

Abstract
The nature of DNA has captivated scientists for more than fifty years. The discovery of the double-helix model of DNA by Watson and Crick in 1953 not only established the primary structure of DNA, but also provided the mechanism behind DNA function. Since then, researchers have continued to further the understanding of DNA structure and its pivotal role in transcription. The demonstration of DNA secondary structure formation has allowed for the proposal that the dynamics of DNA itself can function to modulate transcription. This review presents evidence that DNA can exist in a dynamic equilibrium between duplex and secondary conformations. In addition, data demonstrating that intracellular proteins as well as small molecules can shift this equilibrium in either direction to alter gene transcription will be discussed, with a focus on the modulation of proto-oncogene expression.
Introduction
For over half a century DNA has been widely recognized for its central biological role in heredity, but how does DNA accomplish this intricate function so vital for life? The discovery of the molecular double-helix model of DNA by Watson and Crick provided an explanation for the mode of heredity [1]. The Watson-Crick model established that DNA exists in a form, termed “B-form DNA,” where two complementary strands of DNA consist of hydrogen-bonded nucleotide base-pairs: adenine with thymine and guanine with cytosine [1]. Thus the complementarity of the strands and the distinct base sequence allow for the synthesis and reconstruction of new, identical DNA double helices. In addition, the specific nucleotide sequence serves as a template for transcription that ultimately enables the production of peptides required for cellular function. While the general acceptance is that the majority of DNA within cells consists in the B-form, DNA also has the potential to exist in other conformations.
The continued interest in the structure of DNA and advances in molecular structural techniques have uncovered that the structure of DNA is not limited to the B-form conformation, but extends to both non-B-form and non-helical secondary structures. Alternative helical structures such as the A- and Z-forms of DNA have been shown to exist under different experimental conditions [2–5]. While the presence of these DNA forms within cells remains controversial, the formation of Z-form, or left-handed, DNA has been observed under conditions of negative supercoiling [6]. Interestingly, this is not the only non-B-DNA structure to form in response to negative supercoiling, indicating the important influence of topology on DNA conformation. The formation of unconventional DNA secondary structures, including cruciforms [7], H-DNA (triplexes) [8], G-quadruplexes, and i-motifs [9], has demonstrated that negative supercoiling may induce and stabilize less energetically favorable structures. But how can supercoiling alter DNA structure?
The transition of DNA from the typical B-form to a non-B-form requires the local unwinding of the double helix, which occurs to relieve the stress of negative supercoiling [10]. In nature negative supercoiling can result from nuclear processes such as transcription, replication, recombination, and repair and produce significant torsional stress [11,12]. Specifically, the transient high degree of supercoiling induced during transcription occurs as the RNA polymerase (RNAP) complex moves along the transcriptionally active segment of DNA (Figure 1). In order to accommodate the RNAP and the nascent RNA, DNA downstream of the transcription complex becomes positively supercoiled, or overwound, whereas DNA upstream is negatively supercoiled, or underwound [13]. Although DNA topoisomerases have been implicated as the predominant mechanism for relieving DNA torsional stress, these enzymes may inadequately alleviate this stress in response to the significant demand produced by transcription [14–16]. Rather, local melting of double-stranded DNA upstream of transcriptionally active promoters has been shown to be a consequence of torsional stress [10,17]. This local unwinding results in an open, single-stranded DNA conformation that presumably is sufficient for the formation of DNA secondary structures.
Figure 1.

Model of the transcriptionally induced supercoiling that occurs upstream and downstream of the RNAP. Adapted from Kouzine and Levens [12].
So if DNA supercoiling can induce the formation of a variety of non-B-DNA secondary structures, then what determines whether the DNA will adopt either a cruciform or a G-quadruplex conformation, for example? In addition to topological constraints, these DNA secondary structures exhibit sequence dependency. For example, cruciform structures arise from palindromic sequences within double-stranded DNA [18]. Sequences with contiguous guanine or cytosine runs have the potential to form G-quadruplex or i-motif secondary structures, respectively [19–22]. Specifically, the G-quadruplex building block, the G-tetrad, is formed from a sequence consisting of at least four contiguous runs of two or more guanines (Figure 2A). The four G-tetrad guanines interact through Hoogsteen base-pairing, and the tetrad is stabilized by the presence of a monovalent cation, as shown in Figure 2B [19]. The i-motif structure specified by the complementary C-rich sequence comprises two parallel duplexes with intercalated hemiprotonated cytosine+ –cytosine base pairs (Figure 2C) [20–22].
Figure 2.
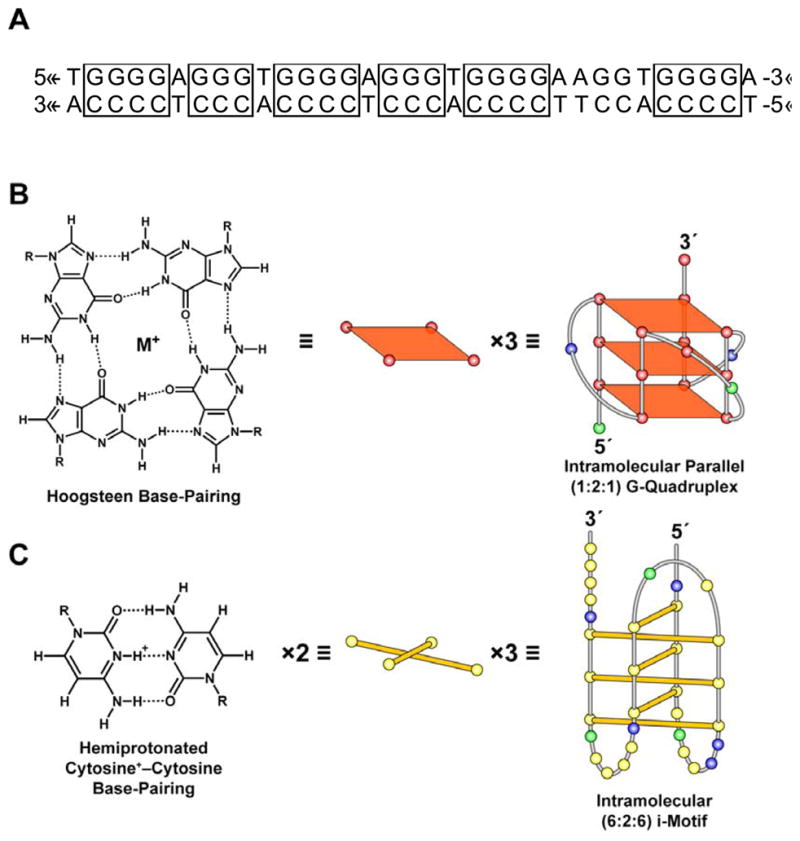
The c-myc GC-rich promoter sequence capable of adopting G-quadruplex and i-motif structures is provided in panel A. The building blocks of these secondary structures are guanine–guanine and cytosine+ –cytosine base pairings that give rise to the G-quadruplex (B) and i-motif (C) secondary structures. The previously proposed c-myc [9] G-quadruplex and i-motif structures formed under conditions of negative supercoiling serve as examples, with the yellow, green, red, and blue circles representing the nucleobases cytosine, adenine, guanine, and thymine, respectively.
The dynamic topology of DNA then poses the question, what is the biological significance of DNA interconverting between duplex and secondary conformations? We, along with many others, have postulated that the ability of duplex DNA to adopt G-quadruplex and i-motif secondary conformations serves as a molecular switch to modulate transcription [23]. If the G-quadruplex and i-motif structures play a role in transcriptional regulation, then, similar to other cis-regulatory elements, these secondary structure-forming sequences must be in close proximity to the transcriptional start site (TSS). In addition, for these structures to be effective transcription modulators they would also need to compete with duplex DNA, be recognized by proteins for remodeling, be targeted by small molecules to induce changes in transcription, and exhibit diversity and complexity among promoter regions.
This review will describe the prevalence, diversity, and complexity of G-quadruplex and i-motif structures as well as observed protein and small molecule interactions to provide evidence for the role of DNA secondary structures in transcriptional regulation. While genome-wide investigations into the occurrences of G-quadruplex-forming sequences will be discussed, emphasis will be placed on the demonstration of stable secondary structure formation within proto-oncogene promoter regions.
G-quadruplex and i-motif forming sequences are extensively found in close proximity to transcriptional start sites
If the G-quadruplex and the i-motif play a role in transcriptional regulation, then one would expect to find sequences capable of forming these structures near the TSS within the promoter regions of various genes. Indeed, recent genomic analysis has revealed that of promoter sequences within 1 kb upstream of the TSS, 43% of genes have the potential to form at least one G-quadruplex structure [24]. In consideration with genomic wide-analyses, which have detected G-quadruplex-forming sequences throughout the genome at ~376,000 sites [25,26], the extensive incidence within close proximity to the TSS demonstrates an enrichment for promoter G-quadruplexes. This enrichment has been postulated to reflect an evolutionary selection for the location of secondary structures and thereby supports the biological role of these structures in transcription [24]. Consistent with quadruplex elements functioning as transcription regulatory units, conservation of potential G-quadruplex-forming sequences near the TSS has been observed across related species. A comparative genome-wide analysis among human, chimpanzee, mouse and rat not only demonstrated at least a 40% prevalence of promoter regions (within ± 1 kb of the TSS) to potentially adopt G-quadruplex conformations, but that over 700 orthologous promoter regions consisted of conserved DNA secondary structure–forming sequences [27]. While the probing of the human genome for potential sites of G-quadruplex formation provides information on the prevalence and localization of these sequences, these studies do not demonstrate the precise capability of the identified regions to form stable G-quadruplex structures. The defined sequence utilized in these searches consisted of four runs of at least three guanines spaced by one to seven bases (G3+N1–7G3+ N1–7G3+ N1–7G3+). To date, the majority of stable G-quadruplex conformations favor smaller loop sizes [28–31]; however, the loop N1–7 search parameters may allow for identification of potential i-motif-forming sequences on the complementary strand since stable i-motif structures tend to exhibit larger loop sizes [9, 32]. Although genome investigations have focused on the detection of G-quadruplex motifs, presumably there is an equal prevalence of potential i-motif-forming sequences in the complementary strand. If there is a significant incidence of these DNA secondary structure–forming sequences within promoter regions, then which types of genes contain these elements? Furthermore, can these potential sequences actually form stable structures?
A review of the gene ontology codes of the promoters that were found to contain G-quadruplex motifs revealed a significant occurrence within genes involved in transcription factor and kinase activity, development, and neurogenesis [24,33,34]. Conversely, olfaction, G-protein signaling, immune response, nucleic acid binding, and protein biosynthesis–related genes were significantly less likely to contain G-quadruplex motifs [24,33,34]. In addition, significantly more proto-oncogenes (69%) were shown to consist of putative G-quadruplex motifs than tumor suppressors [33,34]. Consistent with this prediction, GC-rich elements within several proto-oncogenes and a tumor suppressor gene have been shown to adopt stable G-quadruplex and i-motif conformations. The c-myc proto-oncogene promoter has been extensively studied and has led to the identification of G-quadruplex motifs within the VEGF [35,36], c-kit [37–40], c-myb [41], PDGF-A [42], RET [43], KRAS [44], hif-1α [45], bcl-2 [46–49], hTERT [50], and Rb [51] promoter regions. The wild-type GC-rich element located −142 to −115 upstream of the c-myc P1 promoter has the potential to form an intramolecular parallel G-quadruplex structures with loop sizes of 1:2:1 [9, 52–57] and an intramolecular i-motif structure [58], which, under conditions of negative supercoiling, adopts a 6:2:6 loop conformation [9]. To date, C-rich elements within the VEGF [36], RET [43], bcl-2 [59], and Rb [51] oncogene promoter regions have also been demonstrated to form i-motif structures. The occurrence of DNA secondary structure motifs within oncogene promoter regions in close proximity to the TSS suggests that these alternative DNA conformations serve a regulatory function in transcription that requires resolution of duplex DNA.
DNA secondary structures can compete with duplex DNA
Cellular DNA is mostly present in the B-form, which seemingly presents an obstacle to secondary structure formation that requires single-stranded DNA. For secondary structures to form within B-DNA, local unwinding or melting of the duplex must occur to provide regions of single-stranded DNA [9] and thus create an equilibrium in which there is active transition between duplex and single-stranded DNA (Figure 3). If secondary structure conformations efficiently compete with duplex DNA, then there would be a favorable shift in the equilibrium toward G-quadruplex/i-motif formation. So the question remains, can DNA secondary conformations efficiently compete with duplex DNA? Various investigations into this issue have provided evidence of an equilibrium between duplex and secondary DNA conformations that is context- or environment-dependent. For example, G- and C-rich strands of the telomeric sequence were demonstrated to efficiently compete with duplex DNA under conditions that facilitate the formation of the G-quadruplex and/or the i-motif (pH ≤ 4.9 or high temperatures of 50–64 °C) [60]. Within the pH range of 5.3 to 7.0, high temperatures (64–70 °C) result in the melting of the duplex DNA and conversion to the G-quadruplex. The unfolding of the G-quadruplex and consequent transition back to duplex DNA is favored at temperatures below 64 °C. In contrast, when pH is less than 5.0, low to moderate temperatures (below 55 °C) favor G-quadruplex formation over duplex [60]. Similarly, Li et al. [61] and Kumar et al. [62] have also confirmed the dependency of lower pH and specific cation species on the efficiency of DNA secondary structure and duplex competition in the telomeric or thrombin-binding aptamer sequences, respectively. However, a closer examination of the unfolding and folding rates of the telomeric [63] and c-myc [64] G-quadruplexes in the presence of duplex DNA demonstrated that at low, equivalent molar strand concentrations, duplex conformation is significantly less favorable than G-quadruplex formation, even at neutral pH. While the low, equivalent molar strand concentration achieves more physiological conditions compared to previous studies, these studies did not take into consideration the G-quadruplex/i-motif-flanking sequences or negative supercoiling that would normally be present within the genome.
Figure 3.

The proposed model for the dynamic equilibrium of DNA topology induced by negative supercoiling. Negative supercoiling within duplex DNA (A) induces the local unwinding (B), which facilitates the transition from duplex DNA (A) to single-stranded (C) to G-quadruplex and i-motif DNA secondary formation (D).
As mentioned previously, transcriptionally active genes generate both negative and positive supercoiled sections of DNA, with the former acting as the driving force for unwinding DNA. Recent studies demonstrated a significant increase in negative supercoiling with a concurrent increase in transcriptional activity [10,17]. More importantly, melting of a duplex region 1.2 kb upstream of the inducible c-myc promoter resulted from the negative supercoiling stress [17]. This may be sufficient to provide single-stranded DNA within the GC-rich element capable of adopting G-quadruplex/i-motif conformations (Figure 3). Recently, our group utilized the inherent negative supercoiling within plasmid DNA as a model system to test this hypothesis for the c-myc GC-rich promoter region. Indeed, the stress induced from negative supercoiling is sufficient to cause unwinding of duplex DNA and allow for G-quadruplex and i-motif formation [9]. Unexpectedly, the i-motif conformation not only formed under neutral pH, but also remained present regardless of G-quadruplex formation [9]. In contrast, single-stranded C-rich DNA requires the pH-dependent protonation of cytosines for i-motif formation [58]. Therefore, studies using plasmid DNA provide a more physiologically relevant system in which to observe the dynamics of DNA topology as consequences of negative supercoiling with respect to the formation of DNA secondary structures. It is important to note that flanking sequence length alone is not sufficient to increase G-quadruplex/i-motif competition with duplex DNA, as was shown within the c-kit promoter region [65], further demonstrating the importance of the in vivo dynamics of DNA topology resulting from negative supercoiling. In addition, intracellular proteins may promote and stabilize the G-quadruplex/i-motif conformation and shift the equilibrium away from duplex.
Protein recognition and remodeling of G-quadruplex-forming regions
Protein recognition of G-quadruplex structures with consequent biological effect provides further evidence for the in vivo existence of DNA secondary structures and their role as cis-regulatory elements. Several proteins, including nucleases and helicases, from different organisms have been identified to bind selectively and with high affinity to G-quadruplex structures [66]. These proteins may promote the cleavage, folding/unfolding, or stabilization/destabilization of G-quadruplex structures [66]. More recently, proteins within the heterogeneous ribonucleoprotein (hnRNP) and NM23 families were shown to facilitate the unfolding of the KRAS [66] and c-myc [68,69] G-quadruplexes, respectively, while nucleolin confers stabilization on the c-myc G-quadruplex [70]. Initial affinity column studies using pancreatic nuclear extract and the KRAS G-quadruplex structure revealed three proteins, poly[ADP-ribose] polymerase 1 (PARP-1); ATP-dependent DNA helicase 2, subunit 1 (Ku70); and hnRNP A1, that bound specifically to DNA in the secondary conformation [71]. Further analysis of hnRNP A1 and its derivative Up1 demonstrated not only recognition of the KRAS G-quadruplex, but also a destabilization of the structure that resulted in transition to duplex DNA [67]. Likewise, NM23-H2 facilitates the unfolding of the c-myc G-quadruplex and sequesters the G-rich strand in single-stranded form [68,69]. The expression of NM23-H2 was demonstrated to be inversely correlated with c-myc expression in CaCo-2, HT29, and HCT116 colon cancer cell lines. Furthermore, c-myc expression was significantly increased subsequent to knock-down of NM23-H2 in HeLa cells [68]. Conversely, nucleolin substantially decreases c-myc transcription through recognition and stabilization of the G-quadruplex [70]. This study also demonstrated that the recognition and binding of nucleolin was highly preferential to the c-myc G-quadruplex and, to a lesser extent, the VEGF, RET, and PDGF-A structures. Nucleolin displayed low affinity for other G-quadruplexes, such as those formed in the bcl-2 and c-myb promoters, indicating that nucleolin may specifically recognize parallel G-quadruplex conformations [70]. Collectively, these findings establish protein involvement in the remodeling of DNA topology that allows for the switch from G-quadruplex/i-motif to single-stranded or double-stranded conformations (Figure 4). In the instance of c-myc, the presence of NM23-H2 facilitates unwinding of the G-quadruplex, allowing for transition to duplex DNA, and double-stranded binding transcription factors, such as Sp-1, can bind and initiate transcription (Figure 4). Alternatively, transcription becomes repressed in the presence of nucleolin, which stabilizes the G-quadruplex and shifts the equilibrium toward secondary structure formation (Figure 4). This remains consistent with the ability of DNA secondary structures to actively participate in regulation of transcription. As key players in the transcriptional process, targeting of DNA secondary structures with small molecules should also modulate transcription.
Figure 4.

A proposed model of c-myc transcriptional regulation that involves the resolution of the G-quadruplex by NM23-H2 for duplex DNA formation and subsequent transcriptional activation by Sp1. The binding of hnRNP K and CNBP to single-stranded DNA induced by negative supercoiling also leads to c-myc transcription activation. The stabilization of the G-quadruplex by nucleolin results in negative regulation of c-myc transcription.
Small molecules bind to G-quadruplexes and modulate transcription
The stabilization of DNA secondary structures using small molecules has been well established for both telomeric [72] and c-myc [53,73–75] G-quadruplexes and has led to studies investigating the biological consequences of targeting DNA secondary structures. TMPyP4, a cationic porphyrin known to be a selective G-quadruplex stabilizer [76], stabilizes the c-myc G-quadruplex in a dose-dependent manner and inhibits c-myc promoter transcriptional activity in the Ramos Burkitt’s lymphoma cell line, but not in the CA46 cell line, which lacks one of the alleles of the GC-rich promoter element [53]. Similarly, in a c-kit gastrointestinal stromal tumor cells model, Gunaratnam et al. have demonstrated that a naphthalene diimide derivative stabilizes the c-kit G-quadruplex structures and significantly decreases c-kit transcription [77]. In contrast, a destabilizing G-quadruplex ligand would presumably result in an increase in gene transcription. Accordingly, triarylpyridine ligands have been shown to resolve the c-kit G-quadruplexes and increase c-kit gene expression in the HGC-27 gastric carcinoma cell line [78]. So how does the stabilization/destabilization of DNA in the secondary structure conformation alter gene expression?
Perhaps the formation of DNA secondary structures within promoter regions acts as steric block to the transcriptional machinery. However, for DNA secondary structures to actively participate in transcriptional regulation, either as silencer or activator elements, G-quadruplex/i-motif-forming sequences must localize in close proximity to the TSS (as previously discussed) and also occur in regions of transcription factor and protein recognition sites. The formation of DNA secondary structures would then “mask” these binding sites and prevent their function in transcription activation or repression. It is also likely that there are proteins that recognize the structures themselves to achieve similar biological effects. Recent investigations are beginning to elucidate the mechanism by which G-quadruplex/i-motif elements act as switches to modulate transcription and support this hypothesis. The stabilization of the KRAS and c-myc G-quadruplexes through the use of TMPyP4 hindered the ability of the previously mentioned proteins, hnRNP A1 [67] and NM23-H2 [68,69], to unwind the secondary structures. In these examples small molecule targeting of DNA secondary structures disrupts DNA–protein interactions and results in promoter silencing, suggesting that these particular DNA secondary structures act to negatively regulate their respective promoters.
It is equally likely, however, that in different promoters DNA secondary structures may act as positive regulators. For example, we postulate that the formation of DNA secondary structures within the bcl-2 promoter region results in displacement of WT-1, a known repressor of bcl-2 transcription [79], and activates transcription. While studies involving the stabilization of the bcl-2 G-quadruplex with TMPyP4 and other small molecules have been performed [46,80,81], the effects on transcription have not been assessed. It is important to note that similar molecules, if not the same molecules, have been used to target multiple G-quadruplex structures; however, specificity in molecular targeting will require taking advantage of structural differences among DNA secondary structures. Recent development of a G-quadruplex-specific antibody, hf2, has demonstrated not only selectivity for the c-kit2 G-quadruplex structure over other G-quadruplexes, but also a 3000-fold lower affinity for duplex DNA [82]. Furthermore, interactions of these G-quadruplex-specific antibodies with promoter G-quadruplex-forming sequences throughout the genome have been shown to either increase or decrease transcription of the adjacent gene [83].
Diversity and complexity in G-quadruplex and i-motif structures
As with any cis-regulatory element or targetable group of molecules, conserved features essential to structure and overall function must exist in the presence of diversity and complexity to allow for molecular exploitation. DNA secondary structures exhibit conservation in the core sequences that gives rise to either G-tetrads [28,36] or cytosine+ –cytosine hemiprotonated base pairs [36] for G-quadruplex or i-motif assembly, respectively. In addition, certain features within the loop regions are also conserved to accommodate specific structural features in the G-quadruplex, such as the double-chain reversal. However, the loop regions as well as the number of G-tetrads and cytosine+ –cytosine hemiprotonated base pairs that define a given DNA secondary structure provide a source of diversity and contribute to the complexity of these structures. For a comprehensive discussion on the diversity and complexity of G-quadruplexes readers are directed to a recently published review article [28]. Most commonly, the promoter DNA G-quadruplexes are formed from three G-tetrads; however, PDGF-A (four G-tetrads), Rb (two G-tetrads), and c-Myb (two G-tetrads) deviate from this trend (Table 1). Comparison of the three-tetrad G-quadruplex-forming sequences within the c-myc, KRAS, bcl-2, VEGF, hTERT, hif-1α, RET, and c-kit and promoter regions reveals the conserved single-nucleotide 3′-end loop region (Figure 5, Table 1) [28]. While this nucleotide may vary depending on the promoter, it yields the same structural feature, the double-chain reversal. The stability of a given G-quadruplex structure has been attributed mostly to the incorporation of the double-chain reversal [30,31,43,45,46,55]. With the exception of bcl-2, this double-chain reversal also includes the 5′-end loop. The central loop region of these three-tetrad structures exhibits the greatest flexibility in terms of length and provides for the diverse folding patterns. In addition, further diversity is shown in both the c-Myb [41] and hTERT [50] G-quadruplexes, which consist of two tandem structures separated by a linker sequence.
Table 1.
Proto-oncogene promoter G-quadruplex-forming sequences
| N1–3 |
N2–26 |
N1–2 |
Topology
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| c-myc: | 5′- | GGG | T | GGG | TA | GGG | T | GGG | -3′ | Parallel |
| RET: | 5′- | GGG | C | GGG | GCG | GGG | C | GGG | -3′ | Parallel |
| VEGF: | 5′- | GGG | C | GGG | CCGG | GGG | C | GGG | -3′ | Parallel |
| c-kit-2: | 5′- | GGG | C | GGG | CGCGA | GGG | A | GGG | -3′ | Parallel |
| hif-1α: | 5′- | GGG | A | GGG | GAGAGG | GGG | C | GGG | -3′ | Parallel |
| Mouse KRAS: | 5′- | GGG | A | GGG | AAGGAGGGGA | GGG | C | GGG | -3′ | Parallel |
| hTERT (1:3:1): | 5′- | GGG | A | GGG | GCT | GGG | A | GGG | -3′ | Parallel |
| hTERT (3:26:1): | 5′- | GGG | GCT | GGG | C35–G60 | GGG | C | GGG | -3′ | Parallel/Antiparallel |
| bcl-2: | 5′- | GGG | CGC | GGG | AGGAAGG | GGG | C | GGG | -3′ | Parallel/Antiparallel |
| PDGF-A: | 5′- | GGGG | GG | GGGG | GGGCG | GGGG | CG | GGGG | -3′ | Parallel |
| Rb: | 5′- | GG | GG | GG | TTTT | GG | CG | GG | -3′ | Antiparallel |
| c-Myb: | 5′- | GG | A | GG | A | GG | A | GG | -3′ | Parallel |
Figure 5.

The proposed folding patterns of the G-quadruplex structures formed within the c-myc, VEGF, c-kit-1, c-kit-2, RET, KRAS, hif-1α, bcl-2, and hTERT promoter regions. Structures were either determined by NMR (1) or biophysical methods (2), such as circular dichroism and DMS footprinting or both.
In contrast, promoter i-motif-forming sequences do not display as much similarity as that observed with G-quadruplexes. For example, i-motifs formed in the VEGF, c-myc, and bcl-2 promoters consist of six intercalated cytosine+ –cytosine hemiprotonated base pairs, while RET and Rb i-motifs are formed from five and four base pairs, respectively (Table 2). The loop sizes for the i-motifs also vary significantly with the c-myc [9] and bcl-2 [32] i-motifs having the largest lateral loops, with six and eight nucleotides, and the VEGF [36], RET [43], and Rb [51] i-motifs having maximum lateral loop sizes of three nucleotides. The differences in loop sizes of both i-motif and G-quadruplex structures may offer one structurally diverse feature for small-molecular targeting.
Table 2.
Proto-oncogene promoter i-motif-forming sequences
| N2–8 |
N2–5 |
N2–7 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| VEGF: | 5′- | CCC | GC | CCC | CGG | CCC | G | CCC | -3′ |
| c-myc: | 5′- | CCC | CACCTT | CCC | CA | CCC | TCCCCA | CCC | -3′ |
| RET: | 5′- | CCC | GC | CC | CGC | CCC | GC | CC | -3′ |
| bcl-2: | 5′- | CCC | GCTCCCGC | CCCC | TTTCT | CCC | GCGCCCG | CCCC | -3′ |
| Rb: | 5′- | CC | GC | CC | AAAA | CC | CC | CC | -3′ |
Summary
The rapid increase in recognition of the prevalence of DNA secondary structures within the genome, demonstration of stable structure formation within several promoter regions, and their ability to compete with duplex DNA is consistent with a cis-regulatory function. The close proximity of secondary structure–forming sequences to the TSS allows these regions to be included in the local unwinding that occurs during transcription. This creates a dynamic equilibrium between DNA secondary structures and duplex DNA that can be shifted toward G-quadruplex/i-motif formation upon binding of stabilizing intracellular proteins or small molecules. Likewise, the equilibrium may be shifted in the other direction in the presence of proteins or small molecules that facilitate the unwinding of the DNA secondary structures. More importantly, shifts in this equilibrium result in modulation of transcription that may be exploited for the development of novel anticancer agents.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institutes of Health (CA95060, CA122952, CA009213, GM085585) and the Leukemia & Lymphoma Society (6225-08). We thank David Bishop for his significant contribution in the preparation and editing of the final version of the text and figures displayed in the article.
References
- 1.Watson JD, Crick FHC. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 2.Franklin RE, Gosling R. Nature. 1953;171:740–741. doi: 10.1038/171740a0. [DOI] [PubMed] [Google Scholar]
- 3.Pohl FM, Jovin TM. J Mol Biol. 1972;67:375–396. doi: 10.1016/0022-2836(72)90457-3. [DOI] [PubMed] [Google Scholar]
- 4.Wang AH-J, Quigley GJ, Kolpak FJ, Crawford JL, van Boom JH, van der Marel G, Rich A. Nature. 1979;282:680–686. doi: 10.1038/282680a0. [DOI] [PubMed] [Google Scholar]
- 5.Drew H, Takano T, Tanaka T, Itakura K, Dickerson RE. Nature. 1980;286:567–573. doi: 10.1038/286567a0. [DOI] [PubMed] [Google Scholar]
- 6.Sinden RR, Kochel TJ. Biochemistry. 1987;26:1343–1350. doi: 10.1021/bi00379a021. [DOI] [PubMed] [Google Scholar]
- 7.Lilley DM, Hallam JR. J Mol Biol. 1984;180:179–200. doi: 10.1016/0022-2836(84)90436-4. [DOI] [PubMed] [Google Scholar]
- 8.Htun H, Dahlberg JE. Science. 1989;243:1571–1576. doi: 10.1126/science.2648571. [DOI] [PubMed] [Google Scholar]
- 9.Sun D, Hurley LH. J Med Chem. 2009;52:2863–2874. doi: 10.1021/jm900055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kouzine F, Liu J, Sanford S, Chung HJ, Levens D. Nat Struct Mol Biol. 2004;11:1092–1100. doi: 10.1038/nsmb848. [DOI] [PubMed] [Google Scholar]
- 11.Sinden RR. DNA Structure and Function. Academic Press; San Diego: 1994. [Google Scholar]
- 12.Kouzine F, Levens D. Front Biosci. 2007;12:4409–4423. doi: 10.2741/2398. [DOI] [PubMed] [Google Scholar]
- 13.Liu LF, Wang JC. Proc Natl Acad Sci USA. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Droge P. EMBO J. 1996;15:581–589. [PMC free article] [PubMed] [Google Scholar]
- 15.Koster DA, Croquette V, Dekker C, Shuman SN, Dekker H. Nature. 2005;434:671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- 16.Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kouzine F, Sanford S, Elisha-Feil Z, Levens D. Nat Struct Mol Biol. 2008;15:146–154. doi: 10.1038/nsmb.1372. [DOI] [PubMed] [Google Scholar]
- 18.Lilley DM. Proc Natl Acad Sci USA. 1980;77:6468–6472. doi: 10.1073/pnas.77.11.6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williamson JR, Raghuraman MK, Cech TR. Cell. 1989;59:871–880. doi: 10.1016/0092-8674(89)90610-7. [DOI] [PubMed] [Google Scholar]
- 20.Leroy JL, Gehring K, Kettani A, Gueron M. Biochemistry. 1993;32:6019–6031. doi: 10.1021/bi00074a013. [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Cai L, Zhang X, Rich A. Biochemistry. 1994;33:13540–13546. doi: 10.1021/bi00250a005. [DOI] [PubMed] [Google Scholar]
- 22.Gueron M, Leroy JL. Curr Opin Struct Biol. 2000;10:326–331. doi: 10.1016/s0959-440x(00)00091-9. [DOI] [PubMed] [Google Scholar]
- 23.Brooks TA, Hurley LH. Nat Rev Cancer. 2009 doi: 10.1038/nrc2733. in press. [DOI] [PubMed] [Google Scholar]
- 24.Huppert JL, Balasubramanian S. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huppert JL, Balasubramanian S. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todd AK, Johnston M, Neidle S. Nucleic Acids Res. 2005;33:2901–2907. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma A, Halder K, Halder R, Yadav VK, Rawal P, Thakur RK, Mohd F, Sharma A, Chowdhury S. J Med Chem. 2008;51:5641–5649. doi: 10.1021/jm800448a. [DOI] [PubMed] [Google Scholar]
- 28.Qin Y, Hurley LH. Biochimie. 2008;90:1149–1171. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hazel P, Huppert J, Blasubramanian S, Neidle S. J Am Chem Soc. 2004;126:16405–16415. doi: 10.1021/ja045154j. [DOI] [PubMed] [Google Scholar]
- 30.Risitano A, Fox KR. Nucleic Acids Res. 2004;32:2598–2606. doi: 10.1093/nar/gkh598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bugaut A, Balasubramanian S. Biochemistry. 2008;47:689–697. doi: 10.1021/bi701873c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kendrick S, Akiyama Y, Hecht SM, Hurley LH. J Am Chem Soc. 2009 doi: 10.1021/ja9076292. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eddy J, Maizels N. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huppert JL. Biochimie. 2008;90:1140–1148. doi: 10.1016/j.biochi.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Sun D, Guo K, Rusche JJ, Hurley LH. Nucleic Acids Res. 2005;33:6070–6080. doi: 10.1093/nar/gki917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo K, Gokhale V, Hurley LH, Sun D. Nucleic Acids Res. 2008;36:4598–4608. doi: 10.1093/nar/gkn380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rankin S, Reszka AP, Huppert J, Zloh M, Parkinson GN, Todd AK, Ladame S, Balasubramanian S, Neidle S. J Am Chem Soc. 2005;127:10584–10589. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Blasubramanian S. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phan AT, Kuryavyi V, Burge S, Neidle S, Patel DJ. J Am Chem Soc. 2007;129:4386–4392. doi: 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirude PS, Okumus B, Ying L, Ha T, Balasubramanian S. J Am Chem Soc. 2007;129:7484–7485. doi: 10.1021/ja070497d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palumbo S, Memmott RM, Uribe DJ, Krotova-Khan Y, Hurley LH, Ebbinghaus SW. Nucleic Acids Res. 2008;36:1755–1769. doi: 10.1093/nar/gkm1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin Y, Rezler EM, Gokhale V, Sun D, Hurley LH. Nucleic Acids Res. 2007;25:7698–7713. doi: 10.1093/nar/gkm538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo K, Pourpak A, Beetz-Rogers K, Gokhale V, Sun D, Hurley LH. J Am Chem Soc. 2007;129:10220–10228. doi: 10.1021/ja072185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cogoi S, Xodo LE. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Armond R, Wood S, Sun D, Hurley LH, Ebbinghaus SW. Biochemistry. 2005;44:16341–16350. doi: 10.1021/bi051618u. [DOI] [PubMed] [Google Scholar]
- 46.Dexheimer T, Sun D, Hurley LH. J Am Chem Soc. 2006;128:5404–5415. doi: 10.1021/ja0563861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai J, Dexheimer TS, Chen D, Carver M, Ambrus A, Jones RA, Yang D. J Am Chem Soc. 2006;128:1096–1098. doi: 10.1021/ja055636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai J, Chen D, Jones RA, Hurley LH, Yang D. Nucleic Acids Res. 2006;34:5133–5144. doi: 10.1093/nar/gkl610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Liu Y, Lin S, Yuan G. Chem Eur J. 2009;15:2445–2452. doi: 10.1002/chem.200801922. [DOI] [PubMed] [Google Scholar]
- 50.Palombo SL, Ebbinghaus SW, Hurley LH. J Am Chem Soc. 2009;131:10878–10891. doi: 10.1021/ja902281d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Y, Sugiyama H. Nucleic Acids Res. 2006;34:949–954. doi: 10.1093/nar/gkj485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simonsson T, Sjoback R. J Biol Chem. 1999;274:17379–17383. doi: 10.1074/jbc.274.24.17379. [DOI] [PubMed] [Google Scholar]
- 53.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Proc Natl Acad Sci USA. 2002;66:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phan AT, Modi YS, Patel DJ. J Am Chem Soc. 2004;126:8710–8706. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seenisamy J, Rezler EM, Powell TJ, Tye D, Gokhale V, Joshi CS, Siddiqui-Jain A, Hurley LH. J Am Chem Soc. 2004;126:8702–8709. doi: 10.1021/ja040022b. [DOI] [PubMed] [Google Scholar]
- 56.Ambrus A, Chen D, Dai J, Jones RA, Yang D. Biochemistry. 2005;44:2048–2058. doi: 10.1021/bi048242p. [DOI] [PubMed] [Google Scholar]
- 57.Yang D, Hurley LH. Nucleosides Nucleotides Nucleic Acids. 2006;25:951–968. doi: 10.1080/15257770600809913. [DOI] [PubMed] [Google Scholar]
- 58.Simonsson T, Pribylova M, Vorlickova M. Biochem Biophys Res Commun. 2000;278:158–166. doi: 10.1006/bbrc.2000.3783. [DOI] [PubMed] [Google Scholar]
- 59.Khan N, Avino A, Tauler R, Gonzalez C, Eritja R, Gargallo R. Biochimie. 2007;89:1562–1572. doi: 10.1016/j.biochi.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 60.Phan AT, Mergny JL. Nucleic Acids Res. 2002;30:4618–4625. doi: 10.1093/nar/gkf597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li W, Wu P, Ohmichia T, Sugimotoa N. FEBS Lett. 2002;526:77–81. doi: 10.1016/s0014-5793(02)03118-6. [DOI] [PubMed] [Google Scholar]
- 62.Kumar N, Maiti S. Biochem Biophys Res Commun. 2004;319:759–767. doi: 10.1016/j.bbrc.2004.05.052. [DOI] [PubMed] [Google Scholar]
- 63.Zhao Y, Kan ZY, Zeng ZX, Hao YH, Chen H, Tan Z. J Am Chem Soc. 2004;126:13255–13264. doi: 10.1021/ja048398c. [DOI] [PubMed] [Google Scholar]
- 64.Halder K, Chowdhury S. Nucleic Acids Res. 2005;33:4466–4474. doi: 10.1093/nar/gki750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arora A, Nair DR, Maiti S. FEBS Lett. 2009;276:3628–3640. doi: 10.1111/j.1742-4658.2009.07082.x. [DOI] [PubMed] [Google Scholar]
- 66.Fry M. Front Biosci. 2007;12:4336–4351. doi: 10.2741/2391. [DOI] [PubMed] [Google Scholar]
- 67.Paramasivam M, Membrino A, Cogoi S, Fukuda H, Nakagama H, Xodo LE. Nucleic Acids Res. 2009;37:2841–2853. doi: 10.1093/nar/gkp138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dexheimer TS, Carey SS, Zuohe S, Gokhale VM, Hu X, Murata LB, Maes EM, Weichsel A, Sun D, Meuillet EJ, Montfort WR, Hurley LH. Mol Cancer Ther. 2009;8:1363–1377. doi: 10.1158/1535-7163.MCT-08-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thakur RK, Kumar P, Halder K, Verma A, Kar A, Parent JL, Basundra R, Kumar A, Chowdhury S. Nucleic Acids Res. 2009;37:172–183. doi: 10.1093/nar/gkn919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gonzalez V, Guo K, Hurley L, Sun D. J Biol Chem. 2009;284:23622–23635. doi: 10.1074/jbc.M109.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cogoi S, Paramasivam M, Spolaore B, Xodo LE. Nucleic Acids Res. 2008;36:3765–3780. doi: 10.1093/nar/gkn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cuesta J, Read MA, Neidle S. Mini Rev Med Chem. 2003;3:11–21. doi: 10.2174/1389557033405502. [DOI] [PubMed] [Google Scholar]
- 73.Phan AT, Kuryavyi V, Gaw HY, Patel DJ. Nat Chem Biol. 2005;1:167–173. doi: 10.1038/nchembio723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seenisamy J, Bashyam S, Gokhale V, Vankayalapati H, Sun D, Siddiqui-Jain A, Streiner N, Shin-ya K, White E, Wilson WD, Hurley LH. J Am Chem Soc. 2005;127:2944–2959. doi: 10.1021/ja0444482. [DOI] [PubMed] [Google Scholar]
- 75.Hurley LH, Von Hoff DD, Siddiqui-Jain A, Yang D. Semin Oncol. 2006;33:498–512. doi: 10.1053/j.seminoncol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 76.Han H, Langley DR, Rangan A, Hurley LH. J Am Chem Soc. 2001;123:8902–8913. doi: 10.1021/ja002179j. [DOI] [PubMed] [Google Scholar]
- 77.Gunaratnam M, Swank S, Haider SM, Galesa K, Reszka AP, Beltran M, Cuenca F, Fletcher JA, Neidle S. J Med Chem. 2009;52:3774–3783. doi: 10.1021/jm900424a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waller ZAE, Sewitz SA, Hsu STD, Balasubramanian S. J Am Chem Soc. 2009;131:12628–12633. doi: 10.1021/ja901892u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heckman C, Mochon E, Arcinas M, Boxer LM. J Biol Chem. 1997;272:19609–19614. doi: 10.1074/jbc.272.31.19609. [DOI] [PubMed] [Google Scholar]
- 80.Li H, Liu Y, Lin S, Yuan G. Chem Eur J. 2009;15:2445–2452. doi: 10.1002/chem.200801922. [DOI] [PubMed] [Google Scholar]
- 81.del Toro M, Buck P, Avino A, Jaumot J, Gonzalez C, Eritja R, Gargallo R. Biochimie. 2009;91:894–902. doi: 10.1016/j.biochi.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 82.Fernando H, Rodriquez R, Balasubramanian S. Biochemistry. 2008;47:9365–9371. doi: 10.1021/bi800983u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fernando H, Sewitz S, Darot J, Tavare S, Huppert JL, Balasubramanian S. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp740. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.