Supplementary Figure 1). We note that if a large number of candidate SNPs are screened in a study, this might still result in a large number of false positives.
Next, we attempted to identify novel RA risk loci from a set of SNPs with modest evidence of association from our recent GWAS meta-analysis of 3,392 cases and 12,462 controls1. In our original study, we genotyped SNPs with p<10−4 in the meta-analysis, and found that 6 out of 31 replicated in our independent samples. However, many RA risk alleles have modest effects (e.g., OR <1.2) and will be missed at that significance threshold. We therefore expected that some SNPs at p<0.001 may be risk alleles. After excluding SNPs that were known validated RA risk loci, we identified a total of 370 SNPs from 179 independent regions that obtained p<0.001 (see Methods and Supplementary Note online). The total number of SNPs observed at this threshold is consistent with the approximate number of SNPs expected by chance, suggesting that the majority represent spurious associations and should not be reproducible in an independent case-control study.
For each of the 179 candidate loci we selected the single SNP with the strongest evidence from the GWAS meta-analysis, and then scored it against the 16 validated RA risk loci with GRAIL. If all 179 SNPs were spurious, then ~10 should score ptext<0.01 based on the estimated 5.4% false positive rate. However 22 of the 179 (12.3%) scored ptext<0.01 (see Figure 3A, Supplementary Table 2 online). This represented a significant enrichment compared to random sets of 179 SNPs (p=3.3 × 10−4 by simulation). We therefore expected that of this select subset of 22 SNPs, as many as half might represent true RA risk alleles.
Figure 3.


A. GRAIL identifies 22 SNPs among the 179 candidate SNPs with p<0.001 in a GWAS meta-analysis. We plot a histogram of the 179 SNPs as a function of their GWAS meta-analysis p-value. Gray bars represent the 157 SNPs that were not selected, while colored bars represent the 22 SNPs that were selected; purple indicating SNPs that replicated convincingly in follow-up genotyping (p<0.0023), orange indicating nominally associated SNPs in follow-up genotyping (p<0.05), and yellow indicating genotyped SNPs without any independent evidence of association. 3B. Enrichment of SNPs with z-scores >2 in replication samples. For each of the 22 SNPs tested, we calculated a one-sided CMH z-score statistic from our two-staged replication data. A z-score of 0 corresponds to a p=0.5; a z-score of 1.65 corresponds to a p=0.05; and a z-score of 2.83 corresponds to p=0.0023. For a random collection of unassociated SNPs, this histogram should approximate a normal distribution (dotted line).
In order to identify which of these 22 SNPs represent true RA risk loci, we genotyped them in an independent validation study of 7,957 cases and 11,958 controls from 11 collections from Europe and North America (see Supplementary Table 3). All cases met 1987 American College of Rheumatology classification criteria15 or were diagnosed by a board-certified rheumatologist, and were seropositive for disease-specific autoantibodies (anti-cyclic citrullinated peptide [CCP] antibody or rheumatoid factor [RF]). All individuals were self-described white and of European ancestry. We assessed association with a Cochran-Mantel-Haenszel (CMH) stratified association statistic16. For each SNP we calculated a z-score, where a z>0 indicates the same allele confers risk in both the replication and the meta-analysis samples. To interpret statistical significance, we used a Bonferroni-corrected one-tailed p-value of 0.0023 (=0.05/22, z>2.83). Additionally, we calculated the overall association p-value across all samples (GWAS meta-analysis plus replication).
Strikingly, of the 22 SNPs examined, 19 (86%) obtained z>0 (see Figure 3B). If these SNPs represented spurious associations then only about half should have z>0; the probability of such a positive skew by chance alone is pskew=0.0005 − suggesting the likelihood of a large number of true RA risk loci within this set of 22 SNPs.
Of the 22 SNPs selected by GRAIL, 13 obtained nominal levels of association at p<0.05 (corresponding to z>1.65); whereas no more than 2 might be expected by chance alone. More compellingly, 7 SNPs achieved a Bonferroni-corrected level of significance in replication (p<0.0023, z>2.83).
When we aggregated both GWAS meta-analysis and replication genotype data (see Table 2, Supplementary Table 4 online), we observed the strongest associations at rs11586238 on 1p13.1 near the CD2 and CD58 genes (1.4×10−6 replication, 1.0×10−9 overall), at rs1980422 on 2q33.2 near CD28 (4.7×10−6 replication, 1.3×10−9 overall), and at rs548234 on 6q21 near PRDM1 (1.2×10−5 replication, 2.1×10−8 overall). Based on conservative estimates of genome-wide significance these represent confirmed RA risk alleles.
Table 2. SNPs tested for RA susceptibility.
The first 6 columns list SNP characteristics. The next four columns list GWA meta-analysis results including allele frequencies, a two-tailed p-value for SNP association, and an odds ratio (OR) with respect to the minor allele. The next four columns list similar results for replication genotyping; significance is reported based on stratified one-tailed CMH statistic. The next three columns summarize joint (overall) analysis results. Significance is reported based on stratified two-tailed CMH statistic across all fourteen patient collections. The final column lists the Breslow-Day Test for heterogeneity of odds ratios across all fourteen collections (3 from the meta-analysis and 11 from the replication study).
| SNP | Meta-Analysis | Replication | Joint | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Chr | Pos (HG18) | Gene(s) | Allele | p | OR | Minor Allele | p | OR | Minor Allele | p | OR | Breslow- Day |
|||
| Major | Minor | Control | Case | Control | Case | p | ||||||||||
| Replicated Loci (Uncorrected p<0.0023) | ||||||||||||||||
| *rs11586238 | 1p13.1 | 117,064,661 |
CD2,
IGSF2, CD58 |
C | G | 2.0E-04 | 1.14 | 0.237 | 0.260 | 1.4E-06 | 1.12 | 0.228 | 0.254 | 1.0E-09 | 1.13 | 0.29 |
| *rs1980422 | 2q33.2 | 204,318,641 | CD28 | T | C | 4.2E-05 | 1.16 | 0.230 | 0.255 | 4.7E-06 | 1.11 | 0.237 | 0.255 | 1.3E-09 | 1.13 | 0.81 |
| *rs548234 | 6q21 | 106,674,727 | PRDM1 | T | C | 3.4E-04 | 1.12 | 0.328 | 0.351 | 1.2E-05 | 1.10 | 0.323 | 0.343 | 2.1E-08 | 1.11 | 0.66 |
| *rs394581 | 6q25.3 | 159,402,509 | TAGAP | T | C | 5.6E-04 | 0.89 | 0.302 | 0.269 | 1.5E-04 | 0.92 | 0.286 | 0.270 | 3.8E-07 | 0.91 | 0.63 |
| *rs10919563 | 1q31.3 | 196,967,065 | PTPRC | G | A | 3.8E-04 | 0.84 | 0.128 | 0.108 | 2.6E-04 | 0.90 | 0.132 | 0.117 | 6.7E-07 | 0.88 | 0.64 |
| rs540386 | 11p12 | 36,481,869 |
RAG1, TRAF6 |
C | T | 6.1E-04 | 0.86 | 0.142 | 0.130 | 8.3E-04 | 0.91 | 0.145 | 0.130 | 3.9E-06 | 0.89 | 0.08 |
| *rs12746613 | 1q23.3 | 159,733,666 | FCGR2A | C | T | 9.1E-04 | 1.16 | 0.120 | 0.133 | 0.0022 | 1.10 | 0.124 | 0.130 | 1.5E-05 | 1.12 | 0.25 |
| Nominally Associated Loci (Uncorrected p<0.05) | ||||||||||||||||
| *rs7234029 | 18p11.21 | 12,867,060 | PTPN2 | A | G | 1.9E-04 | 1.16 | 0.158 | 0.179 | 0.013 | 1.06 | 0.164 | 0.172 | 4.4E-05 | 1.10 | 0.61 |
| rs4535211 | 3p24.3 | 17,048,001 | PLCL2 | G | A | 4.4E-04 | 0.90 | 0.489 | 0.457 | 0.015 | 0.96 | 0.474 | 0.461 | 8.9E-05 | 0.94 | 0.524 |
| rs1773560 | 1q24.2 | 165,688,387 | CD247 | A | G | 4.4E-04 | 0.90 | 0.421 | 0.385 | 0.021 | 0.96 | 0.414 | 0.401 | 1.5E-04 | 0.94 | 0.74 |
| rs892188 | 19p13.2 | 10,270,793 |
ICAM1, ICAM3 |
C | T | 4.6E-05 | 1.13 | 0.378 | 0.409 | 0.041 | 1.05 | 0.393 | 0.401 | 4.3E-05 | 1.08 | 0.21 |
| rs4272626 | 1p13.1 | 116,149,950 | NHLH2 | C | T | 3.5E-04 | 1.12 | 0.359 | 0.388 | 0.042 | 1.04 | 0.354 | 0.362 | 1.9E-04 | 1.07 | 0.07 |
| rs231707 | 4p16.3 | 2,664,183 | TNIP2 | G | A | 6.0E-04 | 1.14 | 0.178 | 0.195 | 0.048 | 1.05 | 0.172 | 0.184 | 5.3E-04 | 1.08 | 0.23 |
| Loci that failed to replicate | ||||||||||||||||
| rs2276418 | 11q23.3 | 117,735,474 | CD3G | A | T | 4.0E-04 | 1.16 | 0.142 | 0.161 | 0.077 | 1.04 | 0.155 | 0.155 | 9.5E-04 | 1.08 | 0.11 |
| rs3176767 | 19p13.2 | 10,310,751 |
ICAM1,
ICAM3 |
T | G | 1.0E-04 | 1.15 | 0.224 | 0.245 | 0.09 | 1.03 | 0.229 | 0.233 | 6.9E-04 | 1.07 | 0.60 |
| rs10282458 | 7q36.1 | 149,676,235 | RARRES2 | G | A | 9.1E-04 | 1.12 | 0.259 | 0.282 | 0.23 | 1.02 | 0.260 | 0.266 | 4.4E-03 | 1.06 | 0.045 |
| rs7041422 | 9p21.3 | 21,034,021 | IFNB1 | T | G | 4.7E-04 | 1.12 | 0.300 | 0.331 | 0.24 | 1.02 | 0.297 | 0.301 | 4.4E-03 | 1.06 | 0.86 |
| rs9564915 | 13q22.1 | 72,223,143 | PIBF1 | A | G | 4.3E-04 | 1.12 | 0.319 | 0.341 | 0.27 | 1.01 | 0.317 | 0.315 | 0.008 | 1.05 | 0.14 |
| rs13393256 | 2p21 | 47,140,263 | TTC7A | C | A | 6.9E-04 | 1.13 | 0.210 | 0.227 | 0.44 | 1.00 | 0.221 | 0.221 | 0.014 | 1.06 | 0.14 |
| rs7579737 | 2q12.1 | 102,353,793 | IL1RL1 | A | G | 8.2E-04 | 0.89 | 0.307 | 0.274 | 0.93 | 1.04 | 0.295 | 0.308 | 0.483 | 0.99 | 0.023 |
| rs2614394 | 12q12 | 42,568,433 | IRAK4 | G | A | 9.8E-05 | 0.81 | 0.098 | 0.082 | 0.94 | 1.08 | 0.099 | 0.105 | 0.06 | 0.94 | 0.002 |
| rs9359049 | 6q13 | 74,758,649 | CD109 | T | A | 2.7E-05 | 1.27 | 0.068 | 0.081 | 0.94 | 0.94 | 0.079 | 0.071 | 0.14 | 1.05 | 0.0155 |
These SNPs are close to other loci already associated to autoimmune disease.
Four additional loci replicated; however, aggregate analysis of GWAS meta-analysis and replication genotype data did not exceed a conservative estimate of significance. We observed evidence of association at rs394581 on 6q25.3 near TAGAP (1.5×10−4 replication, 3.8×10−7 overall), rs10919563 on 1q31.3 within a PTPRC intron (2.6×10−4 replication, 6.7×10−7 overall), rs540386 on 11p12 within a TRAF6 intron (8.3×10−4 replication, 3.9×10−6 overall), and rs12746613 on 1q23.3 near FCGR2A (2.2×10−3 replication, 1.5×10−5 overall). These SNP associations likely represent true RA loci, but additional genotyping will be necessary for definitive confirmation.
Interestingly, many of the SNPs picked by GRAIL that validated in independent genotyping were not those with strongest evidence of association in the initial GWAS meta-analysis (see Figure 3A). That is, prioritization based purely on meta-analysis p-values might have missed many of these associations. For example, rs12746613 (FCGR2A) was ranked 163rd of 179 and rs540386 (RAG1/TRAF6) was ranked 110th. Of the 5 SNPs that we genotyped with the most significant GRAIL ptext scores, 3 replicated and 1 demonstrated nominal evidence of association; only rs2614394 (IRAK4) demonstrated no evidence of association.
Many of these seven alleles further implicate genomic regions already associated with autoimmune diseases (see Table 3). At this point none of these RA risk alleles correspond perfectly to any previously established autoimmune allele; but in some cases fine mapping of the region in multiple diseases could clarify the relationship between them. The rs12746613 FCGR2A SNP is 13 kb away from a missense SNP in FCGR2A that has been associated with systemic lupus erythematosus17,18; they are in the same LD block (r2=0.19, D’=1.0). The rs394581 SNP is located in the 5′ untranslated region of TAGAP and is 17 kb away from a SNP associated with celiac disease and with type I diabetes19,20; they are in partial LD (r2=0.32, D’=.73). The rs10919563 PTPRC SNP is 35 kb away from a rare (~1% allele frequency) non-synonymous SNP that alters splicing of PTPRC21; there have been inconsistent reports that it is associated with multiple sclerosis22-24. We also note that the rs7234029, a PTPN2 intronic SNP, is 41 kb away from a SNP associated with both type I diabetes and celiac disease20; they are in the same LD block (r2=0.14, D’=1.0). The rs548234 SNP is located 10 kb downstream from the PRDM1 transcript and is 133 kb away from a SNP previously associated with Crohn’s disease25. The rs11586238 SNP is 50 kb upstream of the CD2 start site, but is also near multiple other key immunological genes including CD58, and IGSF2. It is 159 kb away from a multiple sclerosis associated SNP within a CD58 intron26,27.
Table 3. Tested SNPs near other alleles associated with autoimmune diseases.
Seven of the 22 SNPs tested are near loci already associated with autoimmune diseases. In the first three columns we list the SNPs, cytogenetic location, and the likely candidate gene. In the next three columns we list the published SNP, the attributed gene, and the disease associations. In the final three columns we list the physical distance and measures of LD. For PTPRC the published SNP is rare, and LD cannot be accurately assessed.
| SNP | Published SNP | Proximity | ||||||
|---|---|---|---|---|---|---|---|---|
| ID | Chr | Gene | ID | Gene | Disease Associations |
Distance (kb) |
r2 | D’ |
| rs12746613 | 1q23.3 | FCGR2A | rs1801274 | FCGR2A | Systemic Lupus Erythematosus |
12.7 | 0.19 | 1.00 |
| rs394581 | 6q25.3 | TAGAP | rs1738074 | TAGAP | Celiac Disease, Type I Diabetes |
16.5 | 0.32 | 0.73 |
| rs10919563 | 1q31.3 | PTPRC | rs17612648 | PTPRC | Multiple Sclerosis | 34.5 | - | - |
| rs7234029 | 18p11.21 | PTPN2 | rs478582 | PTPN2 | Type I Diabetes | 41.1 | 0.14 | 1.00 |
| rs1980422 | 2q33.2 | CD28 | rs3087243 | CTLA4 | Type I Diabetes, Rheumatoid Arthritis |
128.5 | 0.04 | 0.40 |
| rs548234 | 6q21 | PRDM1 | rs7746082 | PRDM1 | Crohn’s Disease | 132.8 | 0.01 | 0.08 |
| rs11586238 | 1p13.1 | CD2 | rs2300747 | CD58 | Multiple Sclerosis | 158.9 | 0.01 | 0.29 |
The rs1980422 SNP is located about 10 kb away from the 3′ UTR of CD28 and is 129 kb away from a known RA and type I diabetes risk allele in the CTLA4 region (rs3087243)11. There is minimal LD between these two SNPs (r2=0.04, D’=0.40); conditional analysis confirmed that the two SNPs independently confer RA risk (see Supplementary Table 5 online).
These SNP associations continue to clarify critical biological processes involved in RA pathogenesis, including T-cell activation, NF-κB signaling, and B-cell activation and differentiation. The CD2 protein is a co-stimulatory molecule on the surface of natural killer cells and T-cells; CD2 signaling is mediated by binding PTPRC directly28. Association to CD28, contributes additional evidence of the role of T-cell activation in disease pathogenesis. TRAF6 is involved in downstream NF-κB activation; it binds CD40 directly and is a key component of B-cell activation29. Our study has also implicated novel processes represented by PRDM1 (BLIMP-1), a transcription factor that regulates terminal differentiation of B-cells into immunoglobulin secreting plasma cells30. Functional studies and re-sequencing will be required to confirm that these genes are indeed the truly causal genes.
We examined all 7 replicated RA SNPs along with known RA risk alleles for epistatic interactions (see Supplementary Note online). Despite the functional relationships between these genes, we found no evidence of significant interactions.
Population stratification could result in spurious associations. However, we were careful for each collection either to use (1) epidemiologically matched samples or (2) ancestry informative markers to match cases and controls. We further note that our seven replicated SNP associations demonstrate consistent effects across all 14 collections without evidence of heterogeneity (p>0.05 by Breslow-Day test of Heterogeneity, see Table 2).
In this study we demonstrate the utility of functional information to prioritize SNPs for replication. We did not pre-define pathways, but rather we used GRAIL to look for genes that had relationships to other validated RA genes. We note that GRAIL is limited in its ability to identify disease genes in entirely novel pathways (i.e., pathways not suggested by the 16 previously known RA risk loci). Arguably, those loci could point to truly novel pathogenic mechanisms. Additionally, successful application of GRAIL is contingent on the scientific literature’s comprehensive description of relevant gene relationships. The general application of GRAIL to other diseases will depend critically on the completeness of the validated loci list and the documentation about relevant processes in the literature. Despite these limitations, our study has identified at least three novel RA risk loci, with strong evidence for additional risk loci.
METHODS
Evaluating GRAIL for its ability to identify RA loci
GRAIL is a method that leverages statistical text mining principles to assess whether putative disease loci harbor genes with functional relationships to genes in other associated disease loci2. Two genes are considered similar if the words used to describe them in PubMed abstracts suggest similar functionality. The implementation of GRAIL used here leverages a text database of 250,000 abstracts published prior to December 2006.
To test the ability of GRAIL to distinguish RA risk loci from spurious associations we defined a set of true positive loci that were discovered since December 2006; these loci would not be described in the GRAIL text database. We also approximated a set of spurious associations by randomly selecting 10,000 SNPs from the Affymetrix 500K genotyping array. We tested both SNP sets for relationships to known associated RA loci with GRAIL. Validated SNPs were tested against the other 15 independent loci; spurious SNPs were tested against all 16 loci. The sensitivity was defined as the proportion of true positive associations that GRAIL assigned a ptext<0.01 score; the specificity was defined as the proportion of spurious associations that GRAIL assigned a ptext>0.01 score.
Selecting Nominally Associated SNPs for Follow-up
In order to identify SNPs for follow-up, we examined the results of a recently published meta-analysis of three GWAS studies (see Supplementary Table 3)1. We examined 336,721 SNPs outside the MHC region that passed strict quality control criteria. We identified those SNPs that were nominally associated with RA (p<0.001). We grouped SNPs into independent loci; two SNPs were placed in the same locus if there was evidence of LD (r2>0.1 in CEU HapMap). We removed all loci that overlapped with validated RA risk regions (see Table 1). We also removed loci with p<10−4 that were genotyped in most available patient collections and had failed to validate in a previous study1. From the remaining set of independent loci, we selected the single SNP that demonstrated the greatest evidence of association in the published meta-analysis.
Testing SNPs with GRAIL
We tested 179 candidate SNPs for relationships to genes within the 16 independent loci known to be associated with RA using GRAIL. SNPs that obtained compelling GRAIL scores (ptext<0.01) were selected for follow-up investigation. To assess the degree of enrichment among high scoring SNPs we sampled 100,000 random sets of 179 SNPs and tested these SNP sets with GRAIL. We calculate the proportion of sets with as many or more GRAIL hits to calculate a permutation based p-value. We note that the version of GRAIL that we used is a previous implementation that differs slightly from the published implementation2 - results are not substantially affected when we do the same experiment with the current version of GRAIL (see Supplementary Figure 2).
Patient Collections
Patient collections are described in detail in Supplementary Table 3 and in the Supplementary Note. Each collection consisted only of individuals that were self-described white and of European descent, and all cases either met 1987 ACR classification criteria or were diagnosed by board certified rheumatologists. Informed consent was obtained from each patient, and the Institutional Review Board at each collecting site approved the study.
All cases were autoantibody positive (CCP and/or RF). For most of the collections, matched control samples were collected along with case samples as part of the same study. For some of the collections - where control samples were unavailable - we matched these case collections to shared controls. We used a total of eleven separate patient collections for replication genotyping: (1) CCP positive cases from the Brigham Rheumatoid Arthritis Sequential Study (BRASS)31 and controls from three separate studies on multiple sclerosis32, age-related macular degeneration33, and myocardial infarction34; (2) CCP positive cases from the Toronto area (CANADA)13 and controls recruited from the same site along with additional controls taken from a disease study of lung cancer35; (3) CCP positive cases and controls from Halifax and Toronto (CANADA-II)13; (4) CCP positive cases from Sweden and epidemiologically matched controls (EIRA-II)36; (5) CCP positive Dutch cases and controls collected from the greater Amsterdam region (GENRA)37,38; (6) North American RF positive cases and controls matched on gender, age, and grandparental country of origin from the Genomics Collaborative Initiative (GCI)4; (7) CCP or RF positive Dutch cases and controls from Leiden University Medical Center (LUMC) 39,40; (8) CCP positive cases drawn from North American clinics and controls from the New York Cancer Project (together this collection is called NARAC-II)13,36; (9) CCP positive cases drawn from North American clinics (NARAC-III)13 and publicly available controls taken from a Parkinson’s study41 and study 66 and 67 of the Illumina Genotype Control Database; (10) CCP or RF positive cases identified by chart review from the Nurses Health Study (NHS) and matched controls based on age, gender, menopausal status, and hormone use42; and (11) CCP or RF positive cases recruited at multiple sites in the United Kingdom by the United Kingdom Rheumatoid Arthritis Genetics (UKRAG) collaboration6. We used available SNP data from this and previous studies to identify genetically identical samples from the same country; we assumed these represented duplicated individuals and we removed them.
Genotyping
Detailed description of genotyping is provided in the Supplementary Note. All GWAS meta-analysis genotyping was previously described. We genotyped replication samples at the Broad Institute using a single Sequenom iPlex Pool (for EIRA-II and GENRA collections) and Affymetrix 6.0 (BRASS), the National Institutes of Health using a single Sequenom iPlex Pool (NARAC-II), Analytic Genetics Technology Centre in Toronto using a single Sequenom iPlex Pool (CANADA-II), Epidemiology Unit at The University of Manchester using a single Sequenom iPlex Pool (UKRAG), Celera using kinetic PCR43 (GCI and LUMC), at the Nurses Health Study in Boston using the BioTrove multiplex SNP genotyping assay (NHS), at the Feinstein Institute using the Illumina 317K array (NARAC-III); and at Illumina using the Illumina 370K array (CANADA). For NARAC-III we additionally obtained publicly available shared controls genotyped on a similar platform from two separate studies. In the cases where whole genome data were available we either extracted data for the 22 SNPs (BRASS) or used imputation to estimate genotypes for them (CANADA and NARAC-III).
For each collection we applied stringent quality control criteria. We required that each SNP pass the following criteria for each collection separately: (1) genotype missing rate < 10%, (2) minor allele frequency > 1%, and (3) Hardy-Weinberg equilibrium with p>10−3. We then excluded individuals with data missing for > 10% of SNPs passing quality control.
Population stratification
For each replication collection we corrected for possible population stratification by either (1) using only epidemiologically matched samples when cases and controls were drawn from the same population, or (2) matching at least one control for each case based on ancestry informative markers (see Supplementary Note for details). Since the cases in the NHS, GCI, LUMC, EIRA-II, CANADA-II, UKRAG, and GENRA collections were well matched to controls, we did not pursue further strategies to correct for population stratification. For the BRASS, NARAC-II, CANADA, and NARAC-III, we matched cases and controls with ancestry informative markers and placed them each into a single stratum. For the BRASS cases and shared controls, GWAS data on Affymetrix 6.0 (unpublished data) was available; we used 681,637 SNPs passing strict quality control as ancestry informative markers. For NARAC-II cases and NYCP shared controls, case and controls were matched using genotype data on 760 ancestry informative markers. For the NARAC-III cases and shared controls, we used available Illumina 317K GWAS data for 269,771 SNPs passing stringent quality control criteria. For the CANADA cases and controls, we used available Illumina 317K GWAS data for 269,771 SNPs passing stringent quality control criteria. For each case-control collection, we used these SNPs to define the top 10 principal components and to remove genetically-distinct outliers (sigma threshold = 6 with five iterations) with the software program EIGENSTRAT44. We eliminated vectors that correlated with known structural variants on chromosomes 8 and 17, demonstrated minimal variation, or did not stratify cases and controls. After mapping cases and controls in the space of eigenvectors, we matched cases to controls that were nearest in Euclidean distance as described elsewhere1.
Analysis of Genetic Data
For each SNP we conducted three statistical tests. First, we conducted a one-sided CMH statistical test across eleven strata to assess if RA association was reproducible in the replication collections in the same direction as the GWAS meta-analysis. We set our significance threshold, after correcting for 22 hypothesis tests, to be p<0.0023 (=0.05/22). Second, we conducted a 573 strata joint analysis across all meta-analysis strata and substrata and replication strata; the eleven replication collections were each placed into their own strata, while the meta-analysis samples were partitioned into 562 strata to be consistent with the approach taken in the original analysis to correct for stratification1,36. Third, we calculated a Breslow-Day test of heterogeneity of odds ratios. We performed all analyses in MATLAB.
Supplementary Material
ACKNOWLEDGEMENTS
SR is supported by an NIH Career Development Award (1K08AR055688-01A1) and an American College of Rheumatology Bridge Grant. RMP is supported by a K08 grant from the NIH (AI55314-3), a private donation from the Fox Trot Fund, the William Randolph Hearst Fund of Harvard University, the American College of Rheumatology ‘Within Our Reach’ campaign, and holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. MJD is supported by NIH grants through the U01 (HG004171, DK62432) and R01 (DK083756-1, DK64869) mechanisms. The Broad Institute Center for Genotyping and Analysis is supported by grant U54 RR020278 from the National Center for Research Resources. The BRASS Registry is supported by a grant from Millennium Pharmaceuticals and Biogen-Idec. The NARAC is supported by the NIH (NO1-AR-2-2263 and RO1 AR44422). This research was also supported in part by the Intramural Research Program of the National Institute of Arthritis, Musculoskeletal and Skin Diseases of the National Institutes of Health. This research was also supported in part by grants to KAS from the Canadian Institutes for Health Research (MOP79321 and IIN - 84042) and the Ontario Research Fund (RE01061) and by a Canada Research Chair. We acknowledge the help of C.Ellen van der Schoot for healthy control samples for AMC/UVA and the help of Ben A.C. Dijkmans, Dirkjan van Schaardenburg, A. Salvador Peña, Paul L. Klarenbeek, Zhuoli Zhang, Mike T Nurmohammed, Willem F Lems, Rob R.J. van de Stadt, Wouter H. Bos, Jenny Ursum, Margret G.M. Bartelds, Daniëlle M. Gerlag, Marleen G.H. van der Sande, Carla A. Wijbrandts, and Marieke M.J. Herenius in gathering GENRA patient samples and data. We thank the Myocardial Infarction Genetics Consortium (MIGen) study for the use of genotype data from their healthy controls in our study. The MIGen study was funded by the U.S. National Institutes of Health and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program R01HL087676 and a grant from the National Center for Research Resources. We thank Johanna Seddon Progression of AMD Study, AMD Registry Study, Family Study of AMD, The US Twin Study of AMD, and the Age-Related Eye Disease Study (AREDS) for use of genotype data from their healthy controls in our study. We thank David Hafler and the Multiple Sclerosis collaborative for use of genotype data from their healthy controls recruited at Brigham and Women’s Hospital.
Footnotes
URLs Gene Relationships Across Implicated Loci (www.broad.mit.edu/mpg/grail/
Illumina Genotype Control Database (www.illumina.com)
REFERENCES
- 1.Raychaudhuri S, et al. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet. 2008;40:1216–23. doi: 10.1038/ng.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raychaudhuri S, et al. Identifying Relationships Among Genomic Disease Regions: Predicting Genes at Pathogenic SNP Associations and Rare Deletions. PLOS Genetics. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–13. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 4.Begovich AB, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–7. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plenge RM, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomson W, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki A, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki A, et al. Functional SNPs in CD244 increase the risk of rheumatoid arthritis in a Japanese population. Nat Genet. 2008;40:1224–9. doi: 10.1038/ng.205. [DOI] [PubMed] [Google Scholar]
- 9.Barton A, et al. Rheumatoid arthritis susceptibility loci at chromosomes 10p15, 12q13 and 22q13. Nat Genet. 2008;40:1156–9. doi: 10.1038/ng.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhernakova A, et al. Novel Association in Chromosome 4q27 Region with Rheumatoid Arthritis and Confirmation of Type 1 Diabetes Point to a General Risk Locus for Autoimmune Diseases. Am J Hum Genet. 2007;81:1284–1288. doi: 10.1086/522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plenge RM, et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet. 2005;77:1044–60. doi: 10.1086/498651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isenberg D. Oxford textbook of rheumatology. xxi. Oxford University Press; Oxford ; New York: 2004. p. 1278. [63] p. of plates. [Google Scholar]
- 13.Gregersen PK, et al. REL, encoding a member of the NF-κB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet. 2009;41:820–3. doi: 10.1038/ng.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raychaudhuri S. Computational text analysis for functional genomics and bioinformatics. xxiv. Oxford University Press; Oxford: 2006. p. 288. [12] p. of plates. [Google Scholar]
- 15.Arnett FC, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 16.Kuritz SJ, Landis JR, Koch GG. A general overview of Mantel-Haenszel methods: applications and recent developments. Annu Rev Public Health. 1988;9:123–60. doi: 10.1146/annurev.pu.09.050188.001011. [DOI] [PubMed] [Google Scholar]
- 17.Duits AJ, et al. Skewed distribution of IgG Fc receptor IIa (CD32) polymorphism is associated with renal disease in systemic lupus erythematosus patients. Arthritis Rheum. 1995;38:1832–6. doi: 10.1002/art.1780381217. [DOI] [PubMed] [Google Scholar]
- 18.Harley JB, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunt KA, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smyth DJ, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–77. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tchilian EZ, et al. The exon A (C77G) mutation is a common cause of abnormal CD45 splicing in humans. J Immunol. 2001;166:6144–8. doi: 10.4049/jimmunol.166.10.6144. [DOI] [PubMed] [Google Scholar]
- 22.Barcellos LF, et al. PTPRC (CD45) is not associated with the development of multiple sclerosis in U.S. patients. Nat Genet. 2001;29:23–4. doi: 10.1038/ng722. [DOI] [PubMed] [Google Scholar]
- 23.Jacobsen M, et al. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nat Genet. 2000;26:495–9. doi: 10.1038/82659. [DOI] [PubMed] [Google Scholar]
- 24.Vorechovsky I, et al. Does 77C-->G in PTPRC modify autoimmune disorders linked to the major histocompatibility locus? Nat Genet. 2001;29:22–3. doi: 10.1038/ng723. [DOI] [PubMed] [Google Scholar]
- 25.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Jager PL, et al. The role of the CD58 locus in multiple sclerosis. Proc Natl Acad Sci U S A. 2009;106:5264–9. doi: 10.1073/pnas.0813310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubio JP, et al. Replication of KIAA0350, IL2RA, RPL5 and CD58 as multiple sclerosis susceptibility genes in Australians. Genes Immun. 2008;9:624–30. doi: 10.1038/gene.2008.59. [DOI] [PubMed] [Google Scholar]
- 28.Schraven B, Samstag Y, Altevogt P, Meuer SC. Association of CD2 and CD45 on human T lymphocytes. Nature. 1990;345:71–4. doi: 10.1038/345071a0. [DOI] [PubMed] [Google Scholar]
- 29.Ishida T, et al. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996;271:28745–8. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- 30.Calame K. Activation-dependent induction of Blimp-1. Curr Opin Immunol. 2008;20:259–64. doi: 10.1016/j.coi.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 31.Sato M, et al. The validity of a rheumatoid arthritis medical records-based index of severity compared with the DAS28. Arthritis Res Ther. 2006;8:R57. doi: 10.1186/ar1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Jager PL, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–82. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neale BM, et al. A Genome-Wide Scan of Advanced Age-Related Macular Degeneration Suggests a Novel Role of Lipase-C. In Review. 2009 [Google Scholar]
- 34.Kathiresan S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–41. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amos CI, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–22. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plenge RM, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study. N Engl J Med. 2007;357:1199–209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielen MM, et al. Antibodies to citrullinated human fibrinogen (ACF) have diagnostic and prognostic value in early arthritis. Ann Rheum Dis. 2005;64:1199–204. doi: 10.1136/ard.2004.029389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wijbrandts CA, et al. The clinical response to infliximab in rheumatoid arthritis is in part dependent on pre-treatment TNF{alpha} expression in the synovium. Ann Rheum Dis. 2007 doi: 10.1136/ard.2007.080440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurreeman FA, et al. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007;4:e278. doi: 10.1371/journal.pmed.0040278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wesoly J, et al. Association of the PTPN22 C1858T single-nucleotide polymorphism with rheumatoid arthritis phenotypes in an inception cohort. Arthritis Rheum. 2005;52:2948–50. doi: 10.1002/art.21294. [DOI] [PubMed] [Google Scholar]
- 41.Fung HC, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–6. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 42.Costenbader KH, Chang SC, De Vivo I, Plenge R, Karlson EW. Genetic polymorphisms in PTPN22, PADI-4, and CTLA-4 and risk for rheumatoid arthritis in two longitudinal cohort studies: evidence of gene-environment interactions with heavy cigarette smoking. Arthritis Res Ther. 2008;10:R52. doi: 10.1186/ar2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Germer S, Holland MJ, Higuchi R. High-throughput SNP allele-frequency determination in pooled DNA samples by kinetic PCR. Genome Res. 2000;10:258–66. doi: 10.1101/gr.10.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.