

Abstract
This article describes a computational method to construct gene models by using evidence generated from a diverse set of sources, including those typical of a genome annotation pipeline. The program, called Combiner, takes as input a genomic sequence and the locations of gene predictions from ab initio gene finders, protein sequence alignments, expressed sequence tag and cDNA alignments, splice site predictions, and other evidence. Three different algorithms for combining evidence in the Combiner were implemented and tested on 1783 confirmed genes in Arabidopsis thaliana. Our results show that combining gene prediction evidence consistently outperforms even the best individual gene finder and, in some cases, can produce dramatic improvements in sensitivity and specificity.
Computational identification of complete gene models in eukaryote genomes remains a challenging task (Zhang 2002). In the Arabidopsis genome project (The Arabidopsis Genome Initiative 2000), human experts integrated the output of different gene prediction programs with sequence homology data from searches of protein and transcript databases to construct the published gene models. Difficulties in creating accurate annotation arise for a variety of reasons. Sometimes the evidence for a gene is weak, consisting of just one gene prediction but no sequence homology, or just a single expressed sequence tag (EST) match. In other cases, the evidence is plentiful but contradictory: Different gene finders and protein sequence alignments may indicate many overlapping candidate genes, and more than one of these models may in fact be correct. Thus, the process of constructing final gene predictions, even with a human curator in the loop, is time-consuming and fraught with opportunities for errors. For these reasons, we have developed a computational method that evaluates much of the same information human annotators use, as a means of creating gene models that are both more accurate and more consistent than can be derived from any single computational gene prediction algorithm.
This article describes the Combiner program, a statistical algorithm that uses the output from other annotation software to improve the accuracy on predicted genes while maintaining a good balance between sensitivity (the number of true genes detected) and specificity (the number of gene predictions that are correct). Other efforts to combine gene model evidence have primarily focused on integrating the output from gene prediction programs (Murakami and Takagi 1998; Pavlovic et al. 2002; Rogic et al. 2002). The Combiner algorithm integrates multiple gene prediction programs plus much of the evidence available in a typical annotation pipeline, including evidence from proteins, ESTs, cDNAs, and splice site predictions. Other approaches to incorporating multiple evidence types can be found in the EuGène (Schiex et al. 2001) and GAZE (Howe et al. 2002) systems.
We tested three algorithms for use in the Combiner. The first algorithm is a simple Linear Combiner (LC1) that uses a voting function to combine multiple gene prediction programs. Each gene finder is given equal weight, that is, one vote, in LC1. The algorithm first identifies all possible disjoint intervals separated by signals such as splice sites or start codons, similar to signal-based ab initio gene prediction (Salzberg et al. 1998). A dynamic programming algorithm constructs gene models from candidate signals predicted by any of the gene prediction programs, systematically enumerating all possible combinations of signals (start, stop, donor, and acceptor sites). The sequence intervals between these signals are voted on by the gene finders. Each gene finder must vote for either coding or noncoding, and the highest-scoring combination of intervals is pieced together to form a gene model. The second Combiner (LC2) uses a similar dynamic programming algorithm to LC1, but with two significant enhancements. First, it adds sequence alignments (both DNA and protein) and splice site prediction programs to the inputs. Second, it uses different weights for the different forms of evidence.
Aligning genomic sequence to a transcript or protein database produces matches with widely varying amounts of similarity. Clearly, the similarity of each alignment should be a factor in scoring the quality of a predicted exon. However, the degree of similarity of a match does not directly translate into the likelihood that a region encodes a protein, and the alignment algorithms themselves (e.g., BLAST; Altschul et al. 1990) do not produce such likelihoods. Further complicating matters, LC2 uses splice site predictions to indicate potential exon boundary signals, but because it scores intervals (not points between intervals), it does not include the splice site prediction in the linear weighting function. To address these limitations, we developed a statistical scoring method that uses decision trees (in particular, the randomized oblique decision trees of Murthy et al. 1994) to correlate evidence patterns with candidate gene models. This Statistical Combiner (SC) uses the confidence scores output by the gene finders themselves (when available), which were also used by Rogic et al. (2002) to combine outputs from two gene finders. Instead of a simple linear function combining all the inputs, SC builds a nonlinear model based on a decision tree. A description of each approach is given in the Methods section.
RESULTS
The three Combiners were tested on a data set of 1783 cDNA confirmed genes in Arabidopsis thaliana. These reference genes are based on full-length cDNA sequences that have been aligned to the genome and subsequently examined and defined by human annotators (Haas et al. 2002). This carefully curated data set provides a large and reliable source for evaluating the accuracy of our methods. The results are divided into two sets based on the type of evidence used. The first set consists of gene prediction programs trained prior to the availability of the test genes and includes GlimmerM (Pertea and Salzberg 2002), GeneMark.hmm (Lukashin and Bordovsky 1998), Genscan+ (Burge and Karlin, 1997), and GeneSplicer (Pertea et al. 2001). The second test set includes recent TwinScan (Flicek et al. 2003) predictions available at http://genes.cs.wustl.edu/predictions/Arabidopsis/contig_04_23_03 and a newer version of GlimmerM that includes improved modules to better detect translation start sites and polyadenylation sites. In both test sets, the Combiners using homology data take input from alignments between the genomic DNA and protein sequences from a non-redundant amino acid database aligned by using the dps and nap programs (Huang et al. 1997), as well as gene transcripts (including both EST and cDNA sequences) from the TIGR gene index database (Quackenbush et al. 2001) aligned using dds and gap (Huang et al. 1997). The alignment data was filtered to remove proteins, ESTs, and cDNAs that are included in the 1783 genes, which might bias the Combiner's results. We removed all sequences that align with 100% identity regardless of the source of these sequences.
All programs were run on 515 bacterial artificial chromosomes (BACs) with lengths of ∼100 kbp and collectively span roughly one third of the Arabidopsis genome. The three Combiners were run on each BAC, using the evidence from the other software programs as input, and the predictions were compared with the locations on the BAC corresponding to the 1783 cDNA confirmed genes. The test genes specify complete coding regions from the start codon to the stop codon (including possibly introns) and can occur on any portion of the BAC sequence.
Our primary purpose in these tests is to take an existing set of predictions from gene finders along with the output from other gene evidence and see if we can combine their output to produce a more accurate set of gene models. Because our tests genes are already known from cDNA evidence, the tests may favor the Combiners using sequence alignment evidence, particularly if these genes are more frequently expressed. Therefore, in addition to considering the results of combing only gene finders in LC1, we also report results from the SC using only the gene finders and splice site prediction program as input.
Test Set 1
Our initial test used three single-organism gene finders as inputs: Genscan, GeneMark.hmm, and GlimmerM. Figure 1 shows the overlap among correctly predicted gene models from each of the gene finders, in which “correct” is defined to mean that all coding exons were in perfect agreement with the true gene. Only 178 (10%) of the genes were correctly predicted by all three methods. As the figure makes clear, each of the three gene finders has a set of genes for which it is the only correct method. The most accurate single program, GeneMark.hmm, predicts 799 of 1783 genes (45%), but if we could produce an oracle that always chose the best model from the three gene finders, then it would make 1205 (68%) correct predictions. Thus, if the Combiner can cleverly pick a gene model from among the three gene prediction programs, it should be able to improve on the performance of all three.
Figure 1.

The number of correct and incorrect (number in parentheses) whole gene model predictions shared among the three prediction programs: GlimmerM (GA), Genscan+ (GS), and GeneMark.hmm (GM) from a test set of 1783 genes. Incorrect gene refers to cases in which all coding exons in the gene are in perfect agreement among the gene finders but not with the true gene.
Results for the four Combiners—LC1 and SC-g using only gene finders as input and LC2 and SC using gene finders plus homology data—are listed in Table 1. Because both SC and SC-g require a training set, we trained them by using a separate set of 380 genes (none included in the 1783 test set) from 120 BACs. All genes in both training and test sets were confirmed by full-length cDNA sequences that had been mapped to the genome (Haas et al. 2002), currently the gold standard for determining the correctness of a gene.
Table 1.
Performance of the Gene Predictors on 1783 Genes
| CG | MG | CE | ME | WE | SN | |
|---|---|---|---|---|---|---|
| SC | 1179 (66%) | 31 (2%) | 6625 (88%) | 311 (4%) | 27 | 97% |
| SC-g | 1114 (63%) | 12 (1%) | 6580 (88%) | 220 (3%) | 71 | 98% |
| LC2 | 1078 (61%) | 19 (1%) | 6468 (86%) | 298 (4%) | 44 | 98% |
| LC1 | 967 (54%) | 21 (1%) | 6323 (84%) | 300 (4%) | 57 | 97% |
| GM | 799 (45%) | 31 (2%) | 5947 (79%) | 363 (5%) | 95 | 96% |
| GS | 650 (37%) | 43 (2%) | 5680 (75%) | 722 (10%) | 41 | 92% |
| GA | 557 (31%) | 39 (2%) | 4610 (61%) | 1183 (16%) | 415 | 85% |
SC indicates Statistical Combiner; SC-g, SC combining gene prediction programs only; LC2, Linear Combiner using sequence alignments; LC1, Linear Combiner using gene prediction programs only; GA, GlimmerM; GM, GeneMark.hmm; and GS, Genscan+. The columns are number of whole genes correctly predicted (CG), number of genes completely missed (MG), correctly predicted exons out of the 7510 total (CE), number of exons completely missed (ME), predicted exons overlapping a gene region but do not overlap a true exon (WE), and percentage of protein coding nucleotides correctly detected (SN).
For this initial test, the SC gets 66% of the test gene models exactly correct in contrast to the best individual gene finder, GeneMark.hmm, which gets 45% correct. (Here “exactly correct” means that the entire coding region is correctly predicted, from start to stop, with all intron boundaries correctly identified.) LC1 and LC2 also improved over the individual gene finders, with 54% and 61% correct, respectively. SC-g gets 2% more test genes correct (63%) than does LC2. SC and SC-g identified 88% of the exons correctly (both the 5′ and 3′ ends were correct), whereas the individual gene finders' accuracies ranged from 61%-79%. LC1 and LC2, at 84% and 86%, showed intermediate levels of performance gain. A missed gene (exon) occurs when no predicted exon overlaps the gene's exons (exon) by one base or more. LC1, LC2, and SC missed small and roughly equal numbers of exons (4% of the 7510 total), but SC-g missed 3% of the exons and the LCs and SC-g missed fewer gene models, only 1%, compared with 2% for SC and for the best individual gene finder. Of the 31 whole genes missed by SC, 19 are single exon genes, indicating a possible area for improvement. At the nucleotide level, which measures the percentage of coding bases in the test genes correctly labeled as coding, all four Combiners achieve 97%-98% accuracy. And the number of “wrong exons,” predicted exons that do not overlap a true exon at all, is substantially smaller for SC, with 27 wrong exons, than for any of the gene finders, which had from 41 to 415 wrong exons. The lower overall correct gene count for GlimmerM is due to a tendency to insert short exons (the average exon length is 54 nucleotides).
An additional measure of Combiner performance is its accuracy relative to the agreement among the gene finders. If for example, the Combiner gene model is only correct when it agrees with at least two of the ab initio gene finders, then a simple majority-vote rule might work just as well. Results comparing the performance among all four Combiners with respect to the gene prediction programs are summarized in Table 2. The table shows results divided according to whether each Combiner agreed with zero, one, two, or three gene finders; for example, SC agreed with two of the gene finders on 527 genes, and those predictions were correct for 440 (84%) of them. The Combiners get 97% of the genes correct when all three gene finders agree; these are presumably “easy” genes for automated prediction. (Note that when all three gene finders agree, the Combiners never disagreed with the consensus. Interestingly, there are five genes for which all gene finders agree but for which the reference cDNA alignment indicates a different gene model.) But even when they agree with just one of the gene finders and disagree with the other two, the Combiners get 52% (LC1) to 65% (SC) of the predictions correct. And all four Combiners correctly predict some genes that are missed by all three gene prediction programs. This is possible because the Combiners are able to piece together parts of a gene model from the different inputs to make a new model. As Table 2 shows, LC2, LC1, and SC-g are competitive with SC when at least two gene prediction programs agree with one another, but SC has a distinct advantage when the one or no gene finders make a correct prediction.
Table 2.
Breakdown of Combiner Predictions When Matching Exactly Three, Two, One or Zero Gene Prediction Programs
| Combiner | No. of GP | CG | WG | CG/CG + WG (%) |
|---|---|---|---|---|
| SC | 3 | 178 | 5 | 97 |
| SC-g | 3 | 178 | 5 | 97 |
| LC2 | 3 | 178 | 5 | 97 |
| LC1 | 3 | 178 | 5 | 97 |
| SC | 2 | 440 | 87 | 84 |
| SC-g | 2 | 417 | 70 | 86 |
| LC2 | 2 | 418 | 76 | 83 |
| LC1 | 2 | 401 | 94 | 81 |
| SC | 1 | 419 | 229 | 65 |
| SC-g | 1 | 395 | 308 | 56 |
| LC2 | 1 | 363 | 263 | 58 |
| LC1 | 1 | 307 | 286 | 52 |
| SC | 0 | 142 | 253 | 36 |
| SC-g | 0 | 124 | 282 | 31 |
| LC2 | 0 | 119 | 348 | 26 |
| LC1 | 0 | 81 | 412 | 16 |
The first column (combiner) refers to the four Combiners. The second column refers to the number of matching gene prediction programs. The third column and fourth columns count the number of times the Combiner prediction is correct (CG) and not entirely correct (WG). The fifth column is the percentage of correct predictions.
Test Set 2
We constructed a second set of comparisons by adding the Twin-Scan algorithm, which, unlike any of the other gene finders, uses sequence homology with a related species to inform its gene finding. By using alignments to Brassica oleracea, TwinScan is able to achieve substantial improvements over the best of the gene finders in our original set. Our hypothesis was that the Combiner should improve still further, using the better predictions from TwinScan to boost its performance over the first round of tests. For this test, we used up to five gene finders as inputs: the three from the first test, TwinScan, and a newer version of GlimmerM. Similar to what is shown in the Venn diagram in Figure 1, all five prediction programs predict a different set of correct gene models. Table 3 lists the number of gene models each gene prediction program exclusively identified correctly. In total, 1496 of the 1783 gene models are identified correctly by at least one of the five gene prediction programs. Therefore, each prediction source provides potentially useful information. Because both LC1 and LC2 combine each evidence source by using a linear sum of weights, we expected that they might have difficulty combining the outputs from the highly correlated versions of GlimmerM. Results are summarized in Table 4. Again, we include a version of SC, which combines only the gene finders and splice prediction program.
Table 3.
The Number of Gene Models Each Gene Finder Exclusively Predicts Correctly in Test Set 2
| Program | Correct genes |
|---|---|
| TwinScan | 206 |
| GeneMark.hmm | 59 |
| GlimmerM | 41 |
| GlimmerM2 | 39 |
| Genscan+ | 31 |
Table 4.
Performance for Gene Predictors Including TwinScan and Retrained GlimmerM in Addition to the Programs Listed in Table 1
| CG | MG | CE | ME | WE | SN | |
|---|---|---|---|---|---|---|
| SC-5 | 1385 (78%) | 17 (1%) | 6952 (93%) | 196 (3%) | 22 | 98% |
| SC-3 | 1345 (75%) | 24 (1%) | 6911 (92%) | 194 (3%) | 34 | 98% |
| SC-5g | 1320 (74%) | 16 (1%) | 6878 (92%) | 173 (2%) | 37 | 98% |
| LC2-3 | 1293 (73%) | 12 (1%) | 6810 (91%) | 156 (2%) | 77 | 99% |
| LC1-3 | 1206 (68%) | 14 (1%) | 6692 (89%) | 207 (3%) | 48 | 98% |
| TS | 1200 (67%) | 17 (1%) | 6569 (87%) | 299 (4%) | 66 | 96% |
| GM2 | 563 (32%) | 10 (1%) | 5321 (71%) | 673 (9%) | 386 | 93% |
SC-5 indicates SC using all five gene prediction programs; SC-3, SC using three gene prediction program; SC-5g, SC using five gene prediction programs and no alignment data; LC2-3, LC2 using three gene prediction programs; LC1-3, LC1 using three gene prediction programs; TS, TwinScan; and GM2, newer GlimmerM output. The three prediction programs used by SC-3, LC2-3, and LC1-3 are TwinScan, GeneMark.hmm, and GM2.
TwinScan is the most accurate gene prediction program, with performance that compares favorably to the best Combiner results from the first experiment (Table 1). TwinScan predicts 67% of the gene models and 87% of the exons correctly, completely missing just 17 genes. With the advantage gained from having TwinScan as input, the SC jumps to 78% (1385) correct gene models and 93% correct exons. The SC using only gene finders (SC-5g) also improves, getting 75% of the gene models correct and 92% correct exons. Both LC1 and LC2 improve by using TwinScan, with 68% and 73% of the gene models correct, respectively.
When one of the gene finders is clearly superior to the others, the Combiner should give it a higher weight. This happens automatically in the training of SC, but not in LC1 or LC2. Both LC1 and LC2 would likely improve substantially here if the weights were better tuned to reflect the relative performance among the gene finders. Unlike LC1 and LC2, SC performance improves further when adding one or two more gene prediction programs on top of the first three. Table 4 shows these results as SC-3 for three gene finders and SC-5 for all five.
DISCUSSION
Two of the leading resources for annotation of the human genome are Ensembl and Nation Center for Biotechnology Information (NCBI), in which each applies a different collection of computational tools to construct their gene predictions (Birney et al. 2002). NCBI aligns all known genes from the Reference Sequence database and from GenBank mRNA sequences to the genomic sequence by using MegaBLAST (Zhang et al. 2000), retaining matches with ≥95% identity and covering 50% of the putative transcript (http://www.ncbi.nlm.nih.gov/genome/guide/build.html#gene). Alternate overlapping models are merged into a single gene. Additional genes are reported based on GenomeScan (Yeh et al. 2001) predictions, excluding those that overlap the set of genes identified by alignment. Ensembl takes a similar approach, first aligning known proteins to the genome and using GeneWise (Birney and Durbin 1997) to determine the gene models. Additional predictions come from exons predicted by Genscan, filtered according to whether they correspond to BLAST (Altschul et al. 1990) matches to a protein database. Ensembl also incorporates EST alignments to refine the predicted gene models. Each gene reported from the automated pipeline is supported by sequence alignment evidence, but it is not clear how many of these regions correctly identify each gene model boundary: translation starts, splice sites, and stop sites. Neither of these human genome pipelines incorporate an explicit method for combining multiple gene finders; because our Combiner is open source, it should be easy for these and other annotation providers to include it in their pipelines.
For many organisms, multiple gene finding tools successfully identify protein coding regions in the genome. Our results show that, even in cases in which one program is clearly more accurate, other prediction tools provide useful information, correctly finding some exons and genes that the other programs miss. The difficulty lies in checking each protein coding region to decide when and how to use each piece of evidence. LC2 does surprisingly well considering it only requires that the user assign a weight to each evidence source (see Methods). SC, however, provides a more robust model for incorporating different types of evidence. It uses training data to build its own nonlinear model for combining the evidence. The SC also provides a way to make use of multiple overlapping gene models from a single prediction program, for example, those produced by different parameter settings. This allows the Combiner to decide which alternative model is best supported by sequence alignment evidence, rather than relying on the single best prediction.
An important element of the Combiner approach is to treat each source of evidence as a black box, which enables the use of gene model evidence from any source, as long as the predictions are provided as sequence coordinates. Separating the Combiner from the evidence software allows us to apply the Combiner to each genome sequencing project by using sequence analysis software specific for that organism. The success of the Combiner depends on the accuracy of the underlying evidence and continued improvements in gene prediction algorithms, as illustrated by the TwinScan results in this study, should improve future Combiner results.
METHODS
Linear Combiner
Four types of signals are considered for LC1: start codons, stop codons, splice donors, and splice acceptors. Processing the input sequence from left to right, LC1 computes partial gene models representing the optimal sequence of signals leading to any given signal in the sequence. Signals are linked together in a gene model only when such a link is biologically meaningful; for instance, a donor site (the end of an exon) can only be linked back to a previous acceptor site (the beginning of an exon) or a start codon. In either case, the sequence between the donor site and the previous signal is scored as a protein coding interval. An acceptor site can only be linked to a preceding donor site, and the intervening sequence is scored as a noncoding interval. We use a dynamic programming algorithm that scans the signals from left to right (5′ to 3′ in the DNA sequence) and, for each interval bounded by a pair of signals, computes a separate score for each of the three possible reading frames. (This ensures that all exons in a gene model are in the same frame.) The score is computed as a weighted sum of the evidence. More precisely, let S be an input sequence, Sx be the base at position x, Sx...y be the subsequence spanning the interval from x to y, and Sigx and Sigy be a signal at location x and y, respectively. We compute scores in a dynamic programming matrix D, as follows:
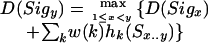 |
1 |
where w(k) is the weight assigned to each evidence source k. We compute D for all three reading frames at each position y. For LC1, which is a simple voting combiner, all weights are set to one. (The evidence for LC1 consists only of gene finders, but we permit other sources in LC2 and in SC.) The function hk(Sx...y) returns the score given by each gene finder (or other evidence type) for the subsequence Sx...y. For protein coding intervals, we compute hk for gene finder k by simply counting the number of bases predicted to be coding by that gene finder; conversely, we count the number of bases predicted to be noncoding for noncoding intervals. Essentially, this formula says that for signal Sigy, we scan back and compute a score for each previous signal by adding together the previous signal's score plus the weighted evidence for the intervening sequence. We then choose the best total score and store it at y. We construct a gene model by tracing the “parse” back through the matrix. Because computation is done only at positions containing one of the signal types, the computational complexity is O(mn2), where m is the number of gene finders and n is the number of signals detected.
Both the SC and LC2 use a modified version of the LC1 algorithm to construct gene models from non-overlapping sequence intervals. The most significant difference is that these algorithms include sequence alignment information as additional evidence types. The other major difference is that rather than computing scores only between predefined signal positions, the algorithm computes scores for subsequence Sx...y, where x and y can be (1) the locations of signals or (2) boundaries of alignment regions. Figure 2 illustrates how a genome is split into sequence intervals where interval I1 = Sx1...(x2 - 1), I2 = Sx2...(x3 - 1), etc. Note that the intervals in Figure 2 can begin or end in the middle of an exon (or intron), because alignments are not constrained by exon boundaries. Figure 3 shows an example in which multiple overlapping gene models occur in a single reading frame. Interval I4 is part of four different candidate gene models: It is alternatively a complete exon, the interior of an exon, the 5′ end of an exon, or the 3′ end of an exon. If these predictions are all in the same reading frame, only the highest-scoring partial gene model will be stored at x5.
Figure 2.

Partitioned output from three evidence types: splice predictions, gene predictions, and sequence alignments. The five sources of evidence (listed in order from top to bottom) are output from a splice prediction program (SP); a gene prediction program (GP1) with exon confidence scores 0.9 and 0.89; a gene prediction program (GP2) with no confidence scores; 89% and 45% identity alignments from a protein database, which make up a single evidence source; and 32% and 20% identity alignments from an EST database. The genome sequence is divided into intervals defined by each potential boundary x1,x2,...,x8. The non-overlapping intervals I1,...,I7 are used to score gene models. The predicted splice site at x5 is associated with I5.
Figure 3.

An example of four overlapping candidate gene models G1 through G4. The exons are assumed to be part of the same coding frame. In this example, if the evidence only predicts G1 and G2, the combiner scores G3 or G4 if either model is optimal.
The evidence for each subsequence Sx...y is captured in a vector v = [v(1), v(2),..., v(m)], for m different types of evidence. We compute the values v(k) by using the scores from the gene finders or the percentage of similarity from the alignment algorithms. For gene finders that do not score each exon, we use a value of one for predicted coding intervals and zero for noncoding intervals. Because many protein (respectively, EST) sequences can align to the same place, we choose the alignment with the highest percentage of similarity to represent the protein (EST) evidence. For example, in Figure 2, interval I1 has evidence from two gene prediction programs (GP1 and GP2), two protein sequence alignments, and one EST alignment. The evidence vector for I1 is v = (GP1, GP2, Protein, EST) = (0.9, 1.0, 0.89, 0.32). Splice site predictions are not included in the LC2 vectors; they are used only to mark potential exon boundaries.
The dynamic programming algorithm for LC2 differs from equation 1 in the way the score is computed for Sx...y. (This change leads directly to the dynamic programming algorithm for SC.) In Figure 2, for example, the score for subsequence Sx2...x5 is the score for interval I2 plus the score for interval I3 plus the score for interval I4. Each interval between Sigx and Sigy is scored according to its evidence vector v. The scoring function b(v), is simply the sum of the scores for each evidence source's entry in the evidence vector v(k), multiplied by the respective weight w(k) and normalized by multiplying by the interval length. A separate score is stored for each signal Sigy (for each reading frame), which maximizes the following:
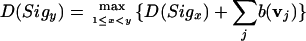 |
2 |
where j iterates over all non-overlapping intervals between Sigx and Sigy. LC2 uses equal weights of 0.3 for each gene prediction program, 0.2 for alignments to gene index entries (ESTs), and 0.21 for protein alignments. EST alignments are given slightly less weight than do proteins because sizable regions of many ESTs correspond to untranslated regions of the mRNA transcript.
Statistical Combiner
The goal of the SC is to identify the most probable set of gene models by using the knowledge gained from a training set. This is accomplished by finding a series of zero or more gene models g1,g2,...,gn with maximal posterior probability given the input evidence E: arg maxg1,g2,...gn P(g1,g2,...,gn E).
Gene models are defined by four exon types: single, initial, internal, and terminal. The four exon types are defined by nine partial and complete exon labels per strand, which are defined by five underlying events: start, coding, donor, acceptor, and stop. In the LCs, only coding and noncoding sequence intervals are scored. The statistical method also scores the evidence at the exon boundaries. For the positive strand (moving left to right in the 5′ to 3′ direction), three states can describe the left boundary of a coding interval: an acceptor (a) site, a start (r) site, or no exon boundary. The three possible states for the right boundary are as follows: donor (d) site, stop (t) site, or no exon boundary. Combining the boundary conditions in all biologically meaningful ways generates nine different complete and partial exon types on the positive strand and a representation for noncoding intervals. Each label is a conjunction of events across an interval and its boundaries. Table 5 lists the events and their corresponding labels. For example, the definition of a beginning internal exon (bn) is an interval such that the left boundary is an acceptor site, a protein coding interval spans the region, there is no start site on the left boundary, no donor site on the right boundary, and no stop site at the right boundary. Stated more intuitively, this interval is a portion of an internal exon that contains the left (5′) boundary but not the right (3′) boundary.
Table 5.
The Set of Labels That Describe Each Sequence Interval and Are Used to Construct Gene Models on the Positive Strand
| Interval labels | Acceptor (a) | Start (r) | Coding (c) | Donor (d) | Stop (t) |
|---|---|---|---|---|---|
| Noncoding (nc) | 0 | 0 | 0 | 0 | 0 |
| Beginning internal (bn) | 1 | 0 | 1 | 0 | 0 |
| Complete internal (cn) | 1 | 0 | 1 | 1 | 0 |
| Complete terminal (ct) | 1 | 0 | 1 | 0 | 1 |
| Partial initial (bi) | 0 | 1 | 1 | 0 | 0 |
| Complete initial (ci) | 0 | 1 | 1 | 1 | 0 |
| Complete single (cs) | 0 | 1 | 1 | 0 | 1 |
| Coding (c) | 0 | 0 | 1 | 0 | 0 |
| Partial terminal (pt) | 0 | 0 | 1 | 0 | 1 |
| Ending internal (en) | 0 | 0 | 1 | 1 | 0 |
Labels reflect partial and complete exons. Each entry asserts whether the condition in that column must be true (1) or false (0). Each of the underlying conditions (acceptor, start, coding, donor, stop) define the type of coding interval and are represented by independent evidence sources.
Evaluating Candidate Gene Models
Each gene model is a series of sequence labels l1,l2,...,lz from Table 5. The probability of a gene model given evidence E = e1,e2,...,ez, is defined as:
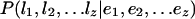 |
where ej is the evidence for interval Ij. Each ej contains five evidence vectors: va,vr,vc,vd,vt, one for each of the five events: acceptor, donor, coding, start, and stop. The vectors representing exon boundary conditions (the splice sites, start and stop codons) are defined by the evidence aligned with the sequence interval's boundary. For example, assuming each evidence type is a valid splice site predictor in Figure 2, the evidence vector representing a possible donor site at the right boundary position of I1 (labeled x2 in the figure), is defined as vd = (SP,GP1, GP2,Protein,EST) = (1,0.9,0,0,0.32).
To compute the probability of a given label lj at interval Ij, we use an approximation because the size of the evidence E is proportional to the sequence length, which can vary. We compute the probability of a gene model by taking the product of probabilities for each label lj, making the simplifying assumption that lj is only dependent on interval Ij and the adjacent intervals Ij - 1 and Ij + 1:
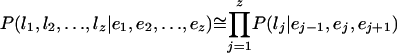 |
Each ej - 1,ej,ej + 1 contains the five vectors: va,vr,vc,vd,vt capturing the evidence from intervals Ij - 1, Ij, and Ij + 1. For interval I1 in Figure 2, each evidence vector encodes the evidence from interval I0, I1, and I2 (for this example, I1 represents the left boundary and I0 represents a zero-valued vector). The donor site adjacent to interval I1 from Figure 2 is vd = (0,0,0,0,0,1,0.9, 0,0,0.32,0,0,0,0.89,0).
By using the five evidence vectors and the independence assumption, the probability of each label from Table 5 is computed from the product of five independent probability values, each conditioned on one of the evidence vectors: va,vr,vc,vd,vt. For example, the beginning internal exon (bn) label is P(lj = bn|ej - 1,ej,ej + 1) = P(a|va) × P(¬r|vr) × P(c|vc) × P(¬d|vd) × P(¬t|vt). Probabilities for each label are computed according to the definitions in Table 5. For each event q ε {a,d,c,r,t}; P(¬q|vq = 1 - P(q vq), and the probability of the noncoding label (nc) is P(lj = nc|ej - 1,ej,ej + 1) = P(¬a|va) × P(¬r|vr) × P(¬c|vc) × P(¬d|vd) × P(¬t|vt).
The most probable set of gene models are found by using the dynamic programming algorithm from LC2 (equation 2), replacing the linear scoring function with the probability estimate that label lj corresponds to interval Ij between signals Sigx and Sigy. Because we use negative log probabilities, we take the minimum rather than the maximum; each Sigy is linked to the previous Sigx that minimizes the score:
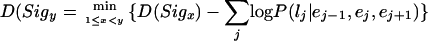 |
Estimating Probabilities by Using Decision Trees
Decision trees are used to compute probabilities for each of the five events: P(a|va), P(r|vr), P(c|vc), P(d|vd), and P(t|vt). For each event model, the list of evidence vectors is generated from the training set. Each evidence vector generated from the training set records the percentage of correct predictions it makes. For example, if a donor evidence vector vd predicts three of the true donor sites but makes two additional predictions, the percentage of correct predictions is 3/5. For the coding evidence vectors (vc), nucleotides are counted instead of the number of occurrences.
Decision trees are constructed by using OC1 (Murthy et al. 1994) to apply the training examples to new data. Using the protein coding model P(c vc) as an example, the entire set of training vectors representing coding intervals are mapped to one of two classes: coding, if more than half of an evidence vector's nucleotides correctly predict protein coding intervals; noncoding, otherwise. Each leaf node represents evidence vectors classified as either coding or noncoding vectors. Traversing the decision tree matches the input to a local region of the vector space. The average percentage of correct predictions from the vectors at the leaf node are the final probability estimate. Separate probabilities are estimated in this way for each of the five event models. An example decision tree is shown in Figure 4. Each leaf stores the individual examples from the training set, which satisfy all of the yes and no conditions, starting from the root of the tree. OC1 decision trees consider both single and multiple conditions at each node. For example, Leaf1 considers two conditions: whether the Genscan prediction is >0.3 and a protein aligns with >50% identity, whereas Leaf2 considers only one condition: whether a prediction is made by GlimmerM.
Figure 4.

An example decision tree for combining gene prediction evidence to predict protein coding intervals. Each leaf x contains a probability value P(c vc,x), computed from the matching examples in the training set.
The criteria for splitting each node in an OC1 tree is nondeterministic in order to consider the wide range of possible solutions. As a result, a different OC1 tree is generated each time the training program is run. Because any one tree may not produce the best results, SC uses 10 decision trees for each of the five event models (acceptor, donor, coding, start, and stop). A single probability value is the average value from the 10 trees. Each decision tree is generated by using the default parameters for the OC1 software.
Program Availability
The original Linear Combiner (LC1) is implemented in Perl, and LC2 and SC are implemented in C++. The software is an open source package and freely available at http://www.tigr.org/software/combiner.
Acknowledgments
This work was supported in part by the National Institutes of Health under grant R01-LM06845. We thank the two anonymous reviewers for their helpful comments.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1562804.
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796-815. [DOI] [PubMed] [Google Scholar]
- Birney, E. and Durbin, R. 1997. Dynamite: A flexible code generating language for dynamic programming methods used in sequence comparison. ISMB 5: 56-64. [PubMed] [Google Scholar]
- Birney, E., Clamp, M., and Hubbard, T. 2002. Databases and tools for browsing genomes. Annu. Rev. Genomics Hum. Genet. 3: 293-310. [DOI] [PubMed] [Google Scholar]
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-84. [DOI] [PubMed] [Google Scholar]
- Flicek, P., Keibler, E., Hu, P., Korf, I., and Brent, M.R. 2003. Leveraging the mouse genome for gene prediction in human: From whole-genome shotgun reads to a global synteny map. Genome Res. 13: 46-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J., Volfovsky, N., Town, C.D., Troukhan, M., Alexandrov, N., Feldmann, K.A., Flavell, R.B., White, O., and Salzberg, S.L. 2002. Full-length messenger RNA sequences greatly improve genome annotation. Genome Biol. 3: RESEARCH0029. [DOI] [PMC free article] [PubMed]
- Howe, K.L., Chothia, T., and Durbin, R. 2002. GAZE: A generic framework for the integration of gene-prediction data by dynamic programming. Genome Res. 12: 1418-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X., Adams, M.D., Zhou, H., and Kerlavage, A.R. 1997. A tool for analyzing and annotating genomic sequences. Genomics 46: 37-45. [DOI] [PubMed] [Google Scholar]
- Lukashin, A.V. and Bordovsky, M. 1998. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 26: 1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, K. and Takagi, T. 1998. Gene recognition by combination of several gene-finding programs. Bioinformatics 14: 665-675. [DOI] [PubMed] [Google Scholar]
- Murthy, S.K., Kasif, S., and Salzberg, S. 1994. A system for induction of oblique decision trees. J. Artificial Intelligence Res. 2: 1-32. [Google Scholar]
- Pavlovic, V., Garg, A., and Kasif, S. 2002. A bayesian framework for combining gene predictions. Bioinformatics 18: 19-27. [DOI] [PubMed] [Google Scholar]
- Pertea, M. and Salzberg, S.L. 2002. Computational gene finding in plants. Plant Mol. Biol. 48: 39-48. [PubMed] [Google Scholar]
- Pertea, M., Lin, X., and Salzberg, S.L. 2001. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 29: 1185-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush, J., Cho, J., Lee, D., Liang, F., Holt, I., Karamycheva, S., Parvizi, B., Pertea, G., Sultana, R., and White, J. 2001. The TIGR gene indices: Analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res. 29: 159-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogic, S., Ouellette, B.F.F., and Mackworth, A.K. 2002. Improving gene recognition accuracy by combining predictions from two gene-finding programs. Bioinformatics 18: 1034-1045. [DOI] [PubMed] [Google Scholar]
- Salzberg, S., Delcher, A.L., Fasman, K.H., and Henderson, J. 1998. A decision tree system for finding genes in DNA. J. Comput. Biol. 5: 667-680. [DOI] [PubMed] [Google Scholar]
- Schiex, T., Moisan, A., and Rouze, P. 2001. EuGène: An eucaryotic gene finder that combines several sources of evidence. In Computational biology (eds. O. Gascuel and M-F. Sagot), pp. 111-125. LNCS 2066. Springer, Heidelberg, Germany.
- Yeh, R.-F., Lim, L.P., and Burge, C.B. 2001. Computational inference of homologous gene structures in the human genome. Genome Res. 11: 803-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M.Q. 2002. Computational prediction of eukaryotic protein-coding genes. Nature Rev. Genet. 3: 698-710. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Schwartz, S., Wagner, L., and Miller, W. 2000. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7: 203-214. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.ensembl.org; ENSEMBL.
- http://www.ncbi.nlm.nih.gov; NCBI.
- http://www.tigr.org; TIGR.
- http://genes.cs.wustl.edu; TWINSCAN.