

Abstract
Glioblastoma multiforme (GBM) can be induced in mice through the combined expression of activated forms of KRas and Akt in glial progenitor cells. We have previously demonstrated that KRas is required for the maintenance of these tumors in vivo as inhibition of KRas expression resulted in apoptotic tumor regression and significantly increased survival. To determine the reliance of these tumors on Akt signaling in vivo, we generated a viral vector that allows the expression of Akt to be controlled post-delivery. Survival rates were compared between those animals with continued Akt expression and animals in which expression of Akt was suppressed. Although a fifth of the tumors were refractory to treatment, inhibition of Akt significantly increased the survival of tumor-bearing mice and nearly a fourth of the mice remained in remission four months after the treatment period. These data suggest that Akt is required for glioblastoma maintenance in the context of activated Ras and that loss of Akt expression results in increased survival; therefore, the PI3K/AKT signaling pathway is a viable therapeutic target in this context.
Keywords: Akt, Ras, glioma, mouse model, tumor maintenance, somatic cell gene delivery, retroviral vector, RCAS, TVA
Introduction
Gliomas are sub-categorized according to their grade by the World Health Organization (WHO) [1]. WHO Grade IV, glioblastoma multiforme (GBM), is the most aggressive type of primary brain tumor and unfortunately is also the most common. The current standard of care for patients with GBM includes surgical resection followed by adjuvant radiotherapy and/or temozolomide chemotherapy. However, treatment is complicated by the location and invasive nature of the tumors and patient prognosis is poor. Median survival is only 14.6 months [2]. Primary GBM (∼90%) develops de novo, whereas secondary GBM (∼10%) progresses from a low-grade glioma to a high-grade glioma through the acquisition of additional genetic changes. While histologically indistinguishable, primary and secondary GBM tumors appear to have distinct genetic alterations [3]. Epidermal growth factor receptor (EGFR) mutation and amplification, phosphatase and tensin homolog deleted on chromosome ten (PTEN) loss, and INK4a/Arf deletion are hallmarks of primary GBM whereas mutations in isocitrate dehydrogenase 1 or 2 (IDH1 or IDH2) [4], overexpression of platelet derived growth factor receptor (PDGFR), mutations in p53, overexpression of cyclin-dependent kinase 4 (CDK4) or loss of RB, and/or PTEN characterize secondary GBM (reviewed in [5]). In both cases, activated receptor tyrosine kinases (RTK) (i.e., EGFR and PDGFR) signal to common downstream effectors including components of the RAS and AKT pathways. RAS is activated in almost all cases of GBM and AKT is activated in ∼70% of GBM tumors [6, 7]. The frequent deregulation of these signaling pathways in cancer has driven significant interest in blocking effectors of these pathways for cancer therapy.
RAS signaling activates a number of pathways but especially important is its ability to activate the canonical mitogen-activated protein kinase (MAPK) pathway (i.e., RAS/RAF/MEK/ERK), which regulates fundamental cellular functions including proliferation, differentiation, and survival (reviewed in [8]). Using an established mouse model of GBM, we previously demonstrated the importance of Ras signaling in the maintenance of KRas and Akt-induced tumors in vivo as inhibition of KRas resulted in apoptotic tumor regression and significantly increased survival [9].
While mutations in AKT have not been observed in human GBM, approximately 40% of GBM tumors show mutation or loss of expression of the tumor suppressor gene PTEN, which functions as a major negative regulator of the phosphati-dylinositol 3-kinase (PI3K)/AKT signaling pathway [3]. In the absence of PTEN, AKT activity is elevated leading to increased proliferation and inhibition of apoptosis. AKT activation has also been documented in GBM as a result of increased PI3K activity due to mutation within the regulatory subunit of PI3K [10]. AKT signaling promotes proliferation and inhibits apoptosis by phosphorylating/inactivating Bad, forkhead transcription factors, and caspase-9. AKT also regulates the cell cycle by preventing GSK-3β mediated phosphorylation and degradation of β-catenin, cyclin D1, cyclin E, p21 CIP1, and Myc (reviewed in [11]). Phosphorylation of TSC2 by activated AKT disrupts its interaction with TSC1, which prevents mTOR inhibition and leads to activation of protein synthesis via p70 S6 kinase and inactivation of the eukaryotic initiation factor 4E binding protein 1 (an inhibitor of translation) [12]. Activated mTOR also induces angiogenesis in both hypoxia inducible factor (HIF)-dependent and independent pathways via vascular endothelial growth factor (VEGF) (reviewed in [13]). A number of agents that inhibit PI3K/AKT/mTOR signaling have recently been developed to determine if targeting this pathway is therapeutic (reviewed in [14]). However, multiple variables exist when testing small molecule inhibitors. If a lack of efficacy is observed, it is often difficult to determine if the target was inappropriate or if the drug was simply ineffective. In this study, we used a genetic approach to examine the role of Akt signaling in the maintenance of KRas and Akt-induced GBM in vivo. Inhibition of Akt expression led to tumor regression and increased survival of tumor-bearing mice. Complete responses were observed in two-thirds of the treated mice but these responses were not durable as subsequent re-expression of Akt induced relapse in the majority of the mice.
Materials and methods
Transgenic mice
Nestin-TVA mice have been described [15]. The mice were maintained on standard food or doxycycline-containing food pellets (Harlan-Teklad, Madison, WI). All experiments were performed in compliance with the guiding principles of the “Care and Use of Animals” (available at www.nap.edu/books/0309053773/html/) and were approved by the IACUC prior to experimentation.
Genotype analysis
DNA was prepared from tail biopsies by proteinase K digestion overnight at 55 °C followed by isopropanol precipitation. PCR of the TVA allele was carried out in 25 μl total volume containing 2.5 μl 10× PCR buffer (Invitrogen, Carlsbad, CA), 1.25 μl MgCl2, 0.5 μl dNTPs, 1.0 μl DMSO, 1.0 μl of 10 μM TVA-386 sense and TVA-786 antisense primers. The following primer sequences were used: TVA-386 sense 5'-AGCTGGTGAGATGGGACTGAAC-3'; TVA-786 antisense 5'-CGAACATTCAAAGCCTCCAG-3' (to detect a 400-bp fragment). Following an initial hot-start at 94 °C for 1 min, samples were amplified for 30 cycles (94 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s).
Isolation of primary astrocytes
Primary astrocytes were isolated from the brains of Nestin-TVA-positive newborn mice. Single cells were obtained by mincing the tissue followed by digestion in 0.05% trypsin (Invitrogen, Carlsbad, CA) for 20 min at 37 °C. The cells were resuspended in RPMI (Invitrogen, Carlsbad, CA) with 5% FBS (Hyclone, Logan, UT) and 1 × penicillin/streptomycin (Invitrogen, Carlsbad, CA). The cells were split 1:3 when confluent.
Vector constructs
The retroviral vectors used in this study were either replication-competent avian leukosis virus (ALV) long terminal repeat (LTR), splice acceptor, and Bryan polymerase-containing vectors of envelope subgroup A [designated RCASBP(A)], or no splice acceptor vectors [designated RCANBP(A)]. RCASBP(A)KRas and RCASBP(A) Tet-off have been described [9]. The RCANBP(A) destination vector [16] and pENTR3C-TRE vector [9] have also been described. To generate pENTR3C-TRE-Akt-HA, RCASBP(A)Akt-HA (a gift of Eric Holland) was digested with Notl and Sall and the Akt-HA fragment was cloned into the pENTR3C-TRE vector digested with Notl and Xhl. RCANBP(A)TRE-Akt-HA was generated by mixing 300 ng of pENTR3C-TRE-Akt-HA with 300 ng of the RCANBP(A) destination vector in the presence of the LR Clonase Enzyme Mix as described [16]. LR reactions were performed for 2 h at room temperature and 1 μl was transformed into 50 μl TOP10-One Shot Competent Cells (Invitrogen, Carlsbad, CA). Transformations were plated on agar plates containing 100 μg/ml ampicillin.
Cell culture
DF-1 cells were grown in DMEM-high glucose media supplemented with 10% fetal bovine serum (BioWhitaker, Walkersville, MD), 1 × penicillin/streptomycin, and maintained at 39°C [17]. The DF-1 cultures were passaged 1:3 when confluent.
Virus propagation
Virus infection was initiated by calcium phosphate transfection of plasmid DNA that contained the retroviral vector in proviral form. In standard transfections, DF-1 cells were plated at 30% confluency, allowed to attach (2-3 h), and 5 μg of purified plasmid DNA was introduced by the calcium phosphate precipitation method previously described [18], followed by a 5-min glycerol shock at 39°C (15% glycerol in the medium). Viral spread was monitored by assaying culture supernatants for ALV capsid protein by ELISA as previously described [19]. Virus stocks were generated from the cell supernatants. The supernatants were cleared of cellular debris by centrifugation at 2000 × g for 10 min at 4°C, filtered through a 0.45-μm filter, and stored in aliquots at -80 °C. Virus was determined to be replication-competent by a reading of 0.200 or greater on ELISA for the viral capsid protein [19].
Viral infections in vitro
Astrocytes were seeded in 6-well plates at a density of 5 × 104 cells/well and were maintained in RPMI with 5% FBS, 1 × penicillin/streptomycin at 37°C. After the cells attached, the medium was removed and replaced with 1 ml of filtered virus-containing medium in the presence of 8 μg/ml polybrene (Sigma, St. Louis, MO) for 2 h at 37°C. The virus was removed and replaced with fresh medium, and the cells were incubated at 37 °C for 1 h. Cells were infected again for another 2 h, after which the virus-containing medium was replaced with fresh medium and the cells were incubated overnight at 37 °C.
Western blotting
Infected astrocytes were washed with PBS and 150 μl SDS-lysis buffer was added to each well of a 6-well dish. The cell lysates were boiled for 10 min and passed through a 26-gauge needle 5 times. Following centrifugation, the proteins were separated on a 10% Tris-glycine poly-acrylamide gel, transferred to nitrocellulose, and incubated for 1 h at room temperature in blocking solution (0.05% Tween-20 in Tris-buffered saline with 5% non-fat dry milk). Blots were immunostained for Akt-HA using an anti-HA monoclonal antibody (Covance, Berkely, CA) and anti-α-tubulin (AB-1) (Oncogene, San Diego, CA) both at a 1:1000 dilution. All antibodies were diluted in blocking solution. Western blots were incubated in the primary antibody for 1 h at room temperature with constant shaking and then washed three times in TBS-T wash buffer (0.05% Tween-20 in Tris-buffered saline). The blots were then incubated with an anti-mouse IgG-HRP secondary antibody diluted 1:3000 (Sigma, St. Louis, MO) for 1 h at room temperature. The blots were washed three times in TBS-T wash buffer, incubated with ECL solutions per the manufacturer's specifications (Amersham, Piscataway, NJ), and exposed to film.
Viral infections in vivo
Infected DF-1 cells from a confluent culture in a 10-cm dish were trypsinized, pelleted, resus-pended in 50 μl PBS, and placed on ice. Mice were injected intracranially 2 mm ventral from bregma (intersection of the coronal and sagittal sutures) with 5 μl of cells using a gas-tight Hamilton syringe. All mice were monitored for tumor development by magnetic resonance imaging (MRI). Censored survival data was analyzed using a log-rank test of the Kaplan-Meier estimate of survival.
Magnetic Resonance Imaging (MRI)
Mice were anesthetized via inhalation of 2% Attane™ isoflurane (Minrad, Orchard Park, NY) with oxygen (0.5 L/min) for 5 min prior to imaging and throughout the procedure. The mice were imaged using a Bruker 7 T USR 20 cm diameter bore (Model 70/20) Biospin MRI running Paravision 4. The 7 T MRI was equipped with a 6 cm inner diameter unshielded gradient coil insert (Model BGA 06) and a 35 mm inner diameter quadrature RF Birdcage coil. The mice were kept warm using a rodent (warm air) heater system (SA Instruments Inc., Stony Brook, NY). Mouse respiration was monitored using an MR-compatible monitoring and gating system (Model 1025; SA Instruments Inc., Stony Brook, NY).
A 100 μl bolus of MultiHance (gadobenate dimeglumine; Bracco Diagnostics Inc., Princeton, NJ) MRI contrast was injected intraperitoneally 15 min prior to imaging. The imaging protocol consisted of a fast gradient-echo three-plane localizer for graphical prescription [Field of View (FOV): 4 cm; Slice Thickness: 1 mm; Matrix: 128 × 128; TR: 100 ms; TE: 6 ms; Flip Angle (FA): 30°, Scan Time: 13 s]. The presence of the chelated gadolinium MR contrast was verified using an axial T1-weighted Rapid Acquisition Relaxation Enhanced (RARE) spinecho sequence [FOV: 2.65 cm; Slice Thickness: 0.75 mm; Matrix: 256 × 256; TR: 800 ms; TE: 7.5 ms; Rare Partitions: 4; Scan Time: 38 s]. Quality contrast-enhanced structural images were acquired using an axial T1-weighted Multi-Slice Multi-Echo (MSME) spin-echo sequence [FOV: 2.65 cm; Slice Thickness: 0.75 mm; Matrix: 256 × 256; TR: 700 ms; TE: 10.7 ms; Scan Time: 3 minutes]. Additional structural images were acquired using an axial T2-weighted TurboRARE sequence to detect edema [Field of View (FOV): 2.65 cm; Slice Thickness: 0.75 mm; Matrix: 256×256; TR: 4200 ms; TE: 12 ms; Rare Factor: 8; Scan Time: 100 seconds]. Fifteen to twenty 0.75 mm thick slices were typically required to cover the brain.
Histology and histochemical staining
Mice were euthanized when they displayed obvious signs of distress or at the times indicated. Brains from all injected mice were fixed in formalin overnight, cut into three sections, and then dehydrated through a graded alcohol series in a Microm STP 420D tissue processor (Thermo Fisher, Kalamazoo, MI). Tissues were paraffin embedded and 5-μm sections were adhered to glass slides. Sections were stained with H&E or left unstained for immunohisto-chemistry. Histological classification was performed independently by two board certified pathologists. Images were captured using a Zeiss Axio microscope equipped with an Axio-Cam ICc3 camera (Zeiss, Thornwood, NY).
Immunohistochemistry
Tissue sections were de-paraffinized and antigen retrieval was performed in ‘Diva Decloaking’ buffer (Biocare Medical, Concord, CA) by boiling for 10 min in a Decloaking Chamber (Biocare Medical). Sections were treated with 3% hydrogen peroxide and blocked in Background Sniper (Biocare Medical) for 10 min. Primary antibodies were diluted in Renaissance background reducing diluent (Biocare Medical). Sections were incubated overnight at 4 °C and probed with Mach 4 rabbit polymer reagent (Biocare Medical) or Mach 4 mouse probe for 15 min followed by Mach 4 polymer for 15 min for mouse monoclonal antibodies. Visualization was carried out with DAB (Biocare Medical). Sections were counterstained with hematoxylin. KRas expression was detected using an antibody to the FLAG epitope (F7425, Sigma) (diluted 1:200). Akt expression was detected using an antibody to the HA epitope (HA.11, Covance) (diluted 1:1000). Cell proliferation was detected using an antibody to Ki67 (M7246, Dako, Carpinteria, CA) (diluted 1:50). Apoptosis was detected by TUNEL staining using the in situ cell death detection kit (Roche, Indianapolis, IN) per the manufacturer's specifications.
Results
Generation of an RCANBP(A)TRE-Akt retroviral vector
Intracranial infection of Nestin-TVA mice with either RCASBP(A)Akt or RCASBP(A)KRas alone is not sufficient to form tumors, but the combination of Akt and KRas induces glioblastomas that are histologically similar to human GBM [7]. To determine the reliance of these tumors on continued Akt signaling, we generated a viral vector in which expression of the inserted gene could be regulated post-delivery using the tetracycline (Tet)-inducible system. We used the RCANBP(A) virus [16], which lacks the splice acceptor at the 3' end of the envelope gene, so that sequences inserted into this region are transcribed from an internal promoter and not the viral long terminal repeat (LTR) (Figure 1A). A tet-responsive element (TRE) was inserted upstream of the Akt gene to generate RCANBP(A)TRE-Akt. The virus was propagated in DF-1 cells, an immortalized chicken embryo fibroblast cell line [17]. Expression from the TRE requires the presence of a tetracycline transcriptional activator (tTA) such as Tet-off or a reverse tTA (rtTA) such as Tet-on. In the Tet-on system, the Tet-responsive gene is only expressed in the presence of doxycycline; in the Tet-off system, the Tet-responsive gene is repressed in the presence of doxycycline [20].
Figure 1.

Schematic representation of the viral vectors and Western blot analysis of Akt expression. A. Diagram of RCASBP(A), RCASBP(A) KRas, RCASBP(A)Tet-off, RCANBP(A), RCANBP(A) TRE-Akt. These vectors are all Gateway compatible to allow for the easy insertion of experimental sequences. LTR, long terminal repeat; Y, packaging signal; SD, splice donor; SA, splice acceptor. B. Nestin-TVA-positive astrocytes were infected in culture with RCASBP(A)Tet-off alone (lanes 1 and 2), RCANBP(A)TRE-Akt alone (lanes 3 and 4) or RCANBP (A)TRE-Akt with RCASBP(A) Tet-off (lanes 5 and 6). The samples in lanes 2, 4, and 6 were treated with 2 μg/ml doxycycline for 48 h. Lys-ates were separated on a 10% Tris-glycine gel, transferred to nitrocellulose, and probed with an antibody against the HA epitope tag on Akt. The membrane was re-probed with an antibody against α-tubulin to ensure equal loading.
Tet-regulated expression of Akt in primary astro-cytes in vitro
To test the ability of Akt expression to be induced from the TRE-Akt virus, Nestin-TVA positive astrocytes were infected in culture with the TRE-Akt virus alone, the Tet-off virus alone, or both the TRE-Akt and Tet-off viruses. The infected cells were cultured in the presence or absence of doxycycline and Akt expression was visualized by Western blot (Figure 1B). Akt expression was observed only in those cells infected with both the TRE-Akt and Tet-off viruses. This expression is tightly regulated, because no Akt expression was detected in cells infected with the TRE-Akt virus alone or in cells infected with both the TRE-Akt and Tet-off viruses in the presence of doxycycline (Figure 1B).
Tumor formation in Nestin-TVA mice infected with KRas, Tet-off, and TRE-Akt viruses
To test the ability of the TRE-Akt virus to induce tumors in vivo, newborn Nestin-TVA mice were injected intracranially with KRas, Tet-off, and TRE-Akt viral producing cells. From three weeks of age, the mice began showing signs of tumor formation (e.g., macrocephaly, lethargy, or cachexia), which was confirmed by MRI. Histo-logical examination revealed tumors that closely resembled human GBM and were similar to those generated by delivery of both RCASBP(A) Akt and RCASBP(A)KRas viruses [7] (Figure 2A). Expression of Akt was confirmed by IHC for the HA epitope tag (Figure 2B) and activity of Akt was detected by IHC for phosphorylated Akt (Figure 2C). Immunohistochemical analysis for the FLAG epitope tag also confirmed virally delivered KRas expression (Figure 2D). Whereas 55% of the Nestin-TVA-positive animals developed tumors detectable at weaning by MRI, no tumors were detected in any of the TVA-negative animals injected with the same viruses. This demonstrates the specificity of the infection to only those cells expressing TVA.
Figure 2.

Brain tumor formation in Nestin-TVA mice injected with viruses expressing KRas, Tet-off and TRE-Akt. A. Hematoxylin and Eosin (H&E) stained 5 μm brain sections showing glioblastoma invading the surrounding normal brain. B. Expression of virally delivered Akt detected by IHC for the HA epitope tag. C. Activity of Akt as detected by IHC for phosphorylated Akt. D. KRas expression as detected by IHC for the FLAG epitope tag. All immunohistochemically stained sections were counterstained with hematoxylin. Scale bar represents 200 μm.
AKT inhibition causes tumor regression and significantly increases survival
To determine the reliance of these tumors on continued Akt expression, tumors were induced by intracranial injection of Nestin-TVA mice with KRas, Tet-off, and TRE-Akt viral producing cells at birth. All injected mice were screened for the presence of tumors by MRI at weaning. Only tumor-bearing mice were used for further experimentation. Doxycycline was administered continuously through the food for 90 days to one cohort of tumor-bearing mice from the time of weaning (∼21 days of age) to suppress Akt expression. A separate cohort of tumor-bearing mice was given standard feed (without doxycycline). As a control, a cohort of mice with tumors induced with KRas and Akt viruses that are not regulated by the Tet system were also given doxycycline impregnated food. Survival rates were compared between untreated mice and mice given doxycycline to determine whether the administration of doxycycline increased survival. The resulting Kaplan-Meier curve demon-strated a significant increase in survival for the mice with reduced Akt expression (P = 1.6 ×10-08; Figure 3A). No difference in survival was detected between untreated mice with tumors induced with KRas, Tet-off, and TRE-Akt viruses and doxycycline treated mice with tumors induced with viruses constitutively expressing KRas and Akt (P = 0.977; Figure 3B). This illustrates that doxycycline has no innate tumor suppressive properties. All further discussion of doxycycline treated mice refers to cohorts with tumors induced with KRas, Tet-off, and TRE -Akt viruses. All of the untreated mice succumbed to disease within ∼50 days of age; the median survival for untreated mice was 28 days. Two-thirds (14/21) of the mice in the doxycycline treated group remained in remission while on doxycycline for the 90 day treatment period while the remaining one-third of the mice (7/21) demonstrated disease progression. Three of these mice died within the first 24 hours, which was prior to complete suppression of Akt expression; therefore, one fifth of the mice (4/21) became resistant while on doxycycline treatment.
Figure 3.

Kaplan-Meier percent survival curves. A. Nestin-TVA mice were injected with KRas, Tet-off, and TRE-Akt viruses at birth. The presence of tumors was confirmed by MRI at weaning (21 days of age). Tumor bearing mice were then randomized into two cohorts one of which was provided with a dietary supplement of doxycycline for 90 days beginning at weaning n = 21 (dashed line) while the untreated cohort remained on a standard diet n = 24 (solid line). Censored survival data was analyzed using a log-rank test of the Kaplan-Meier estimate of survival (P = 1.6 × 10-8). B. Tumors were induced by injection of viruses containing TRE-Akt, KRas, and Tet-off (solid line) or KRas and Akt (dashed line); n = 12 for both groups. The KRas and Akt cohort was administered doxycycline through the diet for the duration of the experiment whereas the TRE-Akt, KRas, and Tet-off cohort remained on a standard diet. Censored survival data was then compared using a log-rank test of the Kaplan-Meier estimate of survival. No difference in survival was detected between doxycycline treated KRas + Akt tumor-bearing mice (dashed line) and untreated TRE-Akt, KRas, and Tet-off tumor-bearing mice (solid line); P = 0.977. Only tumor-bearing mice were included in these analyses and the arrows indicate the start of doxycycline administration for the treated groups (day 21). C. Tumors were induced by injection of viruses containing KRas, Tet-off, and TRE-Akt. The presence of tumors was confirmed by MRI at weaning (21 days of age). Tumor- bearing mice were then provided with a diet containing doxycycline for 90 days at which point doxycycline treatment was withdrawn and the mice were monitored by MRI for disease recurrence. The solid line indicates the duration of doxycycline treatment.
Doxycycline withdrawal permits Akt expression and the majority of the mice relapse
Complete responses were observed in 67% of the mice treated with doxycycline for 90 days. To determine whether Akt suppression induced durable responses in these mice, the mice were monitored for relapse after doxycycline withdrawal. Consistent with the presence of residual microscopic disease, nine of these mice relapsed following doxycycline withdrawal and re-expression of Akt. Five mice remain asymptomatic four months after doxycycline withdrawal with no evidence of disease as assessed by MRI (Figure 3C).
Analysis of tumor growth and regression by MRI
MRI was used to monitor tumor growth or regression at multiple time points throughout the study (Figure 4). Images from a mouse that demonstrated a complete remission are shown in Figure 4A. While some edema remained, no tumor was detectable immediately following 90 days of doxycycline treatment (111 days of age) or four months after doxycycline treatment ended (238 days of age). In contrast, MRI of a resistant mouse showed increased edema and incomplete tumor regression after 16 days of doxycycline treatment (37 days of age) (Figure 4B). This mouse was sacrificed at 57 days of age after 36 days of doxycycline treatment due to disease progression. Mice that had completed the 90 day treatment period were further monitored for disease relapse by MRI after doxycycline withdrawal. Figure 4C shows a mouse with no detectable tumor by MRI immediately following 90 days of doxycycline treatment (111 days of age) but demonstrated re-growth of the tumor 107 days after doxycycline treatment ended (218 days of age).
Figure 4.

Assessment of tumor growth and regression by MRI. Images were acquired on a 7T Bruker MRI using T1-weighted multi-spin-multi-echo (MSME) or axial T2-weighted TurboRARE sequences in the presence of gadolinium-based contrast agent pre-treatment (21 days of age) and at the indicated ages during or after doxycycline treatment. A. MRI data from a representative mouse demonstrating the presence of a tumor pre-treatment, absence of a tumor following 90 days of doxycycline treatment, and continued remission at 238 days of age (127 days after doxycycline withdrawal). B. Representative MRI data of a resistant tumor before and after 16 and 36 days of doxycycline treatment. The numbers in the lower left corner of each image indicate the age of the mouse at the time of the scan. Arrows are pointing to the tumor in each T1 image. C. MRI data from a representative tumor-bearing mouse demonstrating absence of a tumor after 90 days of doxycycline treatment followed by recurrence 107 days later.
Akt expression is required for the maintenance of glioblastoma in vivo
Representative brain sections from both untreated and doxycyline treated tumors are shown in Figure 5A-E. Histologically, the majority of the primary tumors resembled high-grade invasive GBM (WHO Grade IV). The tumors were polymorphic, possessed a diffuse growth pattern, and contained several areas of pseu-dopalisading necrosis that were well-demarcated from the viable tumor. In contrast, the tumors that recurred during doxycycline treatment were relatively monomorphic, did not contain necrosis, and despite their small size were associated with pronounced edema leading to macrocephaly. This may have been in part due to their extension into the area of the choroid plexus. Akt expression was visualized by immunohistochemical staining of the tumors with antibodies to the HA epitope tag on virally delivered Akt. Immunohistochemical analysis of brain tissue in untreated mice confirmed Akt expression in the tumor cells but not in the normal surrounding tissue (Figure 5F). After 24 hours of doxycycline treatment, a few tumor cells remained (Figure 5B) and Akt expression was still visible in these cells (Figure 5G). At later time points, tumor cells were not detectable in mice on doxycycline treatment, which strongly suggests that Akt expression was suppressed as expected. After a latency period of one month, tumor progression was observed in ∼20% of the treated mice while still on doxycycline (Figure 3A). Low level Akt expression was detected in the tumor sections from these mice suggesting that escape from doxycycline regulation may be one mechanism of resistance (Figure 5H). This highlights the importance of Akt expression for tumor growth in this context. Tumors arising after doxycycline withdrawal were generally less invasive but otherwise histologically indistinguishable from the primary tumors (Figure 5D and E) and demonstrated high levels of AKT expression (Figure 5I and J).
Figure 5.
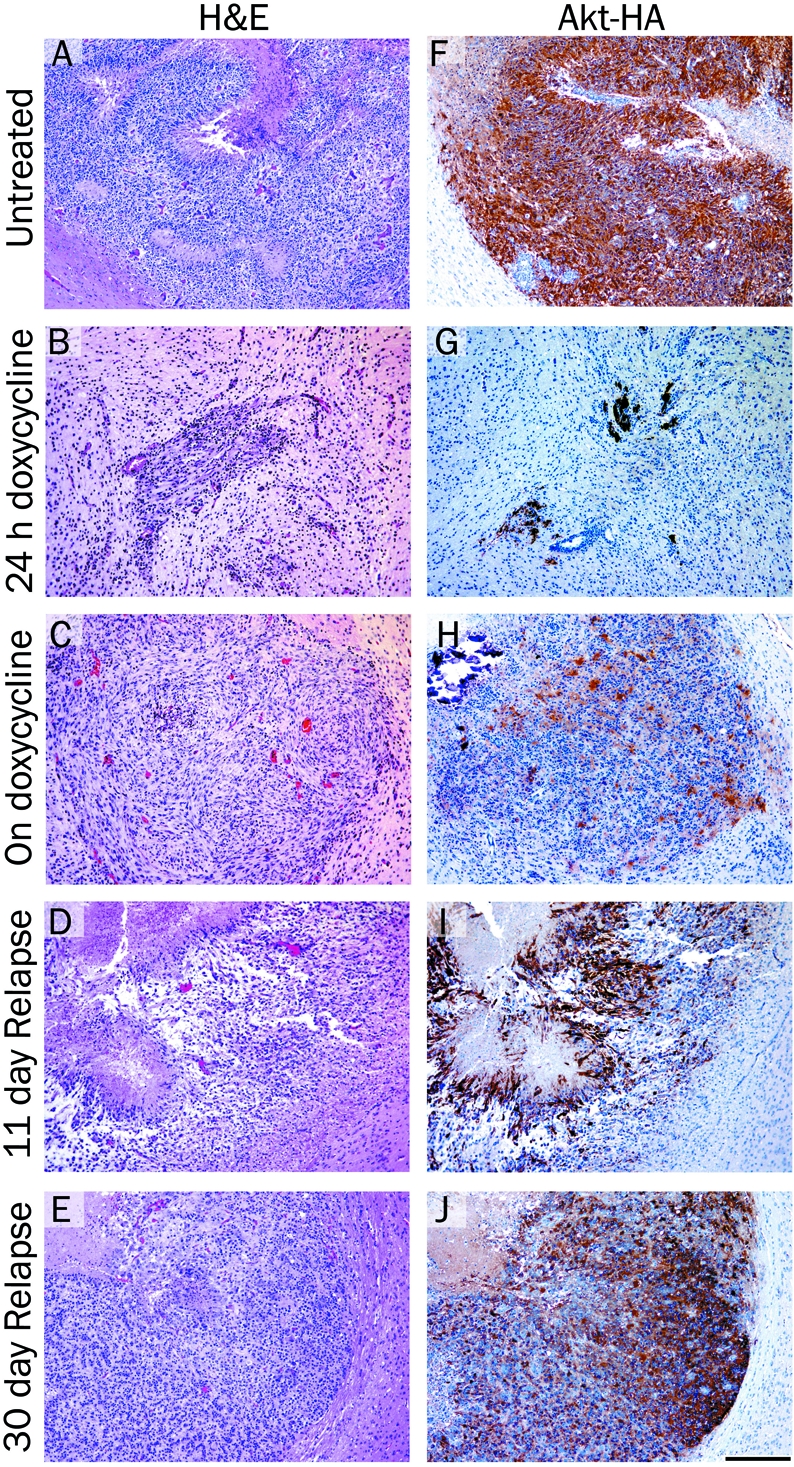
Histological analysis of brain tumors arising in Nestin-TVA mice injected with viruses expressing KRas, Tet-off and TRE-Akt. H&E stained 5 μm brain histology sections from: A. a typical glioma in the brain of an untreated 24 days old mouse; B. a tumor from a mouse treated with doxycycline for 24 hours; C. a resistant tumor from a 57 day old mouse treated with doxycycline for 36 days (corresponds to the MRI shown in Figure 4C); D. a relapsed tumor from a 122 day old mouse 11 days after doxycycline withdrawal; E. a relapsed tumor from a 141 day old mouse 30 days after doxycycline withdrawal. F-J. IHC for the HA epi-tope tag on virally delivered Akt and hema-toxylin counterstain corresponding to the tumors shown in (A-E). Scale bar represents 200 μm.
Inhibition of Akt expression results in decreased proliferation and increased apoptosis
Assessment of cellular proliferation using IHC for the proliferation marker Ki67 demonstrated that doxycycline treatment for 24 hours led to a reduction in proliferating cells compared with tumor cells from untreated mice (compare Figure 6A and B). H&E stained sections are shown for structural reference (Figure 6C and D). Since the tumors from doxycycline-treated mice showed signs of regression, TdT-mediated dUTP nick end labeling (TUNEL) staining was performed to determine if apoptotic cells could be detected within the tumor. TUNEL staining demonstrated a marked increase in the number of apoptotic cells in the tumors treated with doxycycline for 24 hours compared with tumor cells from untreated mice (compare Figure 6E and F). Consequently, Akt inhibition leads to a reduction in proliferation and an increase in apoptosis.
Figure 6.

Inhibition of Akt expression results in decreased proliferation and increased apoptosis. Tumors were induced by injection of KRas, Tet-off and TRE-Akt viruses in the brains of Nestin-TVA mice, which were left untreated (A, C and E) or were treated with doxycycline for 24 hours (B, D and F). (A-B) IHC for the proliferation marker Ki67. (C-D) Representative H&E stained 5 μm brain histology sections. (E-F) TUNEL staining to assess apoptosis. Scale bar represents 100 μm.
Discussion
We previously demonstrated the importance of Ras signaling in the maintenance of KRas and Akt-induced tumors in vivo; inhibition of KRas expression resulted in apoptotic tumor regression and significantly increased survival [9]. In the context of KRas inhibition, none of the tumors developed resistance to doxycycline treatment; however, in the context of Akt inhibition ∼20% of the tumors became refractory to Akt inhibition after a latency period. Since low levels of virally delivered Akt expression was observed in the tumors arising in mice treated with doxycycline, it is likely that a sub-population of tumor cells escaped from doxycycline regulation to repopulate the tumor. Notably, the tumors grew back in the same region of the brain that had previously harbored an Akt-initiated tumor. This strongly suggests that these resistant tumors represent true recurrences rather than de novo tumor formation in the presence of doxycycline.
When doxycycline treatment was withdrawn after treatment for 90 days, tumors were not detectable in any of the mice by MRI yet 64% of the mice experienced disease relapse. Our findings suggest that a significant percentage of mice in which gliomas have regressed beyond detection by MRI following administration of doxycycline harbor residual neoplastic disease and that surviving tumor cells remain dormant for a long period of time in the absence of Akt expression. The mechanism(s) behind this tumor cell dormancy clearly warrant further study as a greater understanding may provide an insight into novel mechanisms for glioma therapy. Interestingly, in the context of KRas inhibition all of the mice eventually succumbed to recurrent tumors following doxycycline withdrawal [9]. In this study, 36% of the mice remain alive with no evidence of disease greater than 4 months after doxycycline withdrawal. This difference may be due to the length of doxycycline treatment as the longest duration of treatment in the KRas study was 45 days compared with 90 days in this study. It is likely that a longer duration of KRas inhibition may produce more durable responses.
The cancer stem cell paradigm proposes that glioblastoma is driven by a component of tumor-initiating cells that retain stem cell-like properties and are primarily responsible for tumor maintenance. These cancer stem-like cells undergo divisions that allow the generation of more stem-like cells, which give rise to differentiated progeny allowing tumor growth [21]. It is also hypothesized that they are more resistant to conventional therapeutic approaches including radiation and chemotherapy [22, 23]. Both AKT and MAPK inhibition have recently been shown to induce differentiation in these stem-like cells [24]. It is therefore possible that prolonged AKT inhibition leads to apoptosis of tumor cells reliant on AKT mediated survival signaling. It also may lead to reduced potency and/ or differentiation of the cancer stem-like cell population; however, unlike hematopoietic cells, mature astrocytes can de-differentiate into neural stem cells [25]; thus the long and variable delay seen in the tumors that relapse could be related to a de-differentiation event transforming a senescent cell into a pluripotent cancer stem-like cell. Thus, inhibiting both the PI3K/Akt and MAPK/Ras pathways may prove to be a more effective combination treatment for glioma. We have recently demonstrated such an approach in vitro [2]; prolonged and combined PI3K/MAPK inhibition synergizes and leads to significant apoptosis in both mouse and human glioma cells. In addition, reducing the number of cancer stem-like cells may improve the efficacy of standard treatment regimens for GBM. In conclusion, our data suggest that a percentage of the cells capable of re-establishing the tumor survived apoptotic cell death following the loss of Akt expression upon doxycycline treatment. Future work will focus on determining effective therapeutic combinations that more effectively eliminate the remaining progenitor cell population to prevent relapse.
Acknowledgments
We thank the Nevada Cancer Institute mouse, imaging, and histology cores for assistance. We also thank Dr. Joseph Khoury for providing pathological analysis of tumors. The histology core is supported by NIH Grant Number P20 RR-016464 from the INBRE Program of the National Center for Research Resources. This work was supported by the Nevada Cancer Institute and RSG-06-198-01-TBE from the American Cancer Society.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson JP, Vanbrocklin MW, Lastwika KJ, McKinney AJ, Brandner S, Holmen SL. Activated MEK cooperates with Ink4a/Arf loss or Akt activation to induce gliomas in vivo. Oncogene. doi: 10.1038/onc.2010.513. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 4.Riemenschneider MJ, Jeuken JW, Wesseling P, Reifenberger G. Molecular diagnostics of gliomas: state of the art. Acta Neuropathol. 2010;120:567–584. doi: 10.1007/s00401-010-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondo Y, Hollingsworth EF, Kondo S. Molecular targeting for malignant gliomas (Review) Int J Oncol. 2004;24:1101–1109. [PubMed] [Google Scholar]
- 6.Guha A, Feldkamp MM, Lau N, Boss G, Pawson A. Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene. 1997;15:2755–2765. doi: 10.1038/sj.onc.1201455. [DOI] [PubMed] [Google Scholar]
- 7.Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 8.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 9.Holmen SL, Williams BO. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005;65:8250–8255. doi: 10.1158/0008-5472.CAN-05-1173. [DOI] [PubMed] [Google Scholar]
- 10.Mizoguchi M, Nutt CL, Mohapatra G, Louis DN. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol. 2004;14:372–377. doi: 10.1111/j.1750-3639.2004.tb00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol. 2005;17:150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006;66:5618–5623. doi: 10.1158/0008-5472.CAN-06-0364. [DOI] [PubMed] [Google Scholar]
- 13.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 14.Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS. Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics. 2009;6:500–512. doi: 10.1016/j.nurt.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell -cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bromberg-White JL, Webb CP, Patacsil VS, Miranti CK, Williams BO, Holmen SL. Delivery of short hairpin RNA sequences by using a replication-competent avian retroviral vector. J Virol. 2004;78:4914–4916. doi: 10.1128/JVI.78.9.4914-4916.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer-Klein J, Givol I, Barsov EV, Whitcomb JM, VanBrocklin M, Foster DN, Federspiel MJ, Hughes SH. The EV-O-derived cell line DF-1 supports the efficient replication of avian leu-kosis-sarcoma viruses and vectors. Virology. 1998;248:305–311. doi: 10.1006/viro.1998.9291. [DOI] [PubMed] [Google Scholar]
- 18.Holmen SL, Salter DW, Payne WS, Dodgson JB, Hughes SH, Federspiel MJ. Soluble forms of the subgroup A avian leukosis virus [ALV(A)] receptor Tva significantly inhibit ALV(A) infection in vitro and in vivo. J Virol. 1999;73:10051–10060. doi: 10.1128/jvi.73.12.10051-10060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith EJ, Fadly A, Okazaki W. An enzyme-linked immunosorbent assay for detecting avian leukosis-sarcoma viruses. Avian Dis. 1979;23:698–707. [PubMed] [Google Scholar]
- 20.Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park DM, Rich JN. Biology of glioma cancer stem cells. Mol Cells. 2009;28:7–12. doi: 10.1007/s10059-009-0111-2. [DOI] [PubMed] [Google Scholar]
- 22.Blazek ER, Foutch JL, Maki G. Daoy medulloblastoma cells that express CD133 are radio-resistant relative to CD133- cells, and the CD133+ sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys. 2007;67:1–5. doi: 10.1016/j.ijrobp.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 23.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sunayama J, Matsuda K, Sato A, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A, Kitanaka C. Crosstalk Between the PI3K/mTOR and MEK/ERK Pathways Involved in the Maintenance of Self-Renewal and Tumorigenicity of Glioblastoma Stem-Like Cells. Stem Cells. 28:1930–1939. doi: 10.1002/stem.521. [DOI] [PubMed] [Google Scholar]
- 25.Mao XG, Xue XY, Zhang X. The potential of the braplasticity implications for de-differentiation of mature astrocytes. Cell Mol Neurobiol. 2009;29:1105–1108. doi: 10.1007/s10571-009-9416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]